



Hemocompatibility of β-Cyclodextrin-Modified (Methacryloyloxy)ethyl Phosphorylcholine Coated Magnetic Nanoparticles

 and
and
Abstract
:1. Introduction
2. Materials and Methods
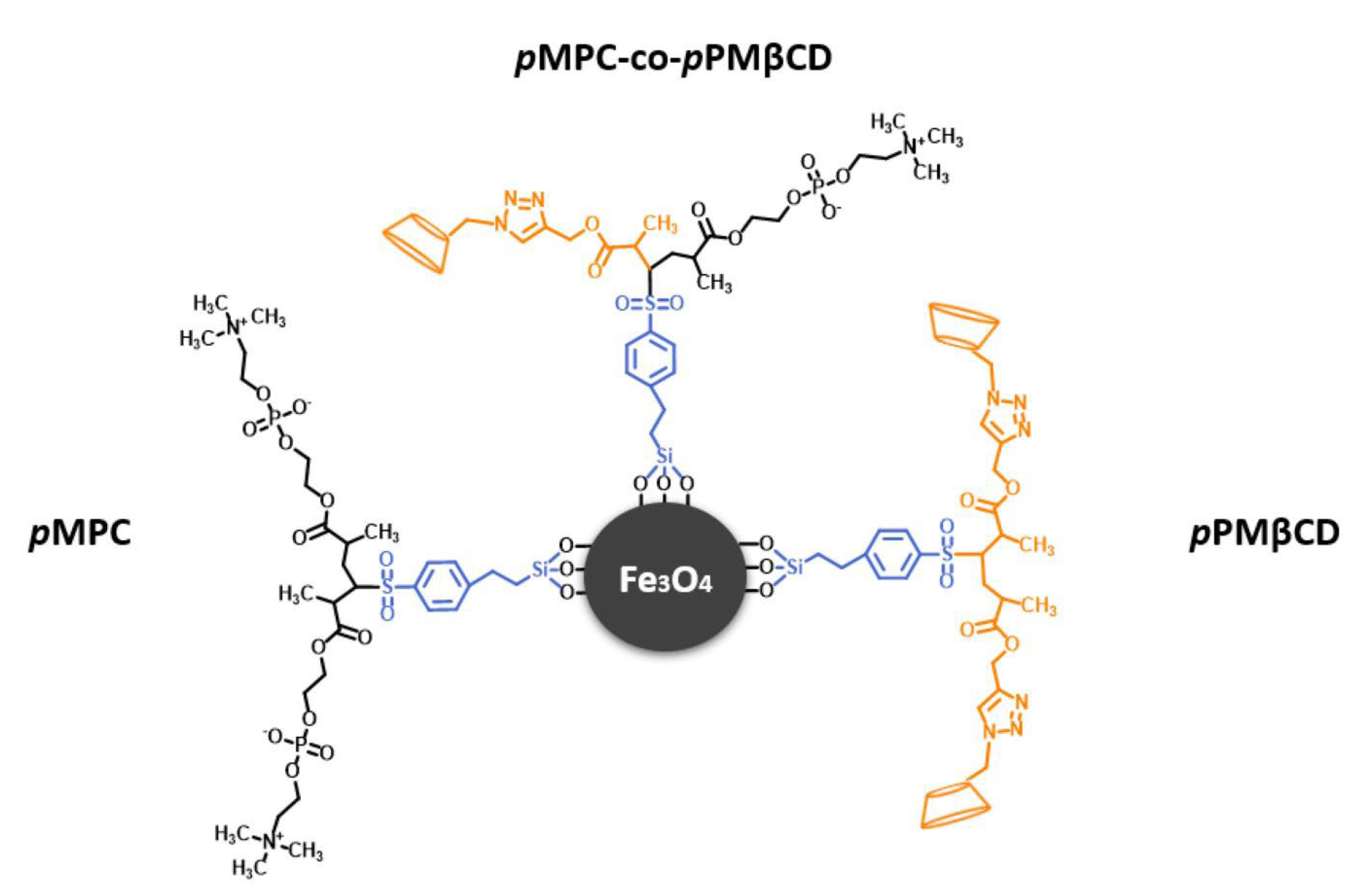
2.1. Polymer Synthesis and Fixation
2.2. Polymer Characterization
2.3. Circular Dichroism
2.4. Fluorescence Spectroscopy
2.5. SDS-PAGE and Immunoblot
2.6. Fibrin Clot Formation in Plasma
2.7. Whole-Blood Hemocompatibility Testing
3. Results and Discussion
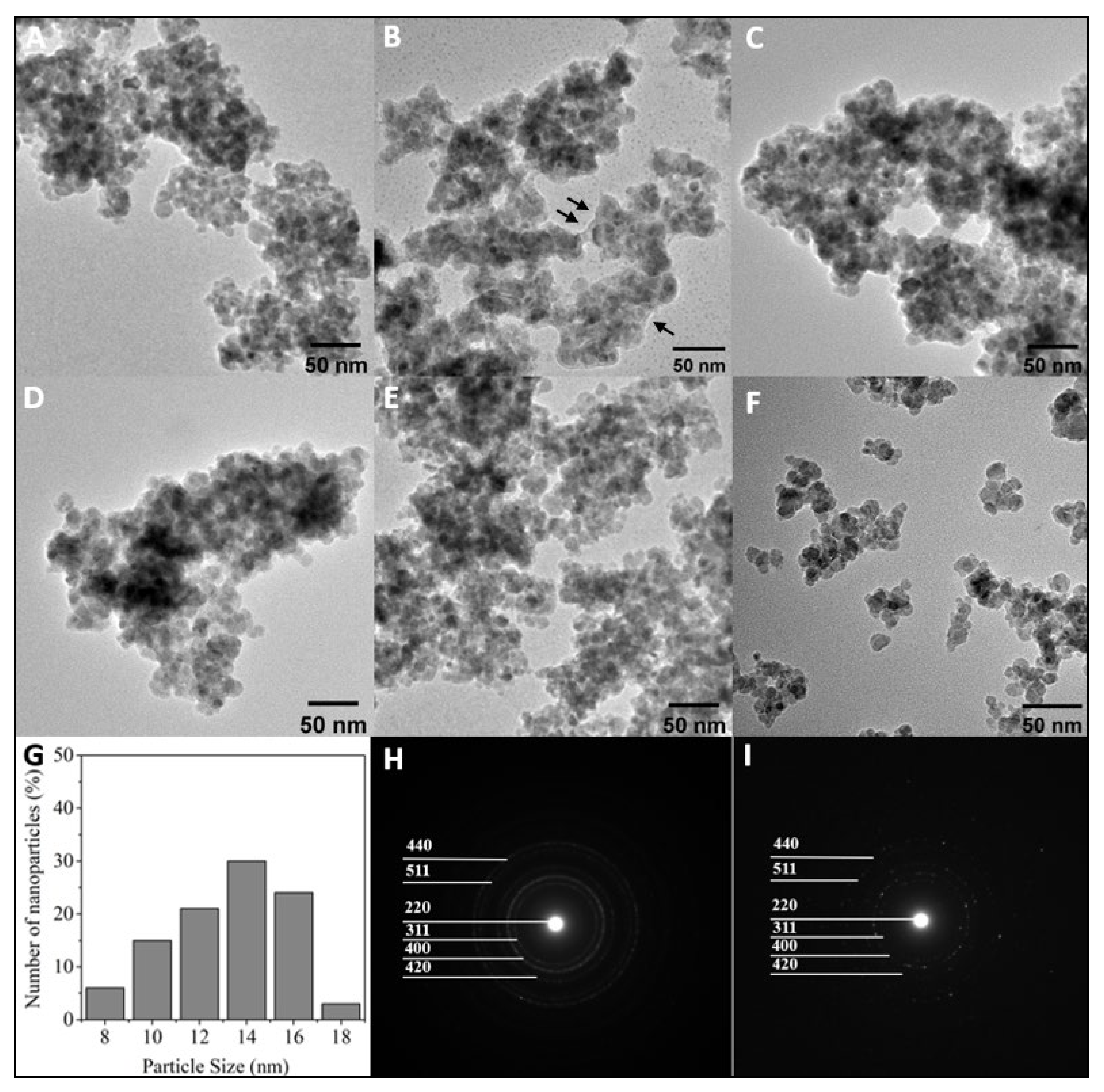
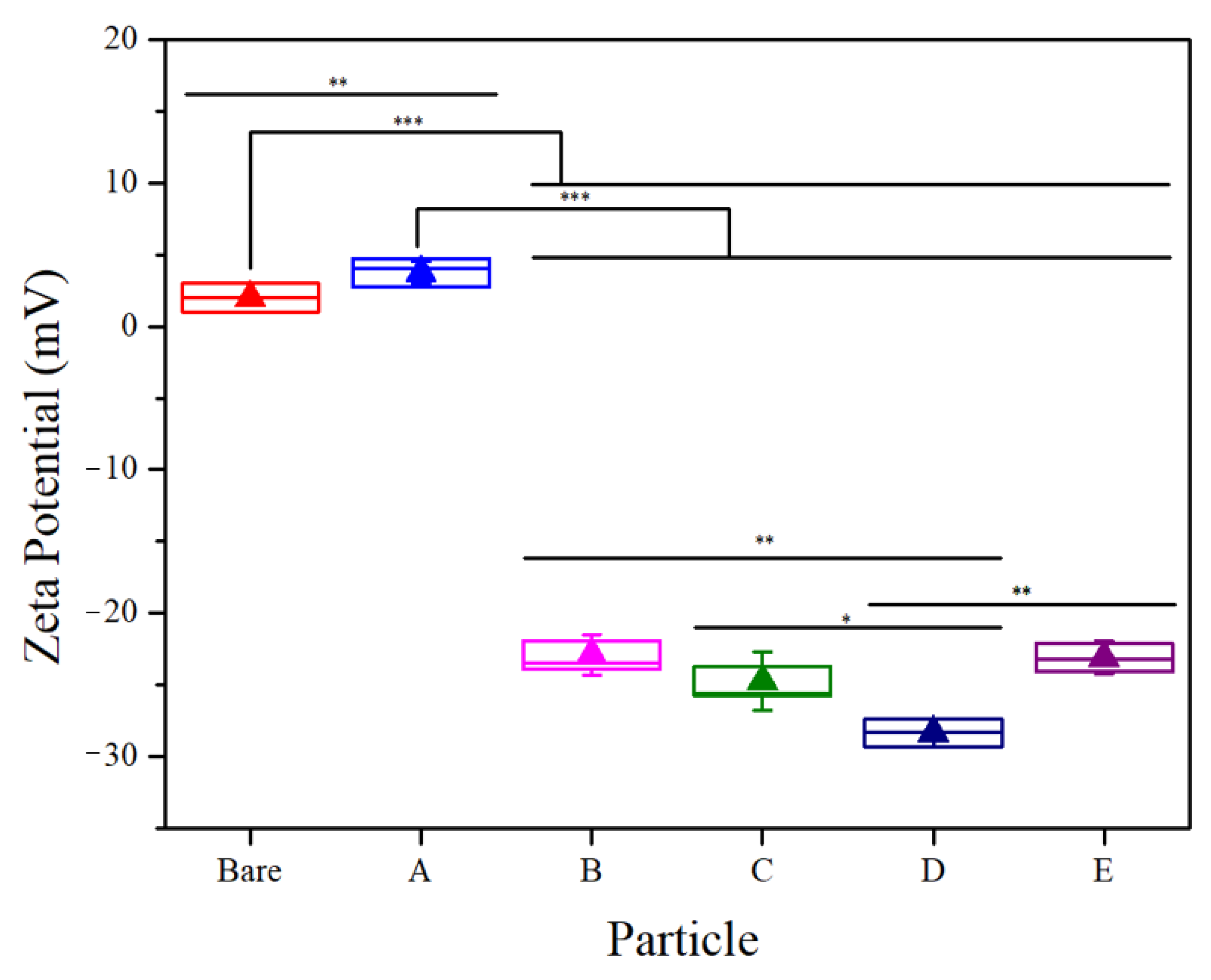
3.1. Properties and Characterization of the Modified MNPs
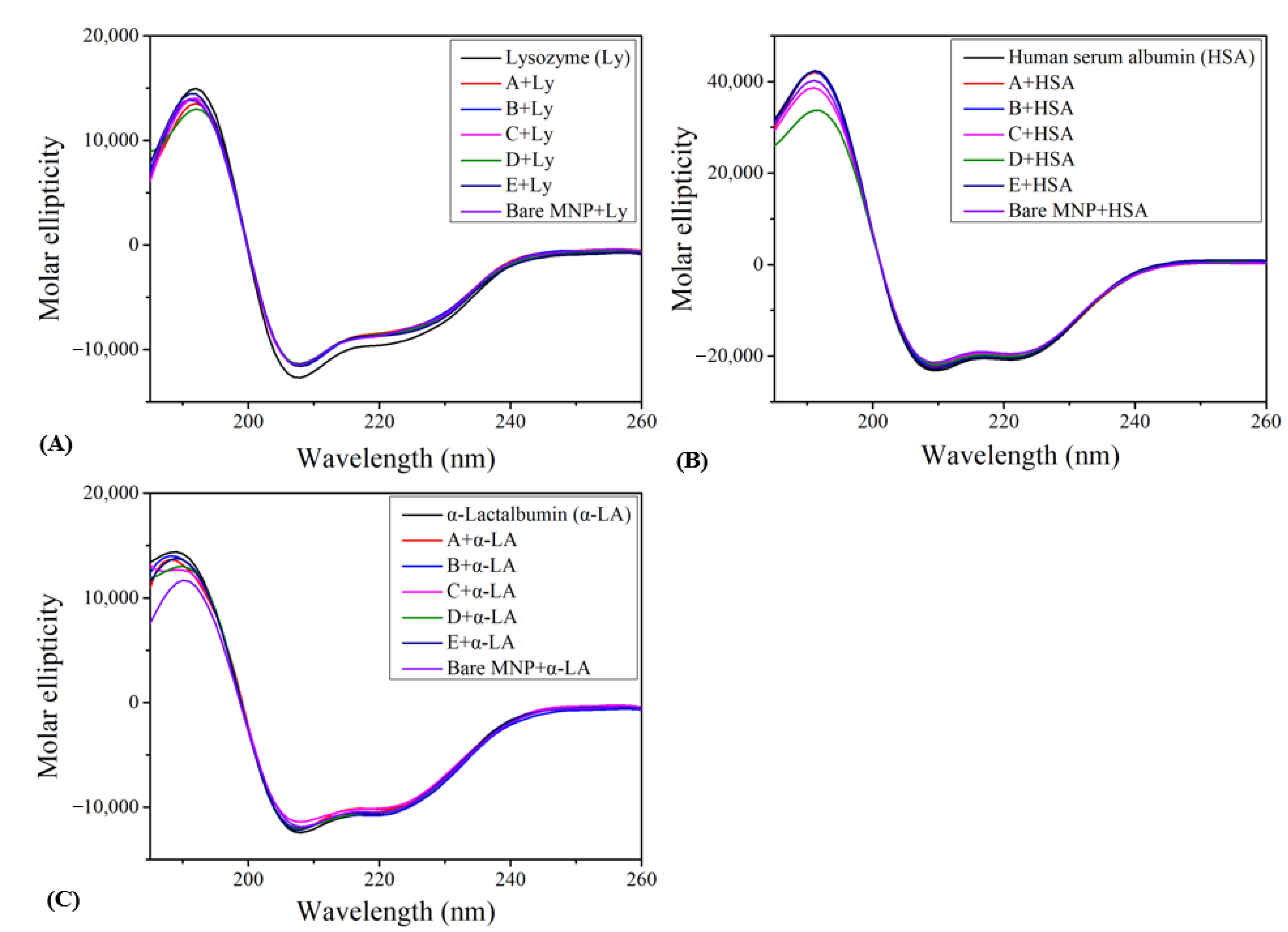
3.2. Circular Dichroism
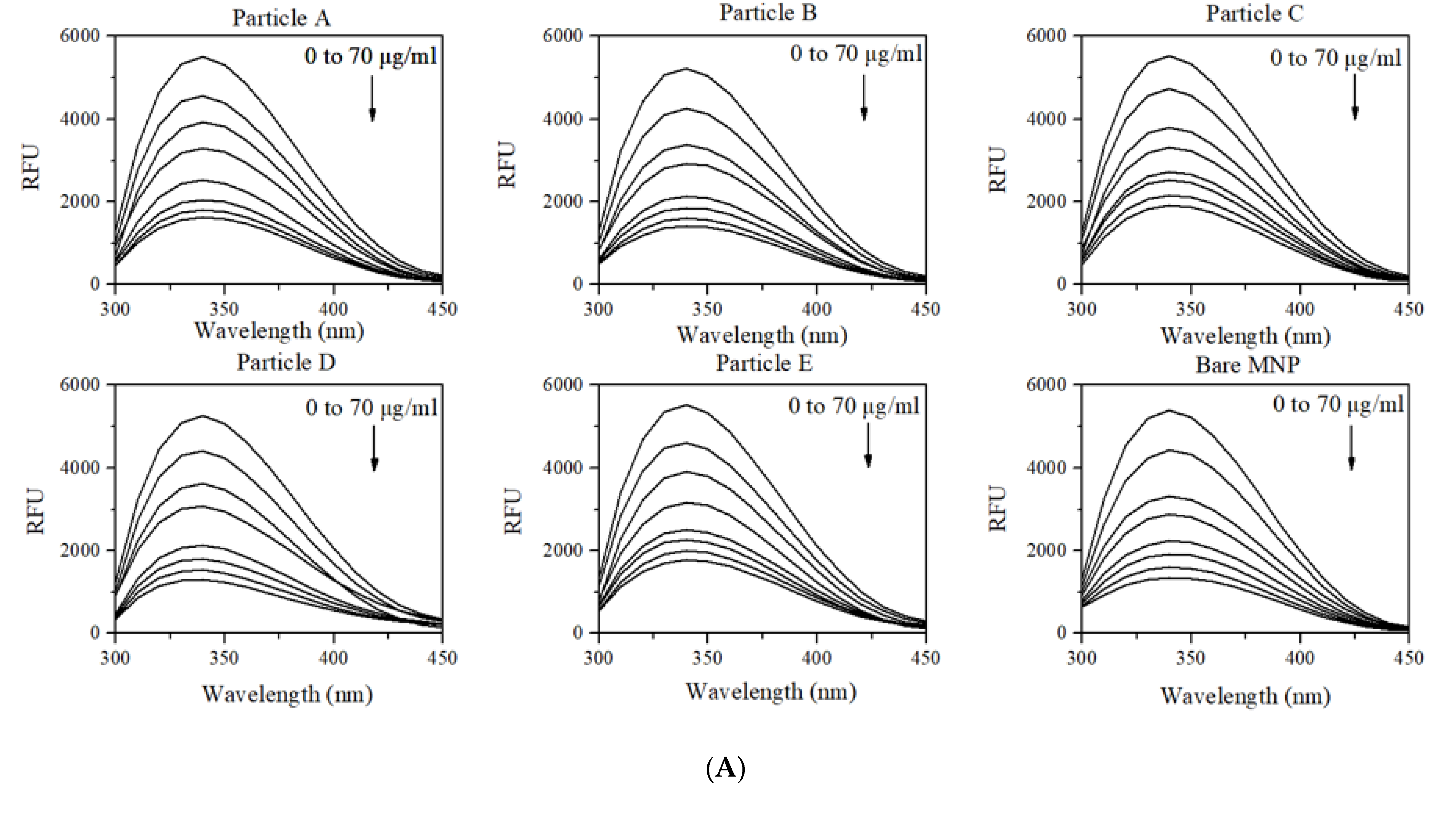
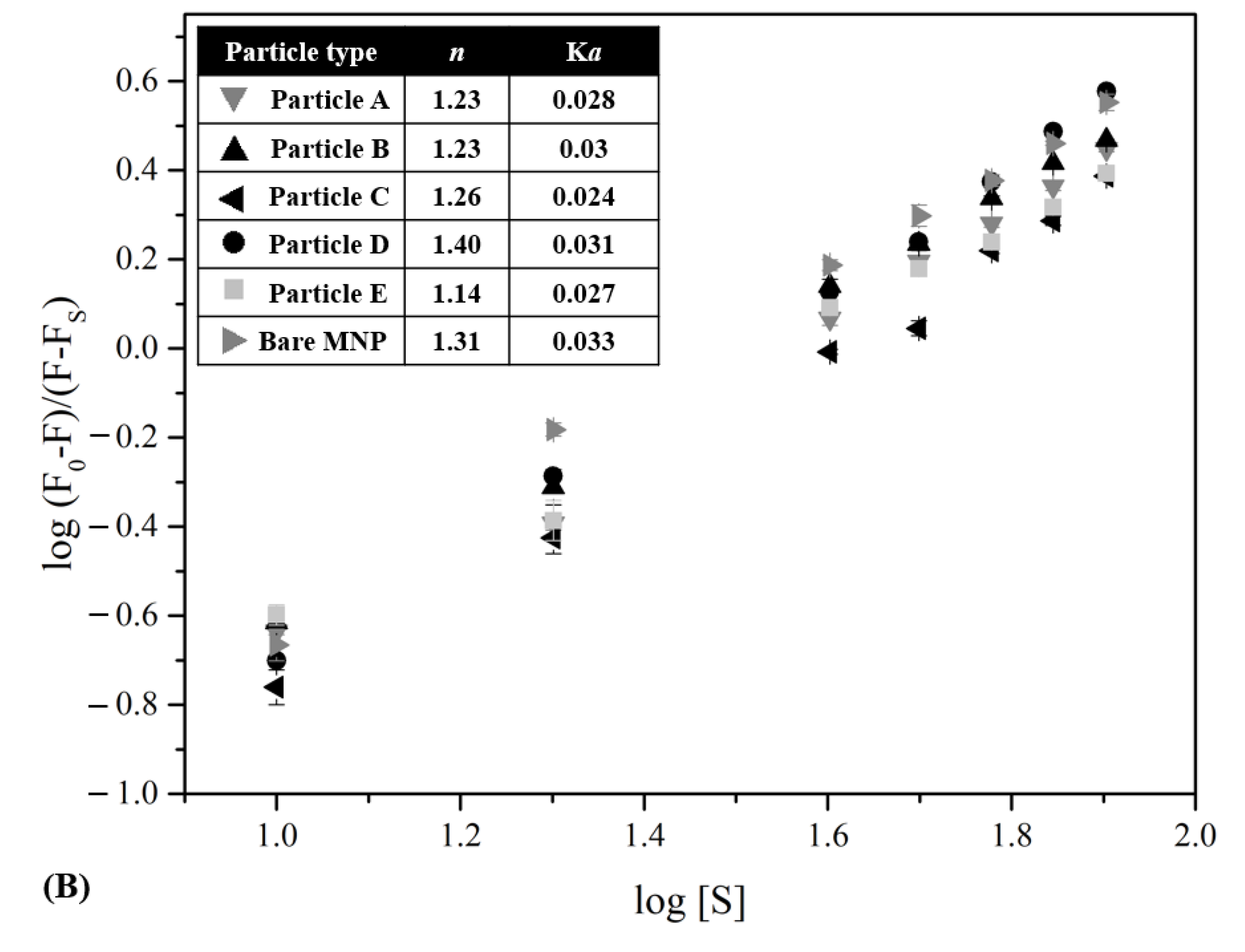
3.3. Changes in Intrinsic Fluorescence of HSA upon Adsorption
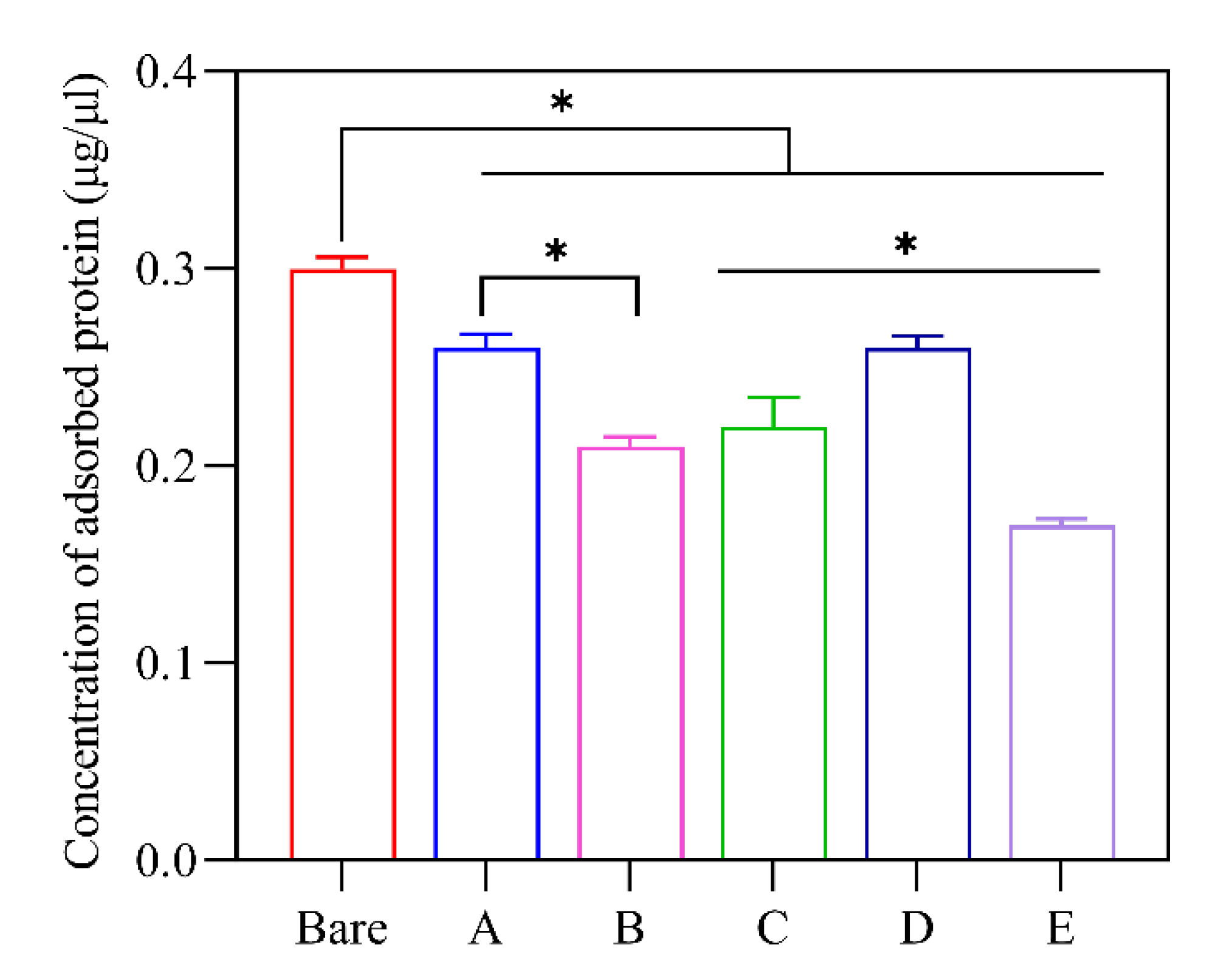
3.4. Total Adsorbed Protein
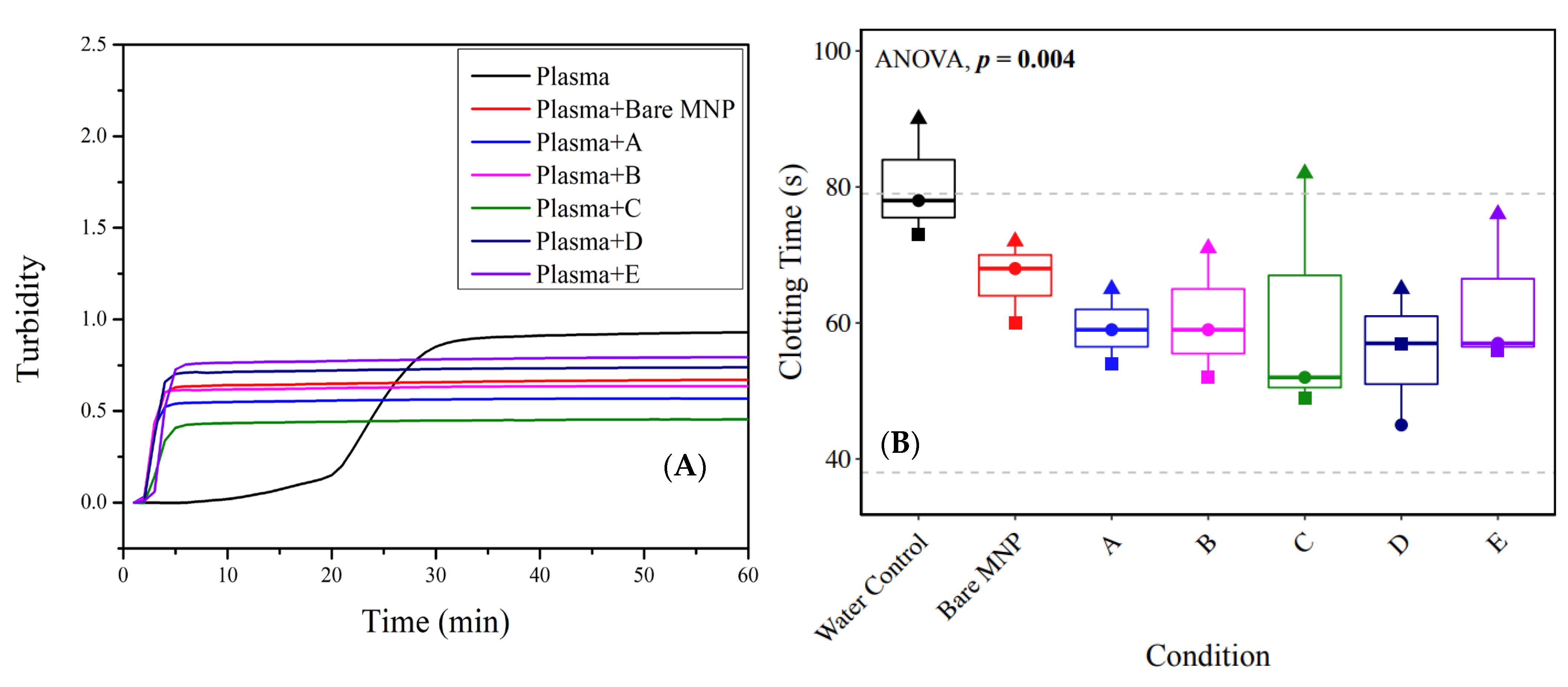
3.5. Plasma Clotting in the Presence of Polymer-Coated Nanoparticles
3.6. Quantification of Protein Adsorption
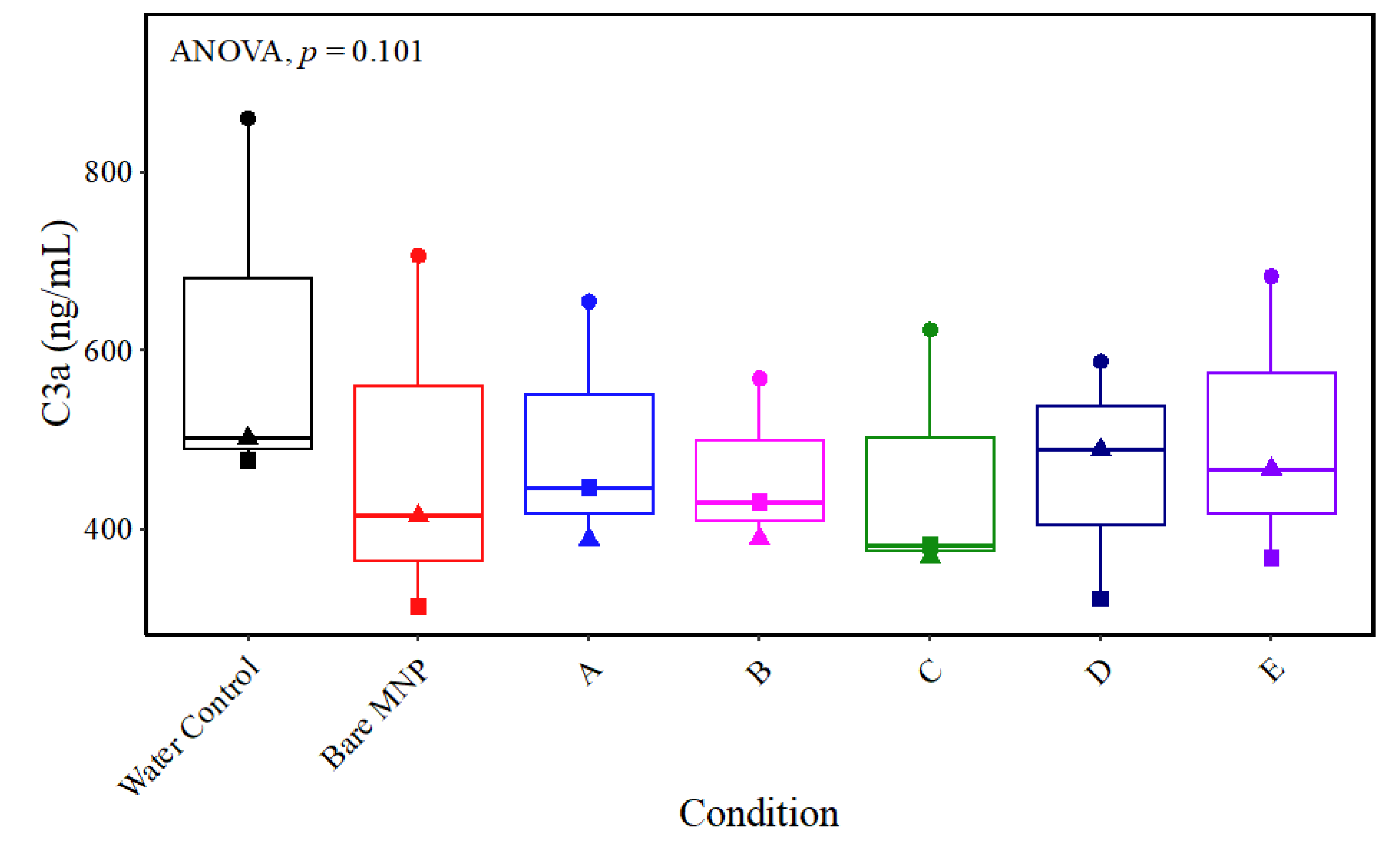
3.6.1. Immune Response-Related Proteins
3.6.2. Coagulation-Related Proteins
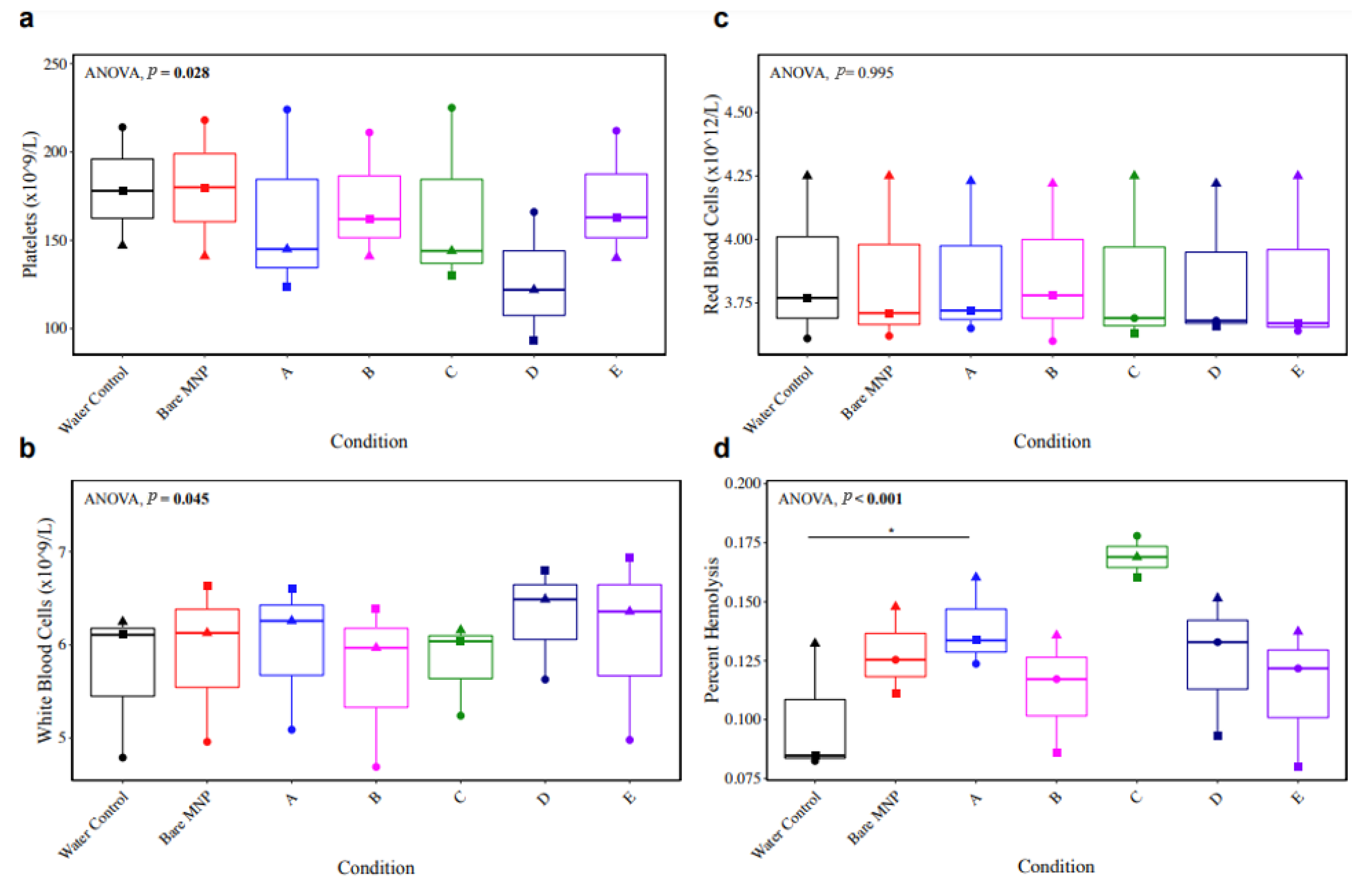
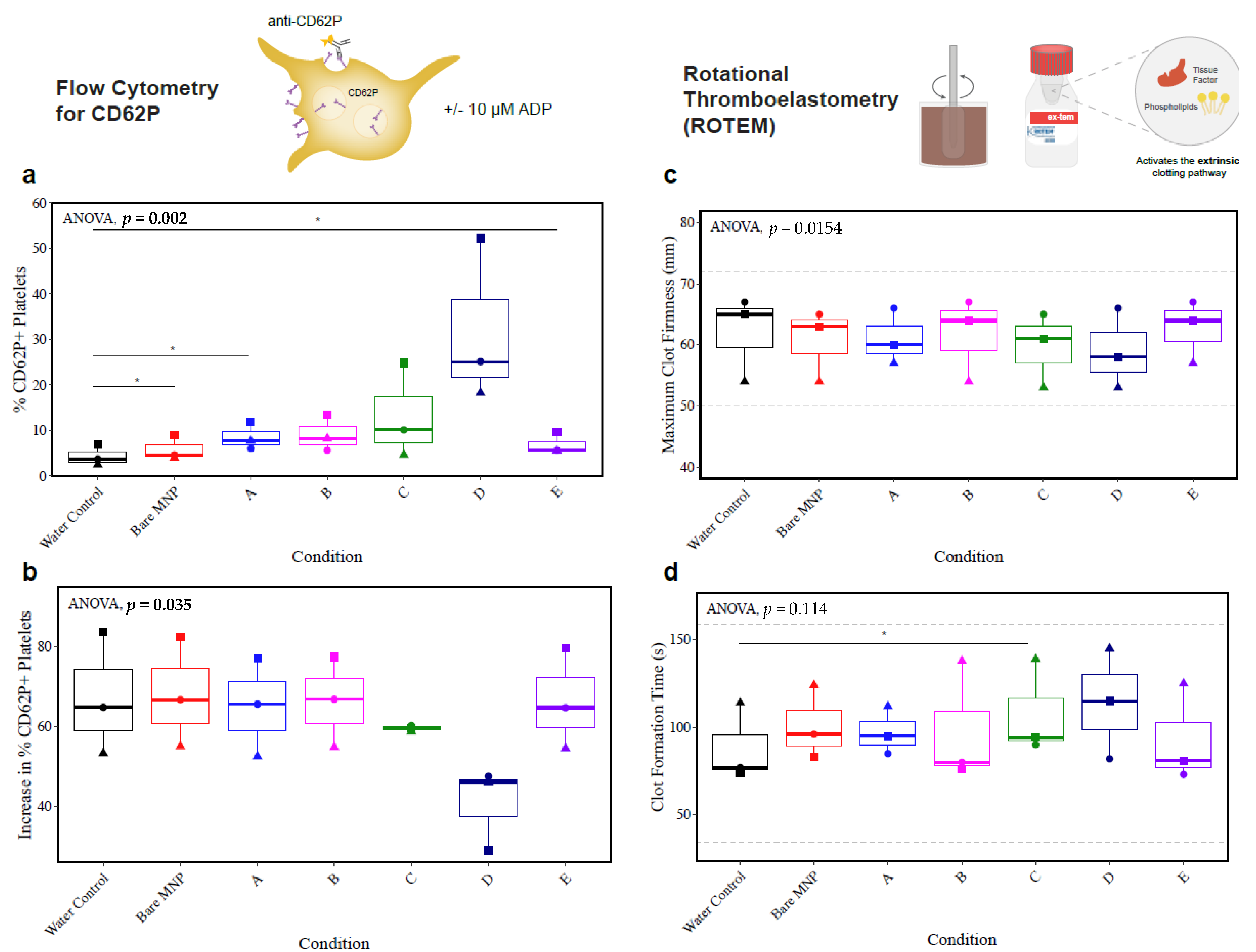
3.7. Whole-Blood Hemocompatibility and Impact on Platelet Function
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Klefter, R.; Nielsen, B.J.A.O. Savings in dialysis treatment? Artif. Organs 2002, 26, 49–54. [Google Scholar] [CrossRef]
- Pavlenko, D.; Giasafaki, D.; Charalambopoulou, G.; Van Geffen, E.; Gerritsen, K.; Steriotis, T.; Stamatialis, D.J.S.R. Carbon adsorbents with dual porosity for efficient removal of uremic toxins and cytokines from human plasma. Sci. Rep. 2017, 7, 14914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lv, J.-C.; Zhang, L.-X. Prevalence and disease burden of chronic kidney disease. Ren. Fibros. Mech. Ther. 2019, 1165, 3–15. [Google Scholar]
- Ma, Y.; Li, S.; Tonelli, M.; Unsworth, L.D.J.M.; Materials, M. Adsorption-based strategies for removing uremic toxins from blood. Microporous Mesoporous Mater. 2021, 319, 111035. [Google Scholar] [CrossRef]
- Bellringer, M.; Smith, T.; Read, R.; Gopinath, C.; Olivier, P.J.F. β-Cyclodextrin: 52-week toxicity studies in the rat and dog. Food Chem. Toxicol. 1995, 33, 367–376. [Google Scholar] [CrossRef]
- Zhou, Y.; He, J.; Lu, J.; Liu, Y.; Zhou, Y. Enhanced removal of bisphenol A by cyclodextrin in photocatalytic systems: Degradation intermediates and toxicity evaluation. Chin. Chem. Lett. 2020, 31, 2623–2626. [Google Scholar] [CrossRef]
- Yu, Q.; Zhang, Y.; Wang, H.; Brash, J.; Chen, H. Anti-fouling bioactive surfaces. Acta Biomater. 2011, 7, 1550–1557. [Google Scholar] [CrossRef]
- Zhang, H.; Chiao, M. Anti-fouling coatings of poly (dimethylsiloxane) devices for biological and biomedical applications. J. Med. Biol. Eng. 2015, 35, 143–155. [Google Scholar] [CrossRef] [Green Version]
- Bhakta, S.A.; Evans, E.; Benavidez, T.E.; Garcia, C.D. Protein adsorption onto nanomaterials for the development of biosensors and analytical devices: A review. Anal. Chim. Acta 2015, 872, 7–25. [Google Scholar] [CrossRef] [Green Version]
- Silva-Bermudez, P.; Rodil, S.J.S.; Technology, C. An overview of protein adsorption on metal oxide coatings for biomedical implants. Surf. Coatings Technol. 2013, 233, 147–158. [Google Scholar] [CrossRef]
- Pinholt, C.; Hartvig, R.A.; Medlicott, N.J.; Jorgensen, L. The importance of interfaces in protein drug delivery “why” is protein adsorption of interest in pharmaceutical formulations? Expert Opin. Drug Deliv. 2011, 8, 949–964. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Li, L.; Zhao, C.; Zheng, J.J.P. Surface hydration: Principles and applications toward low-fouling/nonfouling biomaterials. Polymer 2010, 51, 5283–5293. [Google Scholar] [CrossRef] [Green Version]
- Mohanty, J.; Bhasikuttan, A.C. Cyclodextrins in Anti-Biofouling Applications. In Supramolecular Chemistry in Corrosion and Biofouling Protection; CRC Press: Boca Raton, FL, USA, 2021; pp. 335–346. [Google Scholar]
- Liu, Y.; Zhang, D.; Ren, B.; Gong, X.; Xu, L.; Feng, Z.-Q.; Chang, Y.; He, Y.; Zheng, J. Molecular simulations and understanding of antifouling zwitterionic polymer brushes. J. Mater. Chem. B 2020, 8, 3814–3828. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Gao, K.; Zhou, L.; Jiao, Z.; Wu, M.; Cao, J.; You, X.; Cai, Z.; Su, Y.; Jiang, Z. Zwitterionic materials for antifouling membrane surface construction. Acta Biomater. 2016, 40, 142–152. [Google Scholar] [CrossRef] [PubMed]
- Burns, N.L.; Holmberg, K.; Brink, C. Influence of surface charge on protein adsorption at an amphoteric surface: Effects of varying acid to base ratio. J. Colloid Interface Sci. 1996, 178, 116–122. [Google Scholar] [CrossRef]
- Phillips, S.; Wilson, L.; Borkman, R.F. Acrylamide and iodide fluorescence quenching as a structural probe of tryptophan microenvironment in bovine lens crystallins. Curr. Eye Res. 1986, 5, 611–620. [Google Scholar] [CrossRef]
- Krebs, H.A. Chemical composition of blood plasma and serum. Annu. Rev. Biochem. 1950, 19, 409–430. [Google Scholar] [CrossRef]
- Sperling, C.; Fischer, M.; Maitz, M.F.; Werner, C.J.B. Blood coagulation on biomaterials requires the combination of distinct activation processes. Biomaterials 2009, 30, 4447–4456. [Google Scholar] [CrossRef]
- Fischer, M.; Sperling, C.; Werner, C. Synergistic effect of hydrophobic and anionic surface groups triggers blood coagulation in vitro. J. Mater. Sci. Mater. Med. 2010, 21, 931–937. [Google Scholar] [CrossRef]
- Le, H.T.; Jeon, H.M.; Lim, C.W.; Kim, T.W. Synthesis, cytotoxicity, and phase-solubility study of cyclodextrin click clusters. J. Pharm. Sci. 2014, 103, 3183–3189. [Google Scholar] [CrossRef]
- Diget, J.S.; Städe, L.W.; Nielsen, T.T. Direct synthesis of well-defined zwitterionic cyclodextrin polymers via atom transfer radical polymerization. Eur. Polym. J. 2019, 116, 84–90. [Google Scholar] [CrossRef]
- Goszczyński, T.M.; Gawłowski, M.; Girek, B.; Kowalski, K.; Boratyński, J.; Girek, T. Synthesis of β-cyclodextrin-lysozyme conjugates and their physicochemical and biochemical properties. J. Incl. Phenom. Macrocycl. Chem. 2017, 87, 341–348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, H.; Huang, J.; Koepsel, R.R.; Ye, P.; Russell, A.J.; Matyjaszewski, K.J.B. Recyclable antibacterial magnetic nanoparticles grafted with quaternized poly (2-(dimethylamino) ethyl methacrylate) brushes. Biomacromolecules 2011, 12, 1305–1311. [Google Scholar] [CrossRef]
- Yallapu, M.M.; Othman, S.F.; Curtis, E.T.; Gupta, B.K.; Jaggi, M.; Chauhan, S.C.J.B. Multi-functional magnetic nanoparticles for magnetic resonance imaging and cancer therapy. Biomaterials 2011, 32, 1890–1905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beaven, G.H.; Chen, S.H.; D’albis, A.; Gratzer, W.B. A spectroscopic study of the haemin–human-serum-albumin system. Eur. J. Biochem. 1974, 41, 539–546. [Google Scholar] [CrossRef]
- Chipman, D.M.; Grisaro, V.; Sharon, N. The binding of oligosaccharides containing n-acetylglucosamine and n-acetylmuramic acid to lysozyme: The specificity of binding subsites. J. Biol. Chem. 1967, 242, 4388–4394. [Google Scholar] [CrossRef]
- Bahniuk, M.S.; Alshememry, A.K.; Unsworth, L.D. Human plasma protein adsorption to elastinlike polypeptide nanoparticles. Biointerphases 2020, 15, 021007. [Google Scholar] [CrossRef]
- Bahniuk, M.S.; Pirayesh, H.; Singh, H.D.; Nychka, J.A.; Unsworth, L.D. Bioactive glass 45S5 powders: Effect of synthesis route and resultant surface chemistry and crystallinity on protein adsorption from human plasma. Biointerphases 2012, 7, 41. [Google Scholar] [CrossRef]
- Han, V.; Serrano, K.; Devine, D.V. A comparative study of common techniques used to measure haemolysis in stored red cell concentrates. Vox Sang. 2010, 98, 116–123. [Google Scholar] [CrossRef]
- Mahmoud, W.E.; Al-Hazmi, F.; Al-Noaiser, F.; Al-Ghamdi, A.A.; Bronstein, L.M. A facile method to syntheses monodisperse γ-Fe2O3 nanocubes with high magnetic anisotropy density. Superlattices Microstruct. 2014, 68, 1–5. [Google Scholar] [CrossRef]
- Tombuloglu, H.; Albenayyan, N.; Slimani, Y.; Akhtar, S.; Tombuloglu, G.; Almessiere, M.; Baykal, A.; Ercan, I.; Sabit, H.; Manikandan, A.J.E.S.; et al. Fate and impact of maghemite (γ-Fe2O3) and magnetite (Fe3O4) nanoparticles in barley (Hordeum vulgare L.). Environ. Sci. Pollut. Res. 2022, 29, 4710–4721. [Google Scholar] [CrossRef] [PubMed]
- Al-Harbi, L.; Darwish, M.S.J.H. Functionalized iron oxide nanoparticles: Synthesis through ultrasonic-assisted co-precipitation and performance as hyperthermic agents for biomedical applications. Heliyon 2022, 8, e09654. [Google Scholar] [CrossRef] [PubMed]
- Kmita, A.; Lachowicz, D.; Żukrowski, J.; Gajewska, M.; Szczerba, W.; Kuciakowski, J.; Zapotoczny, S.; Sikora, M.J.M. One-step synthesis of long term stable superparamagnetic colloid of zinc ferrite nanorods in water. Materials 2019, 12, 1048. [Google Scholar] [CrossRef] [Green Version]
- Rösner, H.I.; Redfield, C. The human α-lactalbumin molten globule: Comparison of structural preferences at pH 2 and pH 7. J. Mol. Biol. 2009, 394, 351–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cadenas, E.; Packer, L. Thiol Redox Transitions in Cell Signaling, Part A: Chemistry and Biochemistry of Low Molecular Weight and protein thiols; Academic Press: Cambridge, MA, USA, 2010. [Google Scholar]
- Steudle, A.; Pleiss, J. Modelling of lysozyme binding to a cation exchange surface at atomic detail: The role of flexibility. Biophys. J. 2011, 100, 3016–3024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, T.K.; Foy, S.P.; Erokwu, B.; Dimitrijevic, S.; Flask, C.A.; Labhasetwar, V.J.B. Magnetic resonance imaging of multifunctional pluronic stabilized iron-oxide nanoparticles in tumor-bearing mice. Biomaterials 2009, 30, 6748–6756. [Google Scholar] [CrossRef] [Green Version]
- Dobrovolskaia, M.A.; Patri, A.K.; Zheng, J.; Clogston, J.D.; Ayub, N.; Aggarwal, P.; Neun, B.W.; Hall, J.B.; McNeil, S.E. Interaction of colloidal gold nanoparticles with human blood: Effects on particle size and analysis of plasma protein binding profiles. Nanomed. Nanotechnol. Biol. Med. 2009, 5, 106–117. [Google Scholar] [CrossRef] [Green Version]
- Konno, T.; Kurita, K.; Iwasaki, Y.; Nakabayashi, N.; Ishihara, K.J.B. Preparation of nanoparticles composed with bioinspired 2-methacryloyloxyethyl phosphorylcholine polymer. Biomaterials 2001, 22, 1883–1889. [Google Scholar] [CrossRef] [PubMed]
- Ishihara, K.; Oshida, H.; Endo, Y.; Ueda, T.; Watanabe, A.; Nakabayashi, N. Hemocompatibility of human whole blood on polymers with a phospholipid polar group and its mechanism. J. Biomed. Mater. Res. 1992, 26, 1543–1552. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.-L.J.; Narayanamurthy, R.; Yager, J.Y.; Unsworth, L.D.J.N.T. How does biological sex affect the physiological response to nanomaterials? Nano Today 2021, 41, 101292. [Google Scholar] [CrossRef]
- Davie, E.W.; Fujikawa, K.; Kisiel, W.J.B. The coagulation cascade: Initiation, maintenance, and regulation. Biochemistry 1991, 30, 10363–10370. [Google Scholar] [CrossRef]
- Kratz, F. Albumin as a drug carrier: Design of prodrugs, drug conjugates and nanoparticles. J. Control. Release 2008, 132, 171–183. [Google Scholar] [CrossRef] [PubMed]
- Kowalczyńska, H.M.; Nowak-Wyrzykowska, M.; Szczepankiewicz, A.A.; Dobkowski, J.; Dyda, M.; Kamiński, J.; Kołos, R. Albumin adsorption on unmodified and sulfonated polystyrene surfaces, in relation to cell–substratum adhesion. Colloids Surf. B Biointerfaces 2011, 84, 536–544. [Google Scholar] [CrossRef] [PubMed]
- Dabare, P.R.L.; Bachhuka, A.; Parkinson-Lawrence, E.; Vasilev, K. Surface nanotopography mediated albumin adsorption, unfolding and modulation of early innate immune responses. Mater. Today Adv. 2021, 12, 100187. [Google Scholar] [CrossRef]
- Lin, W.-C.; Liu, T.-Y.; Yang, M.-C. Hemocompatibility of polyacrylonitrile dialysis membrane immobilized with chitosan and heparin conjugate. Biomaterials 2004, 25, 1947–1957. [Google Scholar] [CrossRef] [PubMed]
- Henry, M.; Dupont-Gillain, C.; Bertrand, P. Conformation change of albumin adsorbed on polycarbonate membranes as revealed by ToF-SIMS. Langmuir 2003, 19, 6271–6276. [Google Scholar] [CrossRef]
- Van Dulm, P.; Norde, W. The adsorption of human plasma albumin on solid surfaces, with special attention to the kinetic aspects. J. Colloid Interface Sci. 1983, 91, 248–255. [Google Scholar] [CrossRef]
- Victor, S.P.; Sharma, C.P. Development and evaluation of cyclodextrin complexed hydroxyapatite nanoparticles for preferential albumin adsorption. Colloids Surf. B Biointerfaces 2011, 85, 221–228. [Google Scholar] [CrossRef]
- Walport, M.J. Advances in immunology: Complement (first of two parts). N. Engl. J. Med. 2001, 344, 1058–1066. [Google Scholar] [CrossRef]
- Andersson, J.; Ekdahl, K.N.; Lambris, J.D.; Nilsson, B. Binding of C3 fragments on top of adsorbed plasma proteins during complement activation on a model biomaterial surface. Biomaterials 2005, 26, 1477–1485. [Google Scholar] [CrossRef]
- Melchior, P.; Erlenkötter, A.; Zawada, A.M.; Delinski, D.; Schall, C.; Stauss-Grabo, M.; Kennedy, J.P. Complement activation by dialysis membranes and its association with secondary membrane formation and surface charge. Artif. Organs 2021, 45, 770–778. [Google Scholar] [CrossRef]
- Vidarsson, G.; Dekkers, G.; Rispens, T. IgG subclasses and allotypes: From structure to effector functions. Front. Immunol. 2014, 5, 520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrison, P.M.; Arosio, P. The ferritins: Molecular properties, iron storage function and cellular regulation. Biochim. Biophys. Acta (BBA)-Bioenerg. 1996, 1275, 161–203. [Google Scholar] [CrossRef] [Green Version]
- Ogun, A.S.; Adeyinka, A. Biochemistry, Transferrin; StatPearls Publishing: St. Petersburg, FL, USA, 2022. [Google Scholar]
- Stafford, J.L.; Belosevic, M. Transferrin and the innate immune response of fish: Identification of a novel mechanism of macrophage activation. Dev. Comp. Immunol. 2003, 27, 539–554. [Google Scholar] [CrossRef]
- Bale, M.D.; Wohlfahrt, L.A.; Mosher, D.F.; Tomasini, B.; Sutton, R.C. Identification of vitronectin as a major plasma protein adsorbed on polymer surfaces of different copolymer composition. Blood 1989, 74, 2698–2706. [Google Scholar] [CrossRef] [Green Version]
- Berclaz, P.-Y.; Trapnell, B.C. Rare Childhood Lung Disorders: α1-Antitrypsin Deficiency, Pulmonary Alveolar Proteinosis, and Pulmonary Alveolar Microlithiasis. In Kendig’s Disorders of the Respiratory Tract in Children; Elsevier: Amsterdam, The Netherlands, 2006; pp. 747–761. [Google Scholar]
- Chidambaranathan-Reghupaty, S.; Fisher, P.B.; Sarkar, D. Hepatocellular carcinoma (HCC): Epidemiology, etiology and molecular classification. Adv. Cancer Res. 2021, 149, 1–61. [Google Scholar]
- Charbgoo, F.; Nejabat, M.; Abnous, K.; Soltani, F.; Taghdisi, S.M.; Alibolandi, M.; Shier, W.T.; Steele, T.W.; Ramezani, M. Gold nanoparticle should understand protein corona for being a clinical nanomaterial. J. Control. Release 2018, 272, 39–53. [Google Scholar] [CrossRef] [PubMed]
- Papa, E.; Doucet, J.P.; Sangion, A.; Doucet-Panaye, A. Investigation of the influence of protein corona composition on gold nanoparticle bioactivity using machine learning approaches. SAR QSAR Environ. Res. 2016, 27, 521–538. [Google Scholar] [CrossRef]
- Samir Dekali, A.D.; Kortulewski, T.; Vanbaelinghem, J.; Gamez, C.; Rogerieux, F.; Lacroix, G.; Rat, P. Cell cooperation and role of the P2X7 receptor in pulmonary inflammation induced by nanoparticles. Nanotoxicology 2012, 7, 1302–1314. [Google Scholar] [CrossRef]
- Liu, R.; Jiang, W.; Walkey, C.D.; Chan, W.C.W.; Cohen, Y. Prediction of nanoparticles-cell association based on corona proteins and physicochemical properties. Nanoscale 2015, 7, 9664–9675. [Google Scholar] [CrossRef]
- Armstrong, P.B.; Quigley, J.P. α2-macroglobulin: An evolutionarily conserved arm of the innate immune system. Dev. Comp. Immunol. 1999, 23, 375–390. [Google Scholar] [CrossRef] [PubMed]
- Vandooren, J.; Itoh, Y. Alpha-2-macroglobulin in inflammation, immunity and infections. Front. Immunol. 2021, 12, 803244. [Google Scholar] [CrossRef] [PubMed]
- Levy, J.H.; Welsby, I.; Goodnough, L.T. Fibrinogen as a therapeutic target for bleeding: A review of critical levels and replacement therapy. Transfusion 2014, 54, 1389–1405. [Google Scholar] [CrossRef] [PubMed]
- Lowe, G.D.; Rumley, A.; Mackie, I.J. Plasma fibrinogen. Ann. Clin. Biochem. 2004, 41, 430–440. [Google Scholar] [CrossRef] [Green Version]
- Nava-Ortiz, C.A.; Burillo, G.; Concheiro, A.; Bucio, E.; Matthijs, N.; Nelis, H.; Coenye, T.; Alvarez-Lorenzo, C. Cyclodextrin-functionalized biomaterials loaded with miconazole prevent Candida albicans biofilm formation in vitro. Acta Biomater. 2010, 6, 1398–1404. [Google Scholar] [CrossRef]
- Austen, D. Clinical Biochemistry of Blood Coagulation. In Scientific Foundations of Biochemistry in Clinical Practice; Elsevier: Amsterdam, The Netherlands, 1994; pp. 495–513. [Google Scholar]
- Ashikaga, H.; Chien, K.R. Chapter 28—Blood Coagulation and Atherothrombosis. In Molecular Basis of Cardiovascular Disease, 2nd ed.; Chien, K.R., Ed.; W.B. Saunders: Philadelphia, PA, USA, 2004; pp. 498–518. [Google Scholar]
- Wu, Y. Contact pathway of coagulation and inflammation. Thromb. J. 2015, 13, 17. [Google Scholar] [CrossRef] [Green Version]
- Ortega, V.A.; Bahniuk, M.S.; Memon, S.; Unsworth, L.D.; Stafford, J.L.; Goss, G.G. Polymer-coated nanoparticle protein corona formation potentiates phagocytosis of bacteria by innate immune cells and inhibits coagulation in human plasma. Biointerphases 2020, 15, 051003. [Google Scholar] [CrossRef]
- Ivanov, I.; Verhamme, I.M.; Sun, M.-f.; Mohammed, B.; Cheng, Q.; Matafonov, A.; Dickeson, S.K.; Joseph, K.; Kaplan, A.P.; Gailani, D. Protease activity in single-chain prekallikrein. Blood J. Am. Soc. Hematol. 2020, 135, 558–567. [Google Scholar] [CrossRef]
- Veloso, D.; Colman, R.W. Western blot analyses of prekallikrein and its activation products in human plasma. Thromb. Haemost. 1991, 65, 382–388. [Google Scholar] [CrossRef]
- To, W.S.; Midwood, K.S. Plasma and cellular fibronectin: Distinct and independent functions during tissue repair. Fibrogenesis Tissue Repair 2011, 4, 21. [Google Scholar] [CrossRef] [Green Version]
- Rogers, H.J.; Nakashima, M.O.; Kottke-Marchant, K. Hemostasis and thrombosis. In Hematopathology; Elsevier: Amsterdam, The Netherlands, 2018; pp. 57–105.e104. [Google Scholar]
- Woodhouse, K.; Brash, J. Adsorption of plasminogen from plasma to lysine-derivatized polyurethane surfaces. Biomaterials 1992, 13, 1103–1108. [Google Scholar] [CrossRef] [PubMed]
- Quandt, J.; Garay-Sarmiento, M.; Witzdam, L.; Englert, J.; Rutsch, Y.; Stöcker, C.; Obstals, F.; Grottke, O.; Rodriguez-Emmenegger, C. Interactive Hemocompatible Nanocoating to Prevent Surface-Induced Coagulation in Medical Devices. Adv. Mater. Interfaces 2022, 9, 2201055. [Google Scholar] [CrossRef]
- Nalezinková, M. In vitro hemocompatibility testing of medical devices. Thromb. Res. 2020, 195, 146–150. [Google Scholar] [CrossRef]
- Lippi, G. Systematic assessment of the hemolysis index: Pros and cons. Adv. Clin. Chem. 2015, 71, 157–170. [Google Scholar]
- Thomas, M.; Valette, P.; Mausset, A.-L.; Dejardin, P. High molecular weight kininogen adsorption on hemodialysis membranes: Influence of pH and relationship with contact phase activation of blood plasma. Influence of pre-treatment with poly (ethyleneimine). Int. J. Artif. Organs 2000, 23, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Renaux, J.-L.; Thomas, M.; Crost, T.; Loughraieb, N.; Vantard, G. Activation of the kallikrein-kinin system in hemodialysis: Role of membrane electronegativity, blood dilution, and pH. Kidney Int. 1999, 55, 1097–1103. [Google Scholar] [CrossRef] [Green Version]
- Dobrovolskaia, M.A.; Patri, A.K.; Simak, J.; Hall, J.B.; Semberova, J.; De Paoli Lacerda, S.H.; McNeil, S.E. Nanoparticle size and surface charge determine effects of PAMAM dendrimers on human platelets in vitro. Mol. Pharm. 2012, 9, 382–393. [Google Scholar] [CrossRef] [Green Version]
- Koziara, J.M.; Oh, J.J.; Akers, W.S.; Ferraris, S.P.; Mumper, R.J. Blood compatibility of cetyl alcohol/polysorbate-based nanoparticles. Pharm. Res. 2005, 22, 1821–1828. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| A | B | C | D | E | |
|---|---|---|---|---|---|
| PMβCD:MPC | 4:0 | 3:1 | 2:2 | 1:3 | 0:4 |
| PMβCD (mmol) | 1.25 | 0.9375 | 0.625 | 0.3125 | 0 |
| MPC (mmol) | 0 | 0.3125 | 0.625 | 0.9375 | 1.25 |
| Particle | Helix | Beta-Sheet | Beta-Turn | Random Coil | |
|---|---|---|---|---|---|
| Lysozyme | Blank | 33.8 ± 0.1 | 19.7 ± 0.2 | 18.1 ± 0.0 | 28.4 ± 0.3 |
| A (1:0) | 30.4 ± 0.2 | 22.1 ± 0.1 | 18.0 ± 0.1 | 29.6 ± 0.1 | |
| B (3:1) | 30.7 ± 0.1 | 21.8 ± 0.3 | 18.0 ± 0.0 | 29.5 ± 0.0 | |
| C (1:1) | 31.1 ± 0.1 | 21.3 ± 0.2 | 18.0 ± 0.0 | 29.6 ± 0.3 | |
| D (1:3) | 30.6 ± 0.3 | 21.9 ± 0.2 | 18.0 ± 0.0 | 29.9 ± 0.5 | |
| E (0:4) | 31.4 ± 0.1 | 21.0 ± 0.1 | 17.9 ± 0.1 | 29.7 ± 0.2 | |
| Bare MNP | 30.7 ± 0.2 | 21.8 ± 0.3 | 17.9 ± 0.1 | 29.5 ± 0.1 | |
| Human serum albumin | Blank | 71.0 ± 0.1 | 4.1 ± 0.2 | 12.6 ± 0.0 | 12.3 ± 0.2 |
| A (1:0) | 71.0 ± 0.2 | 4.1 ± 0.1 | 12.6 ± 0.1 | 12.3 ± 0.2 | |
| B (3:1) | 71.1 ± 0.3 | 4.1 ± 0.4 | 12.6 ± 0.0 | 12.3 ± 0.2 | |
| C (1:1) | 67.4 ± 0.1 | 5.4 ± 0.2 | 12.9 ± 0.2 | 14.3 ± 0.3 | |
| D (1:3) | 62.6 ± 0.5 | 6.0 ± 0.1 | 13.4 ± 0.3 | 18.0 ± 0.2 | |
| E (0:4) | 68.7 ± 0.2 | 6.5 ± 0.2 | 13.0 ± 0.2 | 11.8 ± 0.1 | |
| Bare MNP | 70.3 ± 0.1 | 4.2 ± 0.3 | 12.7 ± 0.1 | 12.7 ± 0.1 | |
| α-Lactalbumin | Blank | 31.7 ± 0.2 | 22.1 ± 0.3 | 18.2 ± 0.0 | 28.0 ± 0.3 |
| A (1:0) | 31.7 ± 0.1 | 22.1 ± 0.2 | 18.2 ± 0.1 | 28.0 ± 0.2 | |
| B (3:1) | 30.8 ± 0.3 | 22.4 ± 0.2 | 18.2 ± 0.1 | 28.6 ± 0.1 | |
| C (1:1) | 31.4 ± 0.4 | 21.7 ± 0.2 | 18.1 ± 0.1 | 28.7 ± 0.2 | |
| D (1:3) | 30.0 ± 0.1 | 22.9 ± 0.3 | 18.1 ± 0.0 | 29.0 ± 0.3 | |
| E (0:4) | 31.3 ± 0.2 | 22.1 ± 0.4 | 18.2 ± 0.1 | 28.5 ± 0.2 | |
| Bare MNP | 28.2 ± 0.2 | 22.4 ± 0.3 | 21.1 ± 0.1 | 28.4 ± 0.1 |
| A | B | C | D | E | Bare MNP | Plasma | |
|---|---|---|---|---|---|---|---|
| Turbidity | 0.67 | 0.57 | 0.64 | 0.45 | 0.74 | 0.79 | 0.93 |
| Clotting starting point (min) | 2 | 1 | 1 | 2 | 2 | 2 | 10 |
| Plateau point (min) | 5 | 5 | 5 | 6 | 6 | 6 | 30 |
| Plasma Protein | Fragment Size (kDa) | Bare MNP | A PMβCD:MPC (4:0) | B (3:1) | C (2:2) | D (1:3) | E (0:4) |
|---|---|---|---|---|---|---|---|
| Fibrinogen | 68 | 7 | 8 | 8 | 7 | 7 | 6 |
| 56 | 6 | 8 | 8 | 7 | 4 | 4 | |
| 48 | 6 | 7 | 8 | 6 | 4 | 4 | |
| <48 | 4 | 3 | 5 | 2 | 0 | 0 | |
| α1-Antitrypsin | 54 | 9 | 8 | 9 | 8 | 5 | 5 |
| Prothrombin | 72 | 2 | 2 | 2 | 2 | 1 | 2 |
| Vitronectin | 54 | 8 | 8 | 10 | 8 | 5 | 6 |
| Prekallikrein | 85 50 | 5 10 | 2 8 | 5 9 | 2 7 | 1 2 | 2 5 |
| Antithrombin | 53 | 6 | 6 | 4 | 4 | 3 | 3 |
| IgG | 55 | 7 | 4 | 3 | 8 | 5 | 5 |
| 27 | 8 | 7 | 8 | 9 | 5 | 5 | |
| Albumin | 66 | 9 | 7 | 7 | 8 | 9 | 9 |
| Plasminogen | 91 | 9 | 8 | 7 | 8 | 5 | 3 |
| C3 | 187 | 0 | 0 | 0 | 0 | 0 | 0 |
| 115 | 5 | 4 | 4 | 5 | 3 | 2 | |
| 70 | 10 | 10 | 10 | 9 | 7 | 8 | |
| 42 | 5 | 8 | 8 | 7 | 3 | 3 | |
| Factor XII | 80 | 3 | 2 | 1 | 2 | 1 | 1 |
| Factor XI | 70 | 9 | 8 | 9 | 10 | 8 | 8 |
| Transferrin | 77 | 8 | 8 | 7 | 10 | 10 | 8 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, S.; Sharaf, M.G.; Rowe, E.M.; Serrano, K.; Devine, D.V.; Unsworth, L.D. Hemocompatibility of β-Cyclodextrin-Modified (Methacryloyloxy)ethyl Phosphorylcholine Coated Magnetic Nanoparticles. Biomolecules 2023, 13, 1165. https://doi.org/10.3390/biom13081165
Li S, Sharaf MG, Rowe EM, Serrano K, Devine DV, Unsworth LD. Hemocompatibility of β-Cyclodextrin-Modified (Methacryloyloxy)ethyl Phosphorylcholine Coated Magnetic Nanoparticles. Biomolecules. 2023; 13(8):1165. https://doi.org/10.3390/biom13081165
Chicago/Turabian StyleLi, Shuhui, Mehdi Ghaffari Sharaf, Elyn M. Rowe, Katherine Serrano, Dana V. Devine, and Larry D. Unsworth. 2023. "Hemocompatibility of β-Cyclodextrin-Modified (Methacryloyloxy)ethyl Phosphorylcholine Coated Magnetic Nanoparticles" Biomolecules 13, no. 8: 1165. https://doi.org/10.3390/biom13081165
APA StyleLi, S., Sharaf, M. G., Rowe, E. M., Serrano, K., Devine, D. V., & Unsworth, L. D. (2023). Hemocompatibility of β-Cyclodextrin-Modified (Methacryloyloxy)ethyl Phosphorylcholine Coated Magnetic Nanoparticles. Biomolecules, 13(8), 1165. https://doi.org/10.3390/biom13081165