



Goldilocks Dilemma: LPS Works Both as the Initial Target and a Barrier for the Antimicrobial Action of Cationic AMPs on E. coli

 , , , ,
, , , ,
Abstract
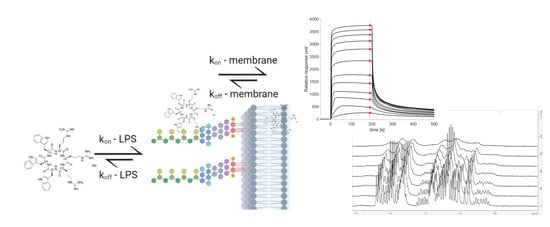
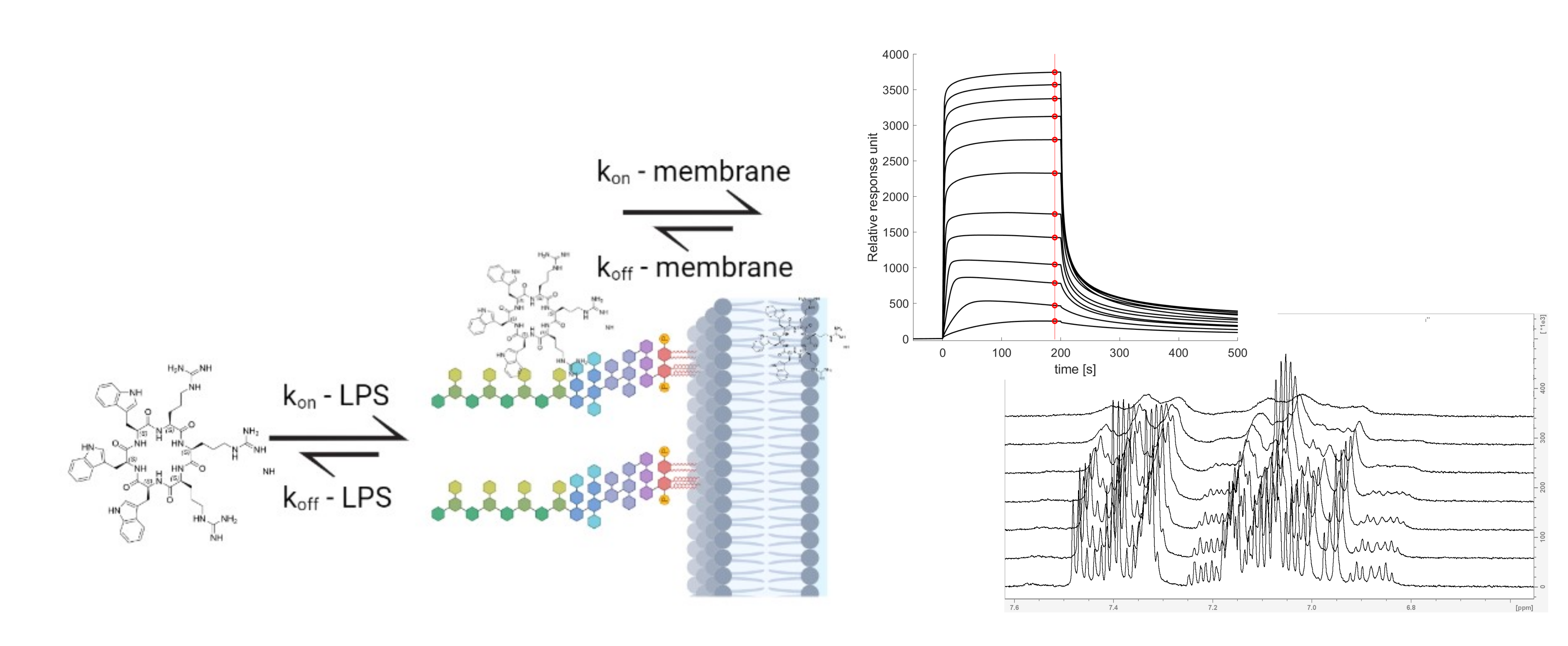
:
1. Introduction
2. Materials and Methods
2.1. Peptide Synthesis
2.2. MIC Testing
2.3. Liposomes Preparation
2.4. SPR
2.5. NMR Experiments
2.5.1. Live Cell NMR
2.5.2. Assignment
3. Results and Discussion
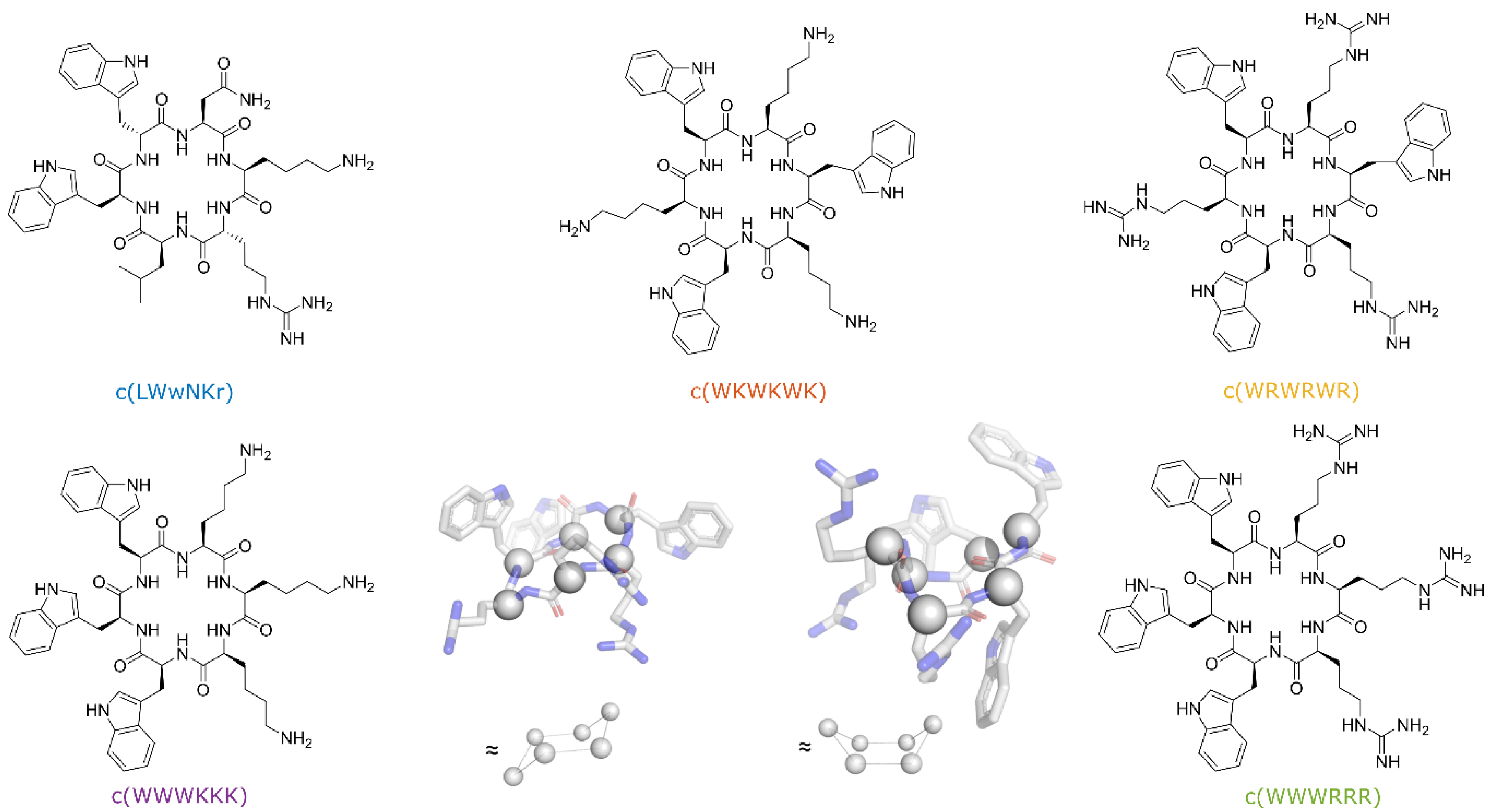
3.1. Cyclic Peptides with Balanced Lipophilic Bulk and Cationic Charge

3.2. Antimicrobial Activity of Peptides

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| AMP | MIC (µg/mL) | |||||
|---|---|---|---|---|---|---|
| P. aeruginosa ATCC 27853 | S. aureus ATCC 9144 | E. coli ATCC 25922 | E. coli CCUG 70662− | E. coli CCUG 70662+ | E. coli NR 698 | |
| Zeta potential | - | - | −21.3 ± 0.6 mV | −21.8 ± 0.8 mV | −14.3 ± 0.5 mV | −21.2 ± 0.6 mV |
| c(LWwNKr) | >250 | >250 | >250 | >250 | >250 | 32 |
| c(WKWKWK) | >250 | 128 | 64 | 64 | 64 | 4 |
| c(WRWRWR) | 64 | 32 | 32 | 16 | 32 | 2 |
| c(WWWKKK) | 32 | 32 | 8 | 4 | 4 | 2 |
| c(WWWRRR) | 16 | 4 | 8 | 8 | 8 | 4 |
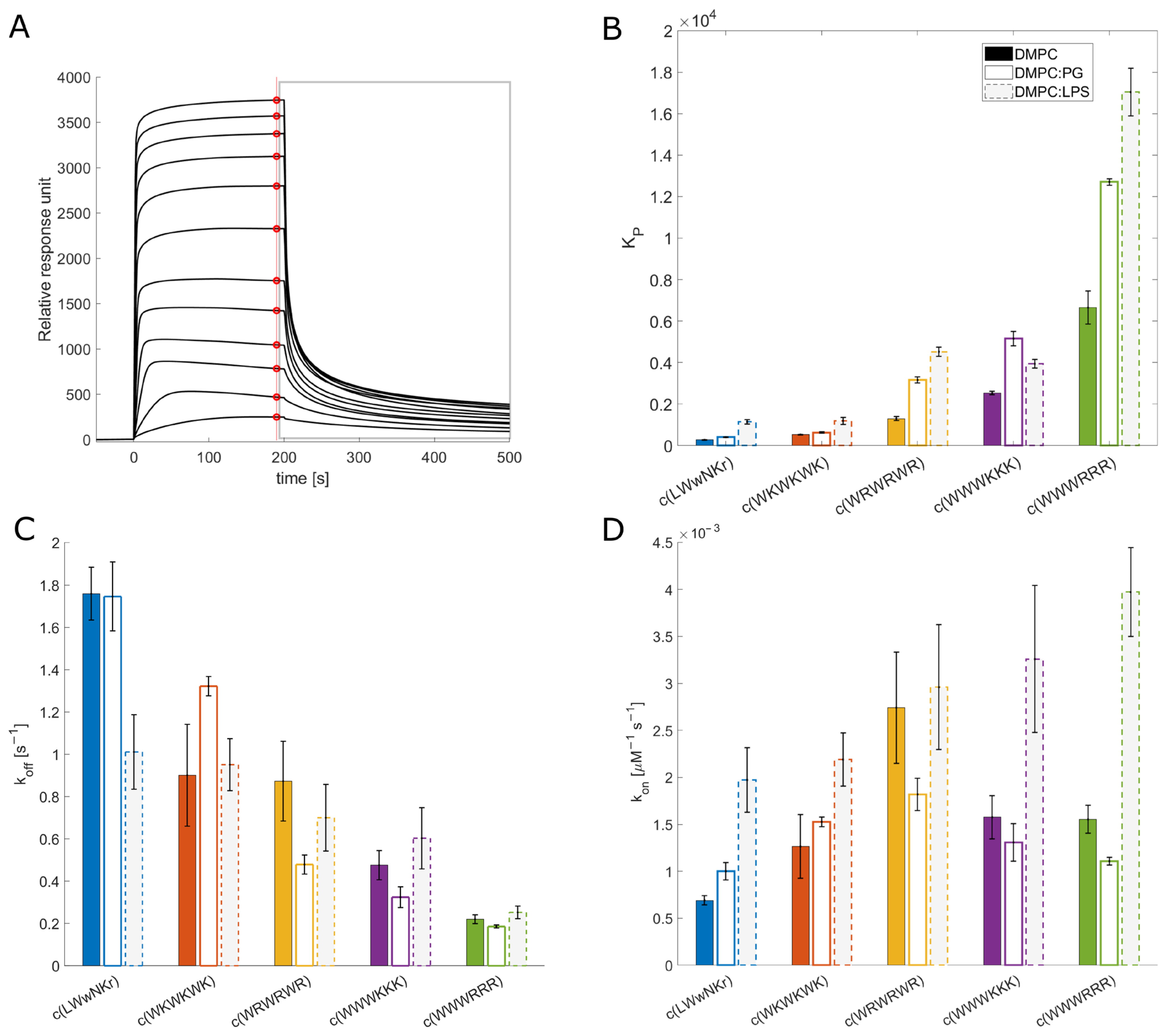
3.3. SPR and Choice of Lipid Models
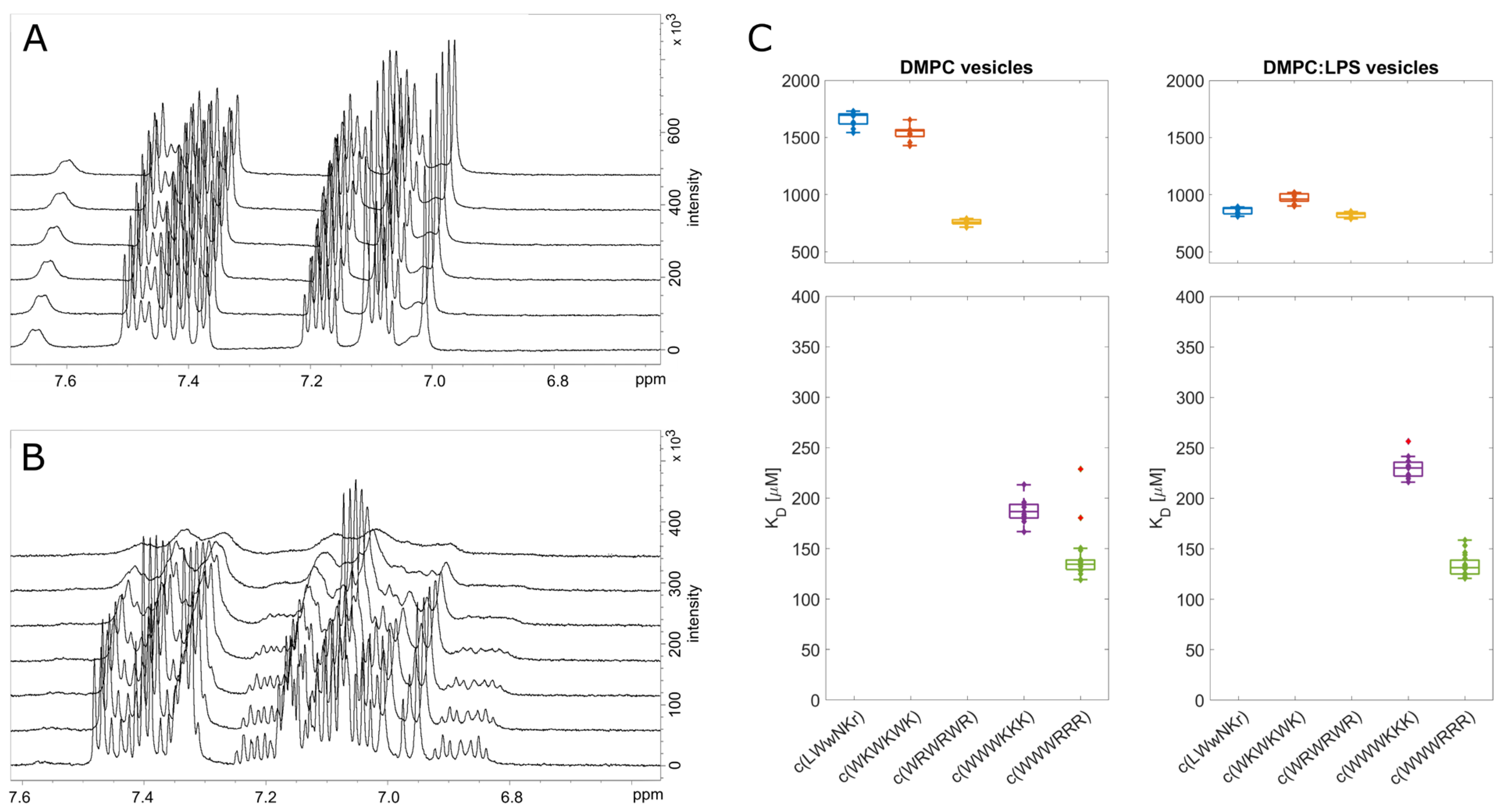
3.4. Antimicrobial Activity Broadly Correlates with KP for DMPC Liposomes
3.5. The Initial Peptide:Membrane Association Is Affected by LPS
3.6. NMR Line Shape Analysis Can Be Used to Probe Binding Strength of AMPs towards Lipid Liposomes
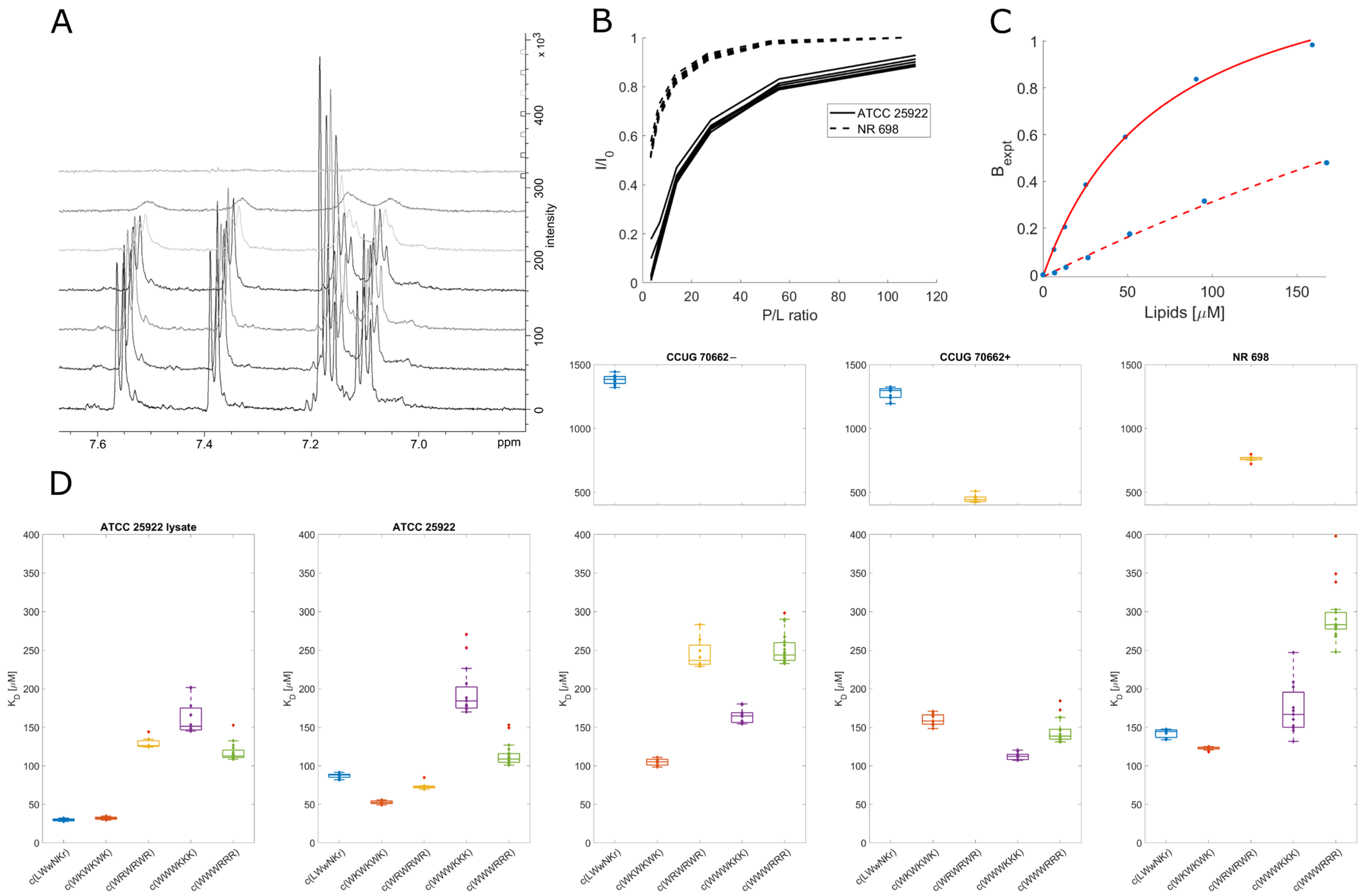
3.7. Live Cell NMR Highlights Strain Differences
3.8. Inactive Peptide c(LWwNKr) Binds Strongly to ATCC 25922 and NR 698
3.9. Active Peptides Are Less Affected by Changes in Membrane Composition
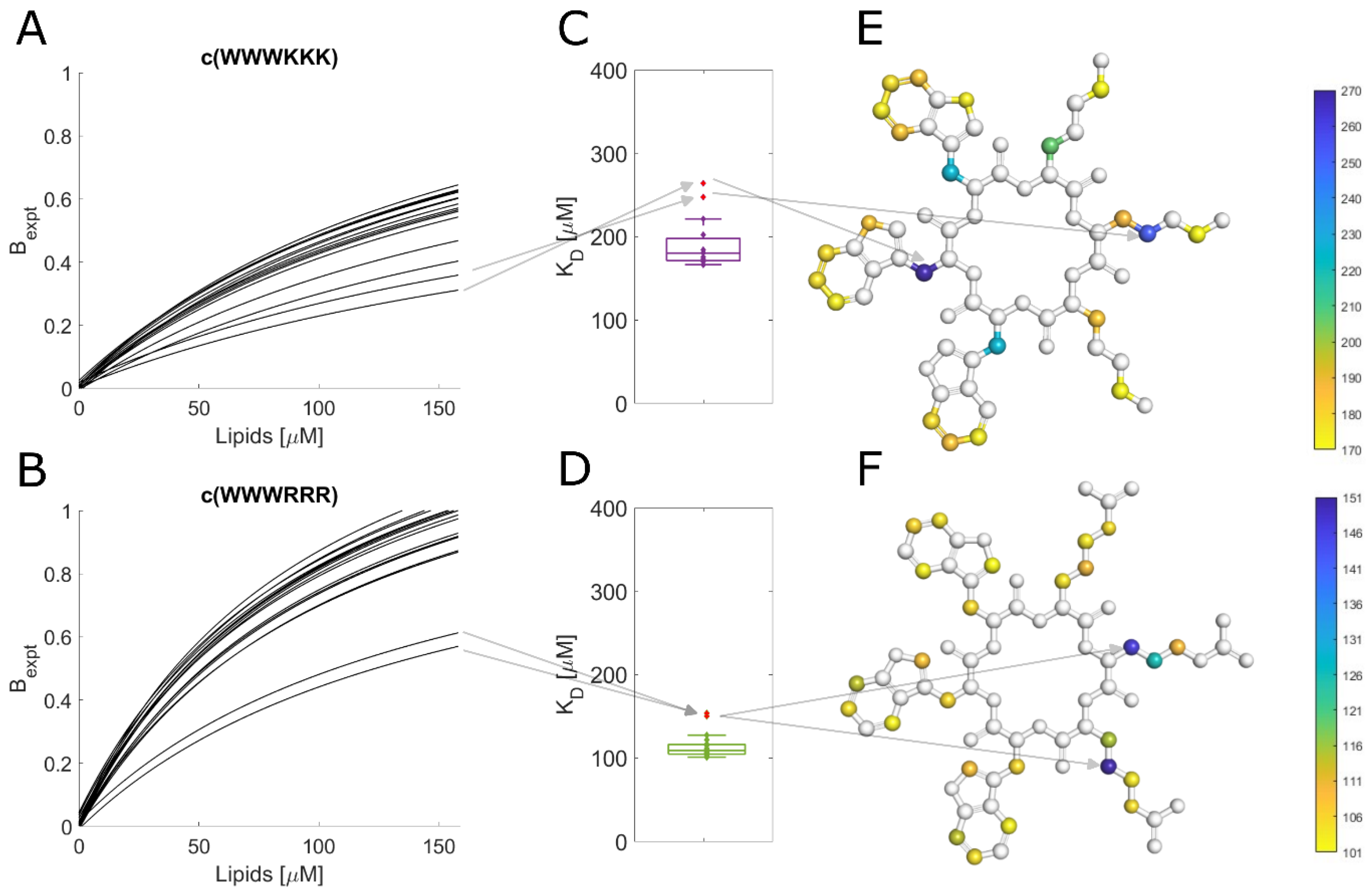
3.10. Different Binding Modes of Hexapeptides towards ATCC 25922
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Aslam, B.; Wang, W.; Arshad, M.I.; Khurshid, M.; Muzammil, S.; Rasool, M.H.; Nisar, M.A.; Alvi, R.F.; Aslam, M.A.; Qamar, M.U.; et al. Antibiotic Resistance: A Rundown of a Global Crisis. Infect. Drug Resist. 2018, 11, 1645–1658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hancock, R.E.W.; Sahl, H.G. Antimicrobial and Host-Defense Peptides as New Anti-Infective Therapeutic Strategies. Nat. Biotechnol. 2006, 24, 1551–1557. [Google Scholar] [CrossRef] [PubMed]
- Ageitos, J.M.; Sánchez-Pérez, A.; Calo-Mata, P.; Villa, T.G. Antimicrobial Peptides (AMPs): Ancient Compounds That Represent Novel Weapons in the Fight against Bacteria. Biochem. Pharmacol. 2017, 133, 117–138. [Google Scholar] [CrossRef]
- Zasloff, M. Antimicrobial Peptides of Multicellular Organisms. Nature 2002, 415, 389–395. [Google Scholar] [CrossRef]
- Chen, Y.; Mant, C.T.; Farmer, S.W.; Hancock, R.E.W.; Vasil, M.L.; Hodges, R.S. Rational Design of α-Helical Antimicrobial Peptides with Enhanced Activities and Specificity/Therapeutic Index. J. Biol. Chem. 2005, 280, 12316–12329. [Google Scholar] [CrossRef] [Green Version]
- Dijksteel, G.S.; Ulrich, M.M.W.; Middelkoop, E.; Boekema, B.K.H.L. Review: Lessons Learned from Clinical Trials Using Antimicrobial Peptides (AMPs). Front. Microbiol. 2021, 12, 616979. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Chen, H.; Song, Y.; Qin, Z.; Xu, L.; He, N.; Tan, Y.; Dessie, W. Advancements, Challenges and Future Perspectives on Peptide-Based Drugs: Focus on Antimicrobial Peptides. Eur. J. Pharm. Sci. 2023, 181, 106363. [Google Scholar] [CrossRef]
- Fjell, C.D.; Hiss, J.A.; Hancock, R.E.W.; Schneider, G. Designing Antimicrobial Peptides: Form Follows Function. Nat. Rev. Drug Discov. 2012, 11, 37–51. [Google Scholar] [CrossRef]
- Zhang, Q.Y.; Yan, Z.B.; Meng, Y.M.; Hong, X.Y.; Shao, G.; Ma, J.J.; Cheng, X.R.; Liu, J.; Kang, J.; Fu, C.Y. Antimicrobial Peptides: Mechanism of Action, Activity and Clinical Potential. Mil. Med. Res. 2021, 8, 48. [Google Scholar] [CrossRef]
- Wimley, W.C. Describing the Mechanism of Antimicrobial Peptide Action with the Interfacial Activity Model. ACS Chem. Biol. 2010, 5, 905–917. [Google Scholar] [CrossRef] [Green Version]
- Pirtskhalava, M.; Amstrong, A.A.; Grigolava, M.; Chubinidze, M.; Alimbarashvili, E.; Vishnepolsky, B.; Gabrielian, A.; Rosenthal, A.; Hurt, D.E.; Tartakovsky, M. DBAASP v3: Database of Antimicrobial/Cytotoxic Activity and Structure of Peptides as a Resource for Development of New Therapeutics. Nucleic Acids Res. 2021, 49, D288–D297. [Google Scholar] [CrossRef] [PubMed]
- Mishra, B.; Wang, G. The Importance of Amino Acid Composition in Natural Amps: An Evolutional, Structural, and Functional Perspective. Front. Immunol. 2012, 3, 31946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- You, Y.H.; Liu, H.Y.; Zhu, Y.Z.; Zheng, H. Rational Design of Stapled Antimicrobial Peptides. Amin. Acids 2023, 55, 421–442. [Google Scholar] [CrossRef] [PubMed]
- Clark, S.; Jowitt, T.A.; Harris, L.K.; Knight, C.G.; Dobson, C.B. The Lexicon of Antimicrobial Peptides: A Complete Set of Arginine and Tryptophan Sequences. Commun. Biol. 2021, 4, 605. [Google Scholar] [CrossRef]
- Cutrona, K.J.; Kaufman, B.A.; Figueroa, D.M.; Elmore, D.E. Role of Arginine and Lysine in the Antimicrobial Mechanism of Histone-Derived Antimicrobial Peptides. FEBS Lett. 2015, 589, 3915–3920. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Brady, A.; Young, A.; Rasimick, B.; Chen, K.; Zhou, C.; Kallenbach, N.R. Length Effects in Antimicrobial Peptides of the (RW)n Series. Antimicrob. Agents Chemother. 2007, 51, 597–603. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Vorobyov, I.; Allen, T.W. The Different Interactions of Lysine and Arginine Side Chains with Lipid Membranes. J. Phys. Chem. B 2013, 117, 11906–11920. [Google Scholar] [CrossRef]
- Chan, D.I.; Prenner, E.J.; Vogel, H.J. Tryptophan- and Arginine-Rich Antimicrobial Peptides: Structures and Mechanisms of Action. Biochim. Biophys. Acta Biomembr. 2006, 1758, 1184–1202. [Google Scholar] [CrossRef] [Green Version]
- Khemaissa, S.; Walrant, A.; Sagan, S. Tryptophan, More Than Just an Interfacial Amino Acid in the Membrane Activity of Cationic Cell-Penetrating and Antimicrobial Peptides. Q. Rev. Biophys. 2022, 55, e10. [Google Scholar] [CrossRef]
- Wang, G.; Li, X.; Wang, Z. APD3: The Antimicrobial Peptide Database as a Tool for Research and Education. Nucleic Acids Res. 2016, 44, D1087. [Google Scholar] [CrossRef] [Green Version]
- Papo, N.; Shai, Y. A Molecular Mechanism for Lipopolysaccharide Protection of Gram-Negative Bacteria from Antimicrobial Peptides. J. Biol. Chem. 2005, 280, 10378–10387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papo, N.; Oren, Z.; Pag, U.; Sahl, H.G.; Shai, Y. The Consequence of Sequence Alteration of an Amphipathic α-Helical Antimicrobial Peptide and Its Diastereomers. J. Biol. Chem. 2002, 277, 33913–33921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Needham, B.D.; Trent, M.S. Fortifying the Barrier: The Impact of Lipid A Remodelling on Bacterial Pathogenesis. Nat. Rev. Microbiol. 2013, 11, 467–481. [Google Scholar] [CrossRef] [PubMed]
- Southey, R. The Story of the Three Bears; W. NICOL: London, UK, 1839. [Google Scholar]
- Joo, H.S.; Fu, C.I.; Otto, M. Bacterial Strategies of Resistance to Antimicrobial Peptides. Philos. Trans. R. Soc. B Biol. Sci. 2016, 371, 20150292. [Google Scholar] [CrossRef] [Green Version]
- Eikås, K.D.R.; Krupová, M.; Kristoffersen, T.; Beerepoot, M.T.P.; Ruud, K. Can the Absolute Configuration of Cyclic Peptides Be Determined with Vibrational Circular Dichroism? Phys. Chem. Chem. Phys. 2023, 25, 14520–14529. [Google Scholar] [CrossRef]
- Clinical and Laboratory Standards Institute. Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically; Clinical and Laboratory Standards Institute: Berwyn, PA, USA, 2006; ISBN 1562386263. [Google Scholar]
- Torchilin, V.; Weissig, V. Liposomes: A Practical Approach; Oxford University Press: Oxford, UK, 2003; ISBN 0-19-963654-0. [Google Scholar]
- Palusińska-Szysz, M.; Zdybicka-Barabas, A.; Luchowski, R.; Reszczyńska, E.; Śmiałek, J.; Mak, P.; Gruszecki, W.I.; Cytryńska, M. Choline Supplementation Sensitizes Legionella Dumoffii to Galleria Mellonella Apolipophorin III. Int. J. Mol. Sci. 2020, 21, 5818. [Google Scholar] [CrossRef]
- Jakubec, M.; Bariås, E.; Furse, S.; Govasli, M.L.; George, V.; Turcu, D.; Iashchishyn, I.A.; Morozova-Roche, L.A.; Halskau, Ø. Cholesterol-containing Lipid Nanodiscs Promote an A-synuclein Binding Mode That Accelerates Oligomerization. FEBS J. 2020, 288, 1887–1905. [Google Scholar] [CrossRef]
- Jakubec, M. GitHub—Martin Jakubec. Available online: https://github.com/MarJakubec (accessed on 21 June 2023).
- Figueira, T.N.; Freire, J.M.; Cunha-Santos, C.; Heras, M.; Gonçalves, J.; Moscona, A.; Porotto, M.; Salomé Veiga, A.; Castanho, M.A.R.B. Quantitative Analysis of Molecular Partition towards Lipid Membranes Using Surface Plasmon Resonance. Sci. Rep. 2017, 7, 45647. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Yin, F.; Zhao, X.; Guo, Y.; Wang, W.; Wang, P.; Zhu, H.; Yin, Y.; Wang, X. Colistin Resistance Gene Mcr-1 Mediates Cell Permeability and Resistance to Hydrophobic Antibiotics. Front. Microbiol. 2020, 10, 3015. [Google Scholar] [CrossRef] [Green Version]
- Beal, J.; Farny, N.G.; Haddock-Angelli, T.; Selvarajah, V.; Baldwin, G.S.; Buckley-Taylor, R.; Gershater, M.; Kiga, D.; Marken, J.; Sanchania, V.; et al. Robust Estimation of Bacterial Cell Count from Optical Density. Commun. Biol. 2020, 3, 512. [Google Scholar] [CrossRef]
- Lee, S.; Bayley, H.; Lee, S.; Bayley, H. Reconstruction of the Gram-Negative Bacterial Outer-Membrane Bilayer. Small 2022, 18, 2200007. [Google Scholar] [CrossRef] [PubMed]
- Shortridge, M.D.; Hage, D.S.; Harbison, G.S.; Powers, R. Estimating Protein-Ligand Binding Affinity Using High-Throughput Screening by NMR. J. Comb. Chem. 2008, 10, 948–958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lau, Q.Y.; Ng, F.M.; Cheong, J.W.D.; Yap, Y.Y.A.; Tan, Y.Y.F.; Jureen, R.; Hill, J.; Chia, C.S.B. Discovery of an Ultra-Short Linear Antibacterial Tetrapeptide with Anti-MRSA Activity from a Structure-Activity Relationship Study. Eur. J. Med. Chem. 2015, 105, 138–144. [Google Scholar] [CrossRef] [PubMed]
- Gopal, R.; Kim, Y.J.; Seo, C.H.; Hahm, K.S.; Park, Y. Reversed Sequence Enhances Antimicrobial Activity of a Synthetic Peptide. J. Pept. Sci. 2011, 17, 329–334. [Google Scholar] [CrossRef]
- Jwad, R.; Weissberger, D.; Hunter, L. Strategies for Fine-Tuning the Conformations of Cyclic Peptides. Chem. Rev. 2020, 120, 9743–9789. [Google Scholar] [CrossRef]
- Feng, S.; Liang, W.; Li, J.; Chen, Y.; Zhou, D.; Liang, L.; Lin, D.; Li, Y.; Zhao, H.; Du, H.; et al. MCR-1-Dependent Lipid Remodelling Compromises the Viability of Gram-Negative Bacteria. Emerg. Microbes Infect. 2022, 11, 1236–1249. [Google Scholar] [CrossRef] [PubMed]
- Krüger, M.; Richter, P.; Strauch, S.M.; Nasir, A.; Burkovski, A.; Antunes, C.A.; Meißgeier, T.; Schlücker, E.; Schwab, S.; Lebert, M. What an Escherichia Coli Mutant Can Teach Us About the Antibacterial Effect of Chlorophyllin. Microorganisms 2019, 7, 59. [Google Scholar] [CrossRef] [Green Version]
- Aghapour, Z.; Gholizadeh, P.; Ganbarov, K.; Bialvaei, A.Z.; Mahmood, S.S.; Tanomand, A.; Yousefi, M.; Asgharzadeh, M.; Yousefi, B.; Kafil, H.S. Molecular Mechanisms Related to Colistin Resistance in Enterobacteriaceae. Infect. Drug Resist. 2019, 12, 965–975. [Google Scholar] [CrossRef] [Green Version]
- Rhouma, M.; Beaudry, F.; Thériault, W.; Letellier, A. Colistin in Pig Production: Chemistry, Mechanism of Antibacterial Action, Microbial Resistance Emergence, and One Health Perspectives. Front. Microbiol. 2016, 7, 1789. [Google Scholar] [CrossRef] [Green Version]
- Elias, R.; Duarte, A.; Perdigão, J. A Molecular Perspective on Colistin and Klebsiella Pneumoniae: Mode of Action, Resistance Genetics, and Phenotypic Susceptibility. Diagnostics 2021, 11, 1165. [Google Scholar] [CrossRef]
- Rainsford, P.; Rylandsholm, F.; Jakubec, M.; Juskewitz, E.; Silk, M.; Engh, R.A.; Isaksson, J. Label-Free Measurement of Antimicrobial Peptide Interactions with Lipid Vesicles and Nanodiscs Using Microscale Thermophoresis. Sci. Rep. 2023. preprint. [Google Scholar] [CrossRef]
- Juskewitz, E.; Mishchenko, E.; Dubey, V.K.; Jenssen, M.; Jakubec, M.; Rainsford, P.; Isaksson, J.; Andersen, J.H.; Ericson, J.U. Lulworthinone: In Vitro Mode of Action Investigation of an Antibacterial Dimeric Naphthopyrone Isolated from a Marine Fungus. Mar. Drugs 2022, 20, 277. [Google Scholar] [CrossRef] [PubMed]
- Malanovic, N.; Lohner, K. Gram-Positive Bacterial Cell Envelopes: The Impact on the Activity of Antimicrobial Peptides. Biochim. Biophys. Acta Biomembr. 2016, 1858, 936–946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rietschel, E.T.; Kirikae, T.; Schade, F.U.; Mamat, U.; Schmidt, G.; Loppnow, H.; Ulmer, A.J.; Zähringer, U.; Seydel, U.; Padova, F.D.; et al. Bacterial Endotoxin: Molecular Relationships of Structure to Activity and Function. FASEB J. 1994, 8, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Snyder, S.; Kim, D.; McIntosh, T.J. Lipopolysaccharide Bilayer Structure: Effect of Chemotype, Core Mutations, Divalent Cations, and Temperature. Biochemistry 1999, 38, 10758–10767. [Google Scholar] [CrossRef]
- Waudby, C.A.; Alfonso, I. An Introduction to One- and Two-Dimensional Lineshape Analysis of Chemically Exchanging Systems. J. Magn. Reson. Open 2023, 16–17, 100102. [Google Scholar] [CrossRef]
- Luchinat, E.; Barbieri, L.; Cremonini, M.; Nocentini, A.; Supuran, C.T.; Banci, L. Drug Screening in Human Cells by NMR Spectroscopy Allows the Early Assessment of Drug Potency. Angew. Chemie Int. Ed. 2020, 59, 6535–6539. [Google Scholar] [CrossRef] [Green Version]
- Pandit, G.; Biswas, K.; Ghosh, S.; Debnath, S.; Bidkar, A.P.; Satpati, P.; Bhunia, A.; Chatterjee, S. Rationally Designed Antimicrobial Peptides: Insight into the Mechanism of Eleven Residue Peptides against Microbial Infections. Biochim. Biophys. Acta Biomembr. 2020, 1862, 183177. [Google Scholar] [CrossRef]
- Waudby, C.A.; Ramos, A.; Cabrita, L.D.; Christodoulou, J. Two-Dimensional NMR Lineshape Analysis. Sci. Rep. 2016, 6, 95–109. [Google Scholar] [CrossRef] [Green Version]
- Komuro, T.; Murai, T.; Kawasaki, H. Effect of Sonication on the Dispersion State of Lipopolysaccharide and Its Pyrogenicity in Rabbits. Chem. Pharm. Bull. 1987, 35, 4946–4952. [Google Scholar] [CrossRef] [Green Version]
- Angyal, S.J.; Warburton, W.K. The Basic Strengths of Methylated Guanidines. J. Chem. Soc. 1951, 2492–2494. [Google Scholar] [CrossRef]
- Harms, M.J.; Schlessman, J.L.; Chimenti, M.S.; Sue, G.R.; Damjanović, A.; Damjanović, D.; García, B.; García-Moreno, G. A Buried Lysine That Titrates with a Normal PKa: Role of Conformational Flexibility at the Protein–Water Interface as a Determinant of PKavalues. Protein Sci. 2008, 17, 833–845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koo, Y.S.; Kim, J.M.; Park, I.Y.; Yu, B.J.; Jang, S.A.; Kim, K.S.; Park, C.B.; Cho, J.H.; Kim, S.C. Structure–Activity Relations of Parasin I, a Histone H2A-Derived Antimicrobial Peptide. Peptides 2008, 29, 1102–1108. [Google Scholar] [CrossRef]
- Mitchell, D.J.; Steinman, L.; Kim, D.T.; Fathman, C.G.; Rothbard, J.B. Polyarginine Enters Cells More Efficiently than Other Polycationic Homopolymers. J. Pept. Res. 2000, 56, 318–325. [Google Scholar] [CrossRef]
- Åmand, H.L.; Fant, K.; Nordén, B.; Esbjörner, E.K. Stimulated Endocytosis in Penetratin Uptake: Effect of Arginine and Lysine. Biochem. Biophys. Res. Commun. 2008, 371, 621–625. [Google Scholar] [CrossRef]
- Zou, G.; De Leeuw, E.; Li, C.; Pazgier, M.; Li, C.; Zeng, P.; Lu, W.Y.; Lubkowski, J.; Lu, W. Toward Understanding the Cationicity of Defensins: Arg and Lys Versus Their Noncoded Analogs. J. Biol. Chem. 2007, 282, 19653–19665. [Google Scholar] [CrossRef] [Green Version]
- Savini, F.; Loffredo, M.R.; Troiano, C.; Bobone, S.; Malanovic, N.; Eichmann, T.O.; Caprio, L.; Canale, V.C.; Park, Y.; Mangoni, M.L.; et al. Binding of an Antimicrobial Peptide to Bacterial Cells: Interaction with Different Species, Strains and Cellular Components. Biochim. Biophys. Acta Biomembr. 2020, 1862, 183291. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jakubec, M.; Rylandsholm, F.G.; Rainsford, P.; Silk, M.; Bril’kov, M.; Kristoffersen, T.; Juskewitz, E.; Ericson, J.U.; Svendsen, J.S.M. Goldilocks Dilemma: LPS Works Both as the Initial Target and a Barrier for the Antimicrobial Action of Cationic AMPs on E. coli. Biomolecules 2023, 13, 1155. https://doi.org/10.3390/biom13071155
Jakubec M, Rylandsholm FG, Rainsford P, Silk M, Bril’kov M, Kristoffersen T, Juskewitz E, Ericson JU, Svendsen JSM. Goldilocks Dilemma: LPS Works Both as the Initial Target and a Barrier for the Antimicrobial Action of Cationic AMPs on E. coli. Biomolecules. 2023; 13(7):1155. https://doi.org/10.3390/biom13071155
Chicago/Turabian StyleJakubec, Martin, Fredrik G. Rylandsholm, Philip Rainsford, Mitchell Silk, Maxim Bril’kov, Tone Kristoffersen, Eric Juskewitz, Johanna U. Ericson, and John Sigurd M. Svendsen. 2023. "Goldilocks Dilemma: LPS Works Both as the Initial Target and a Barrier for the Antimicrobial Action of Cationic AMPs on E. coli" Biomolecules 13, no. 7: 1155. https://doi.org/10.3390/biom13071155
APA StyleJakubec, M., Rylandsholm, F. G., Rainsford, P., Silk, M., Bril’kov, M., Kristoffersen, T., Juskewitz, E., Ericson, J. U., & Svendsen, J. S. M. (2023). Goldilocks Dilemma: LPS Works Both as the Initial Target and a Barrier for the Antimicrobial Action of Cationic AMPs on E. coli. Biomolecules, 13(7), 1155. https://doi.org/10.3390/biom13071155