
Local Control Model of a Human Ventricular Myocyte: An Exploration of Frequency-Dependent Changes and Calcium Sparks




Abstract
:1. Introduction
2. Materials and Methods
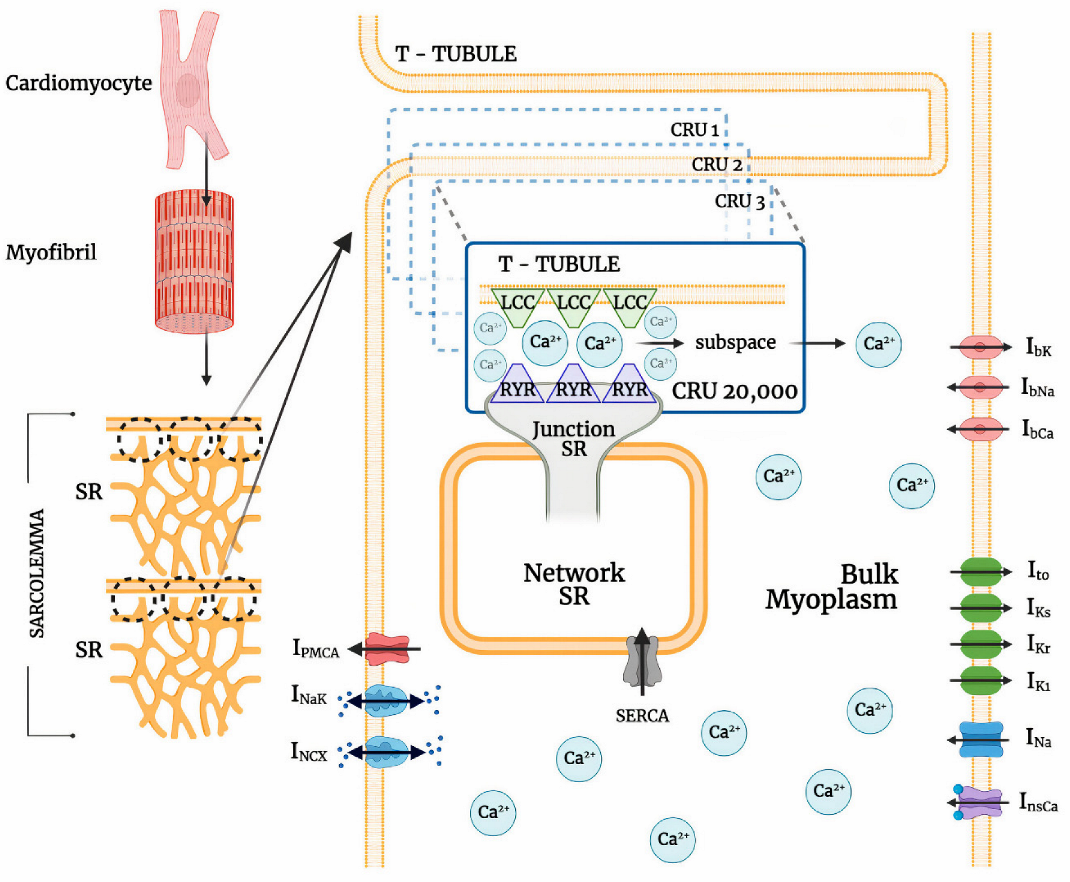
2.1. Model Formulation and Development
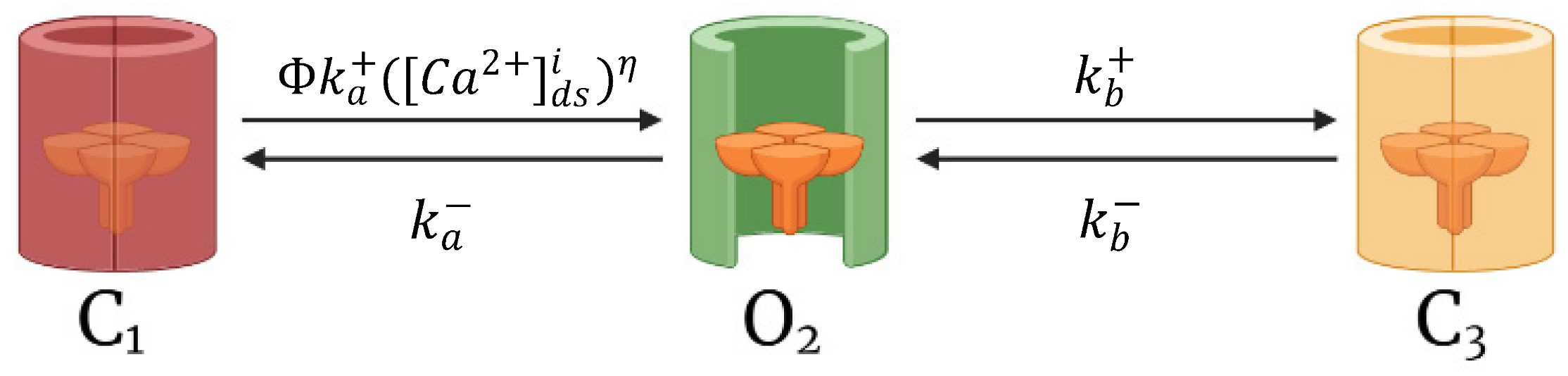
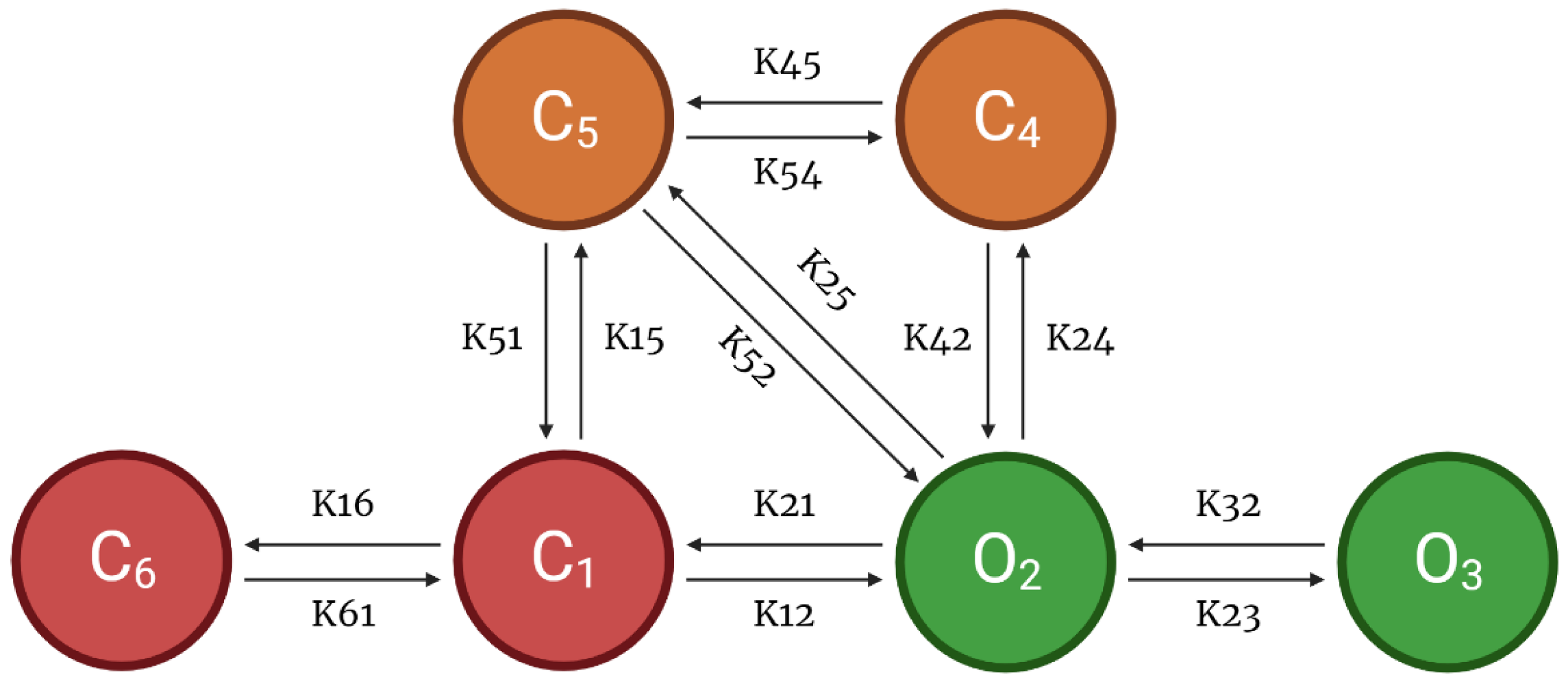
2.2. Calcium-Release Units
2.3. L-Type Calcium Channel Permeability
2.4. Modified Ito
2.5. Numerical Methods
2.6. Pacing Protocols
2.7. Predicted Force
3. Results
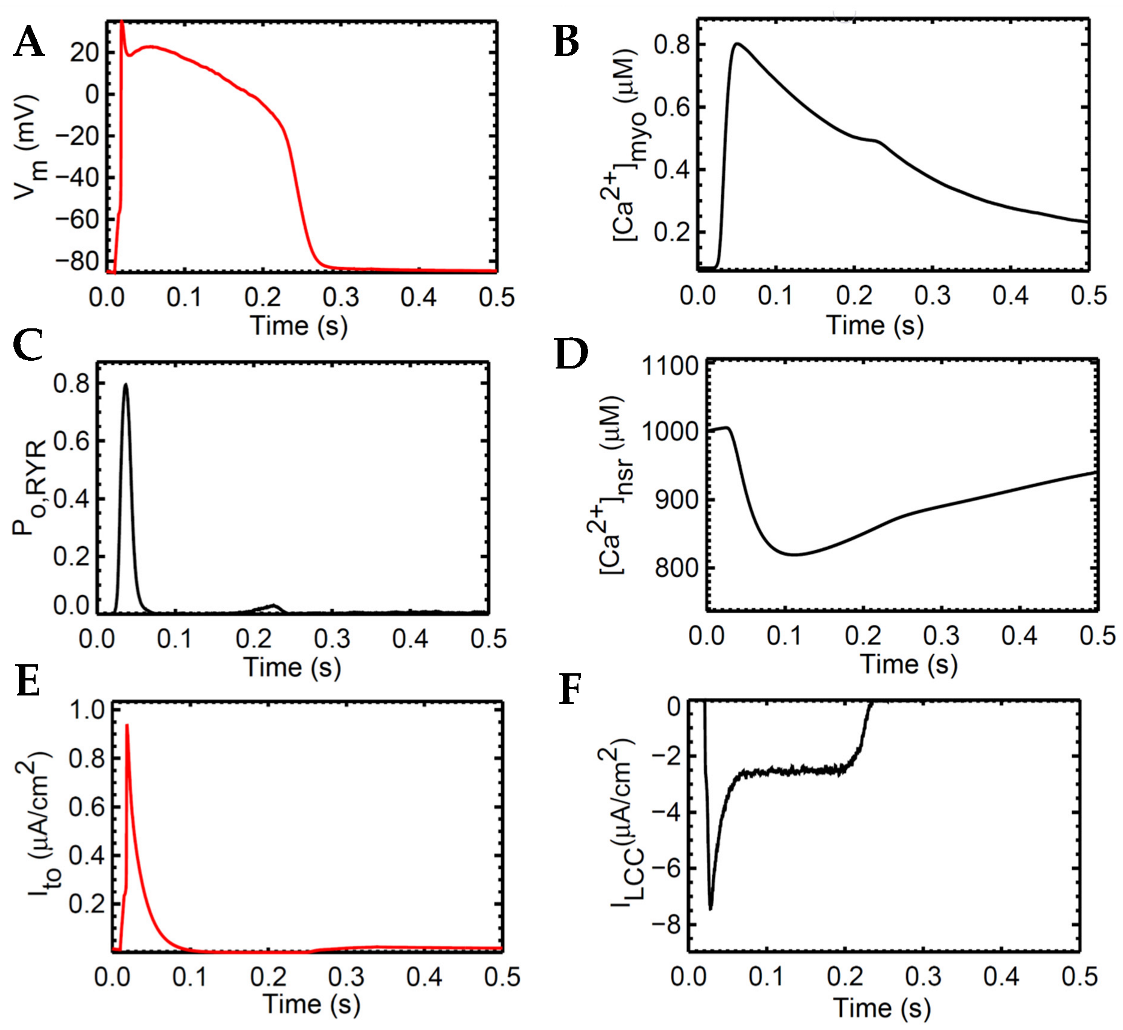
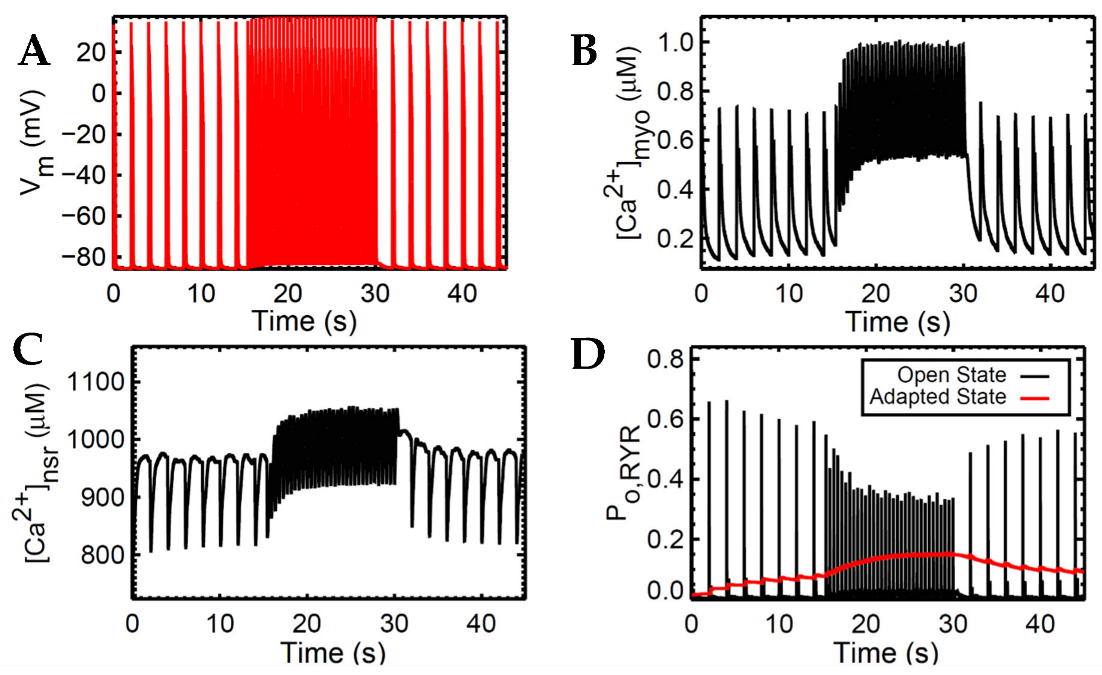
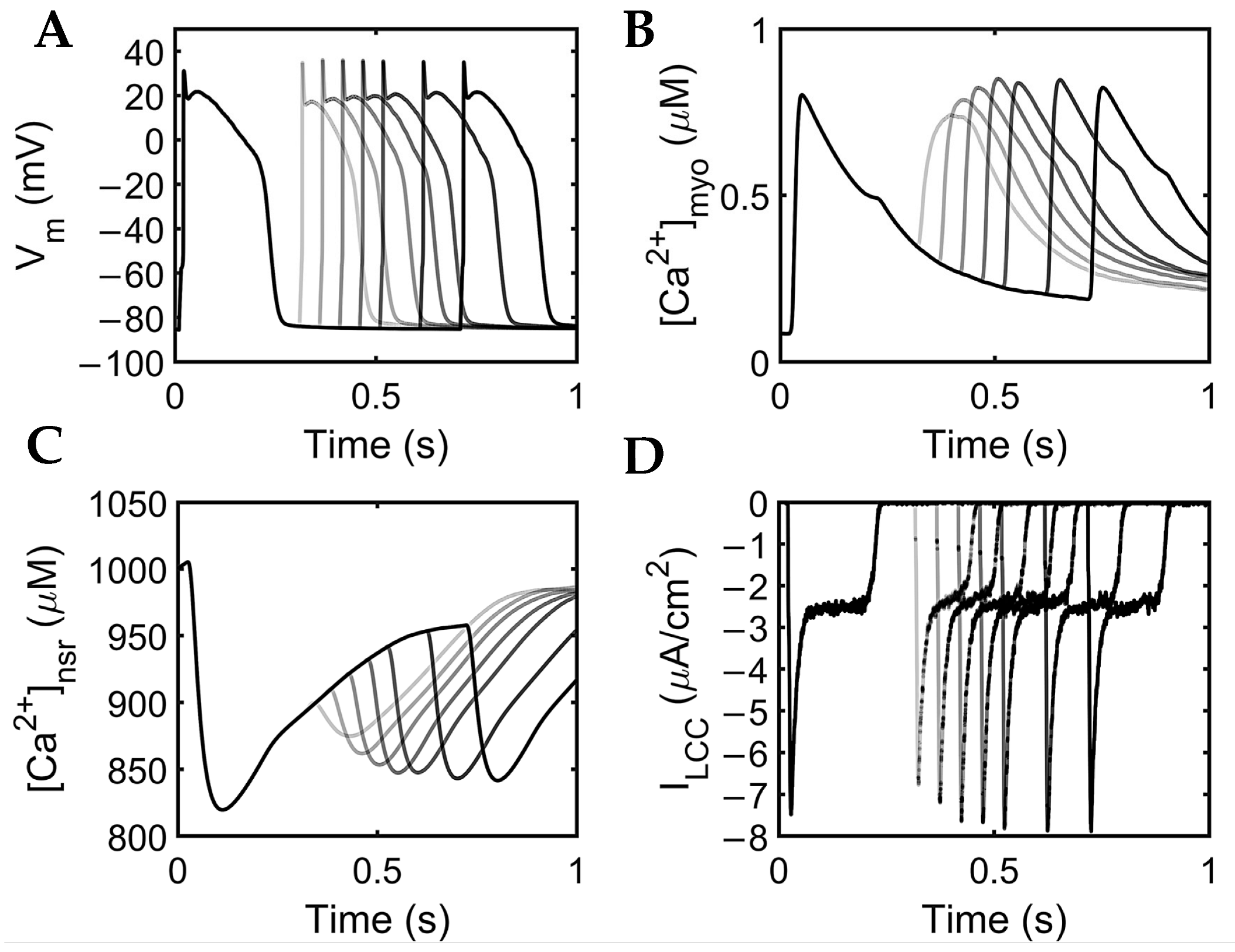
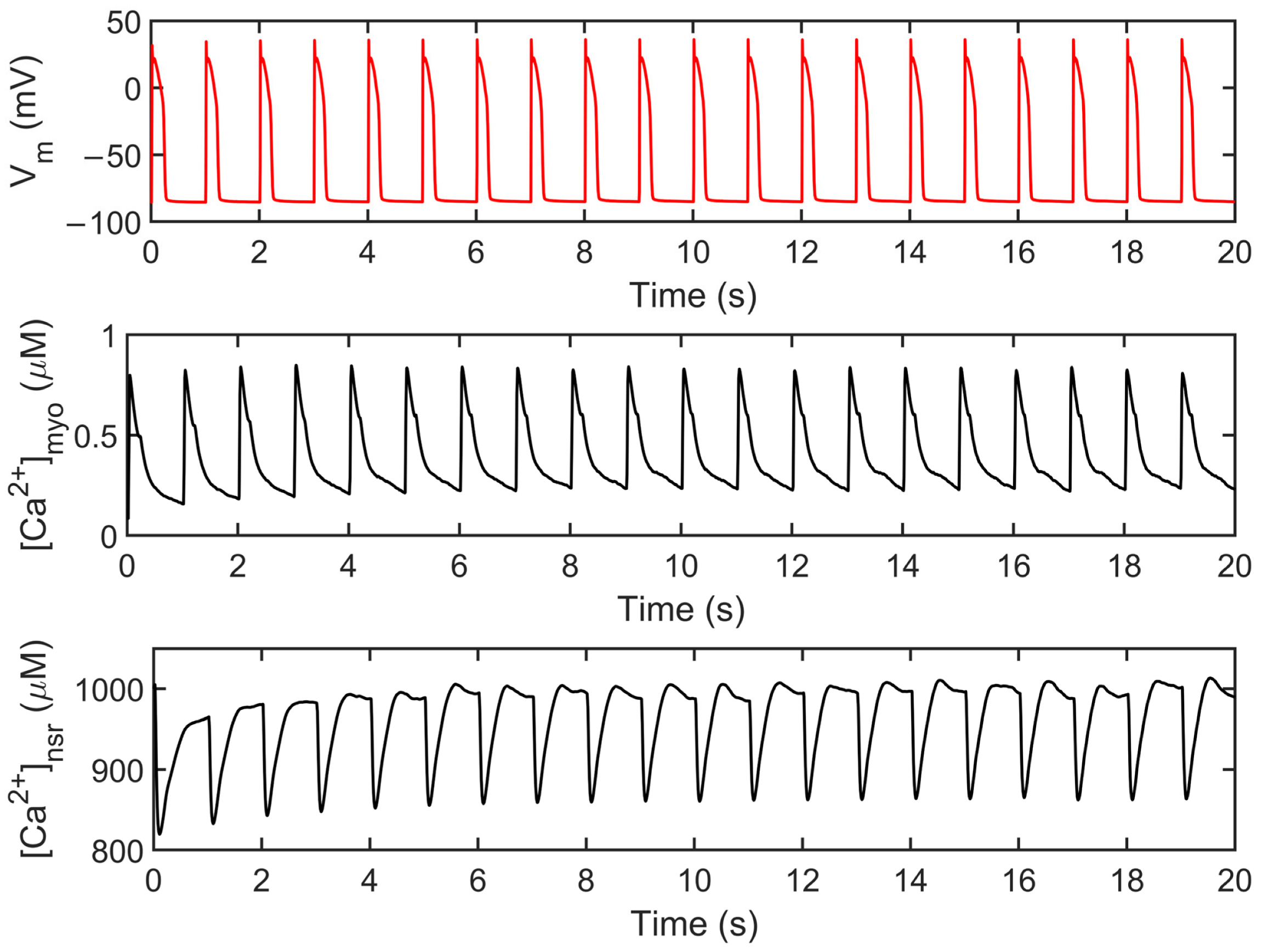
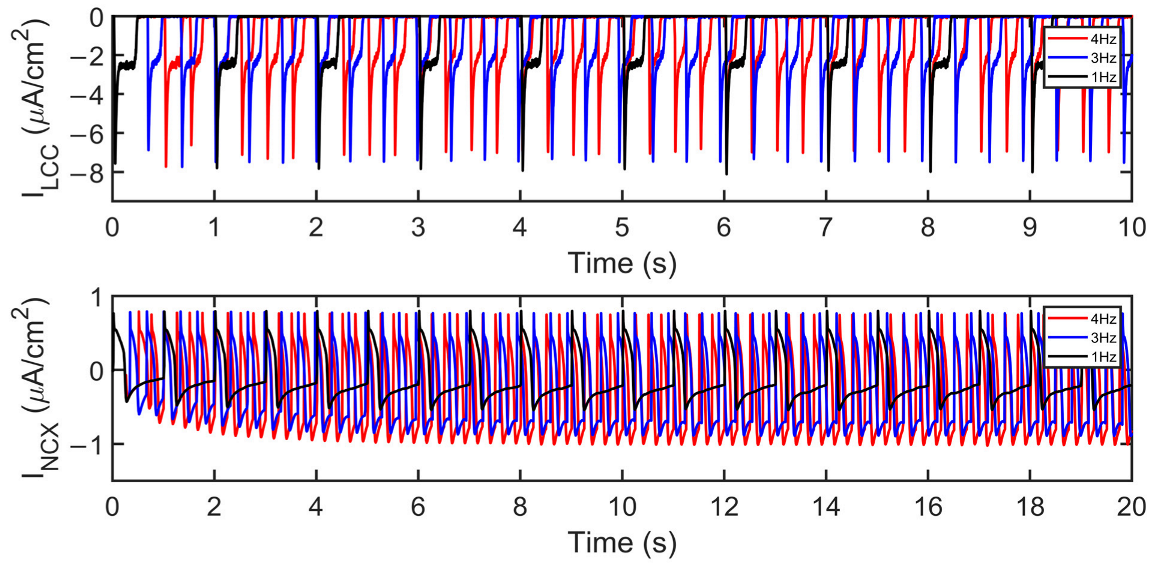
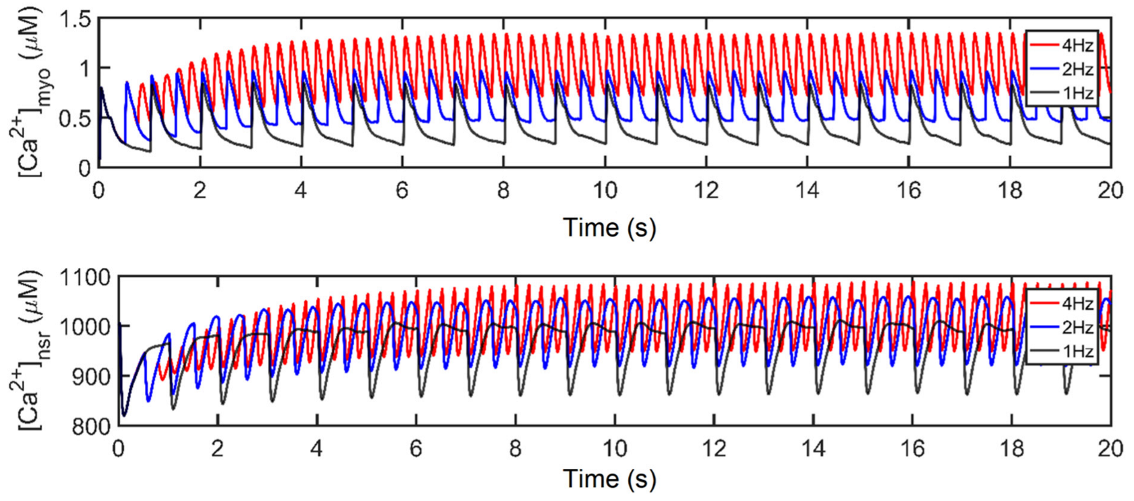
3.1. Excitation–Contraction Coupling Dynamics: 1 Hz Simulations
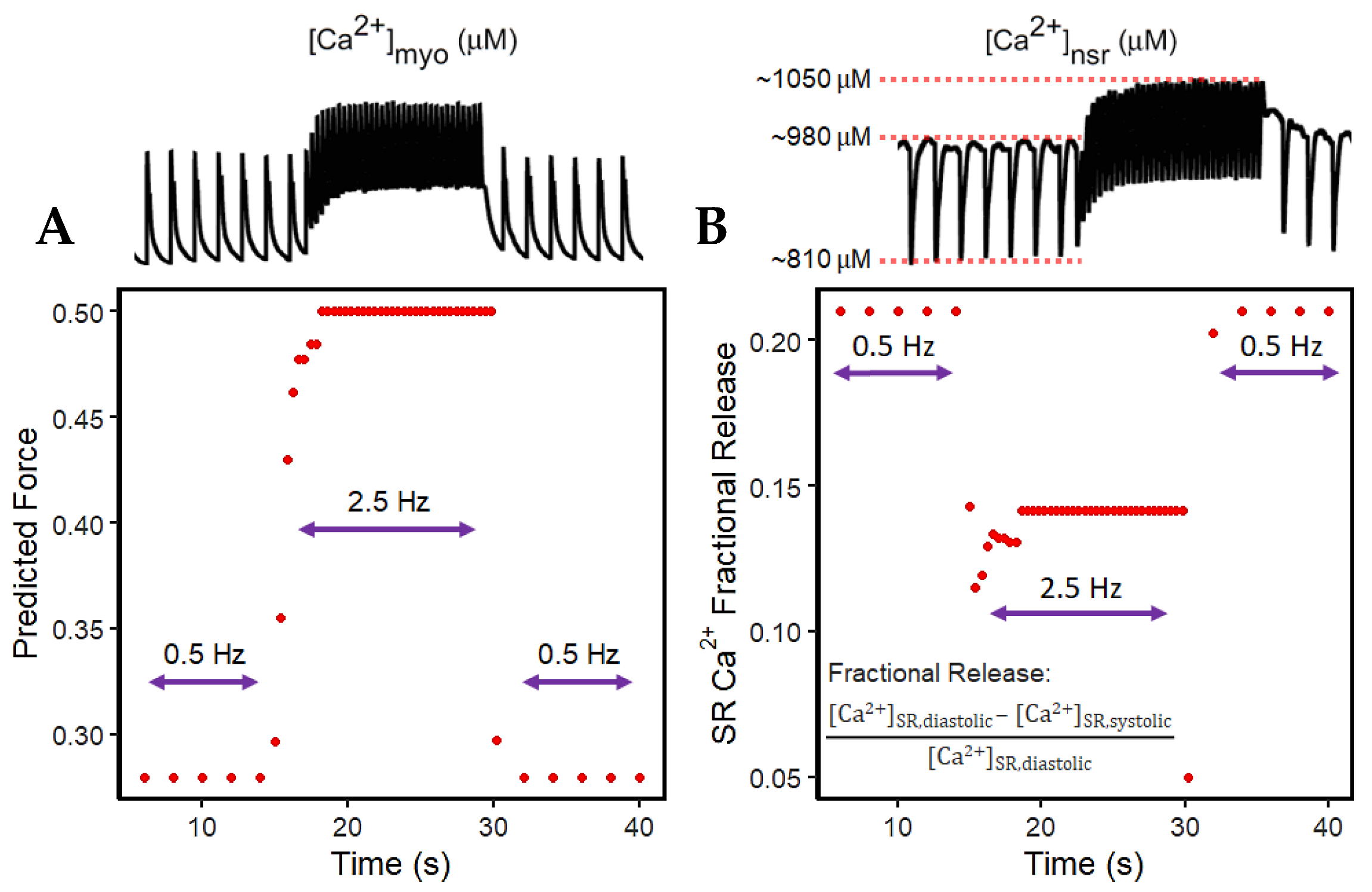
3.2. Interval–Force Relations and the Force–Frequency Relationship
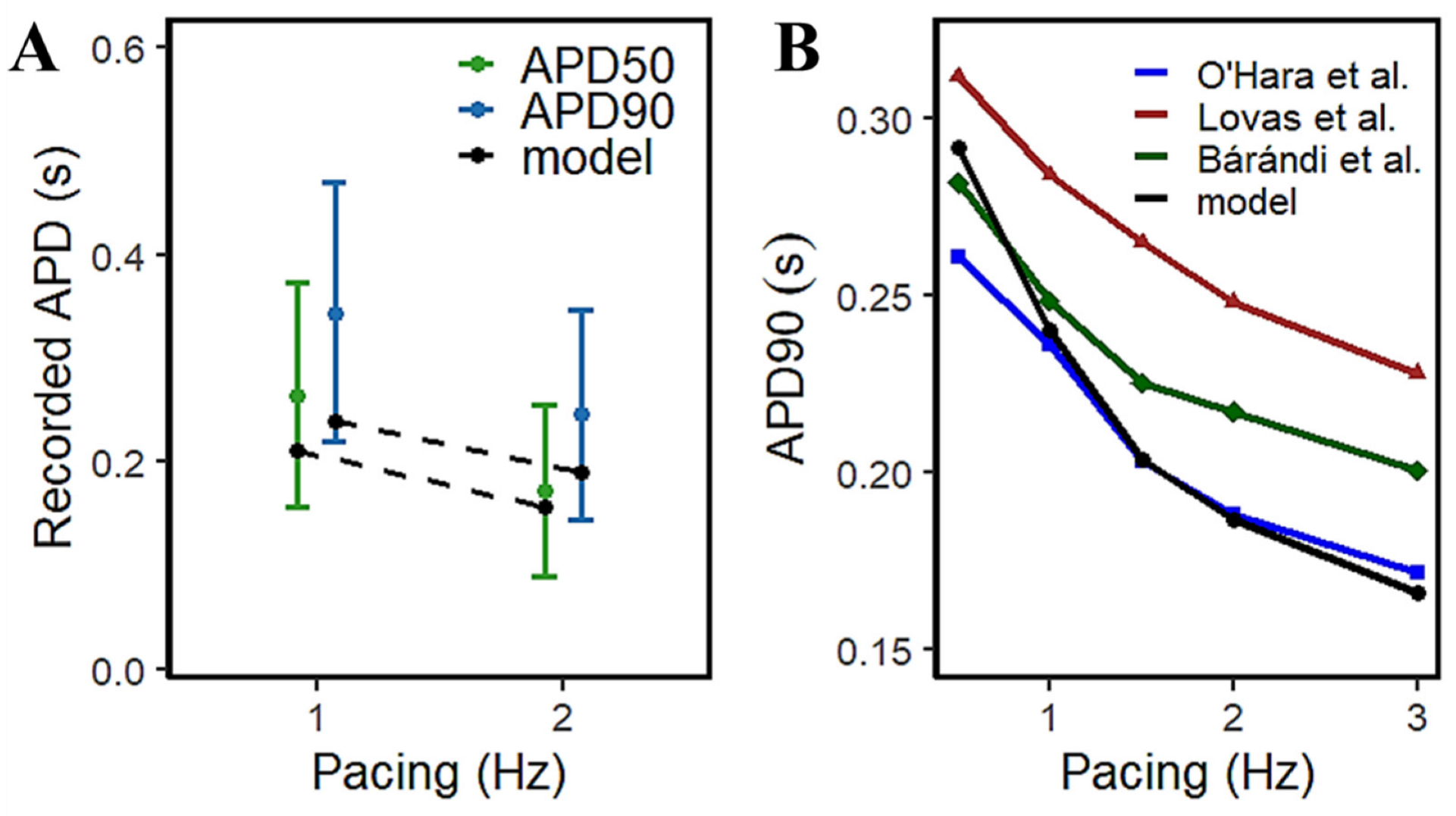
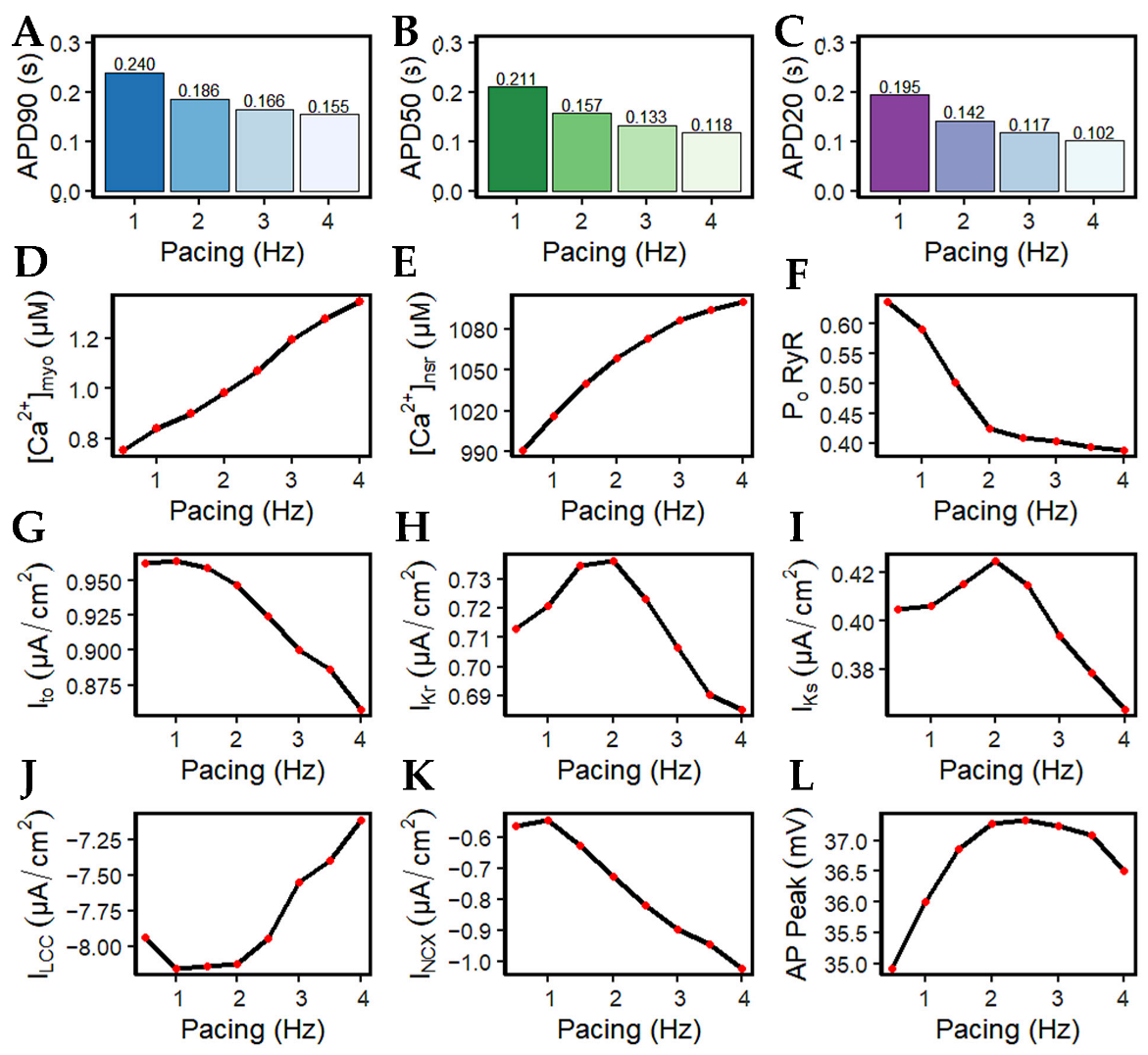
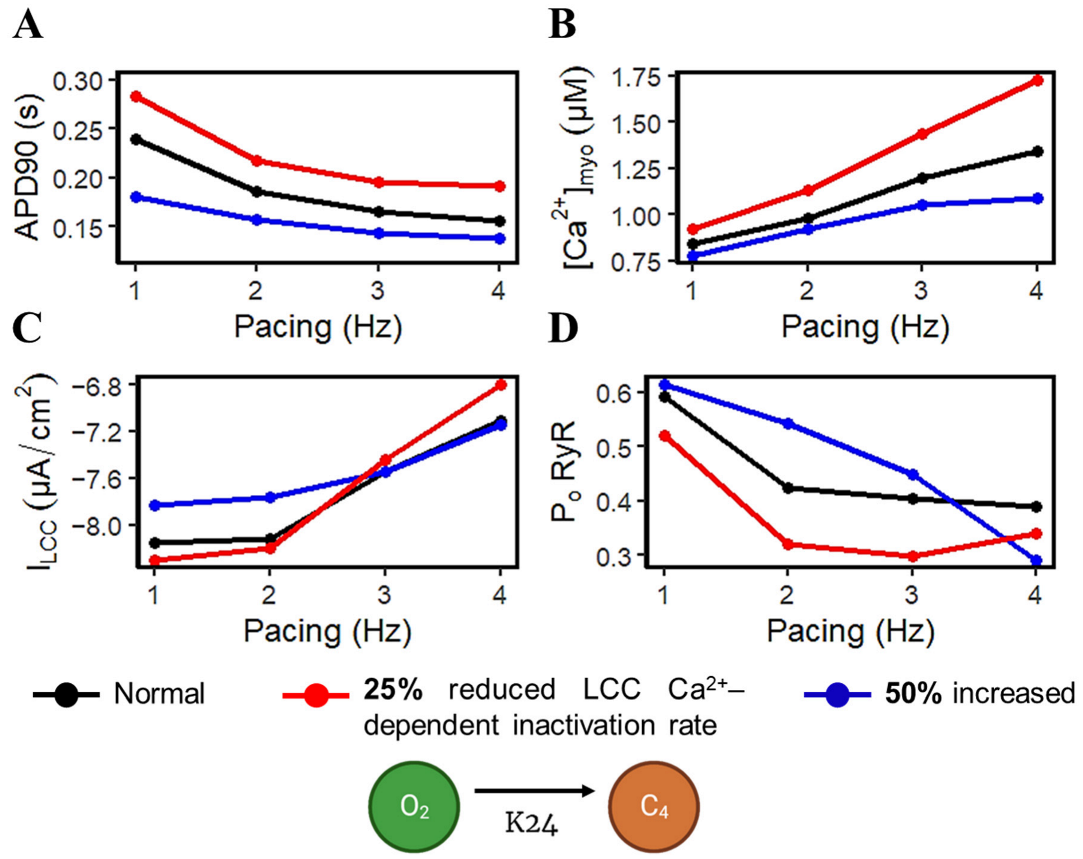
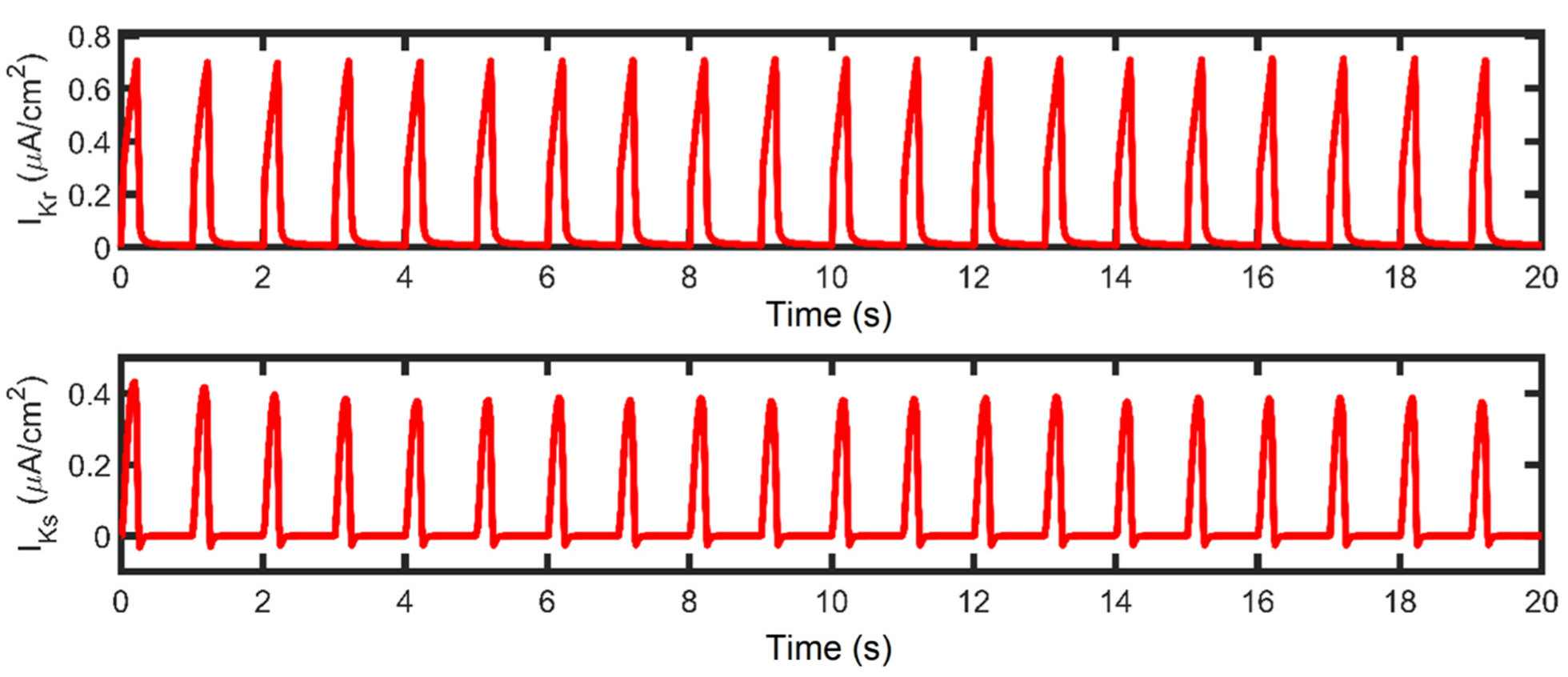
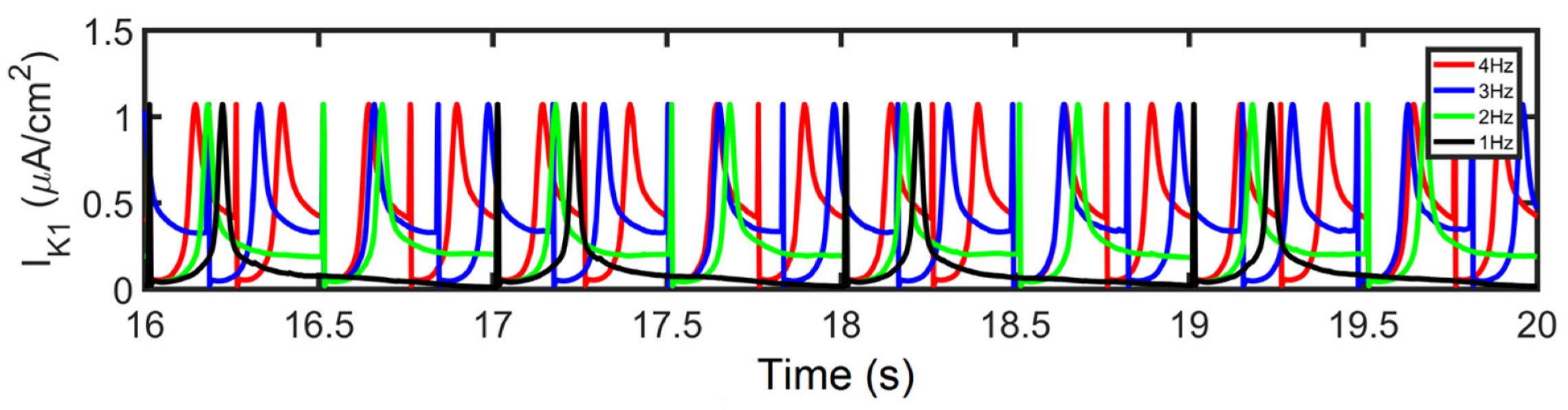
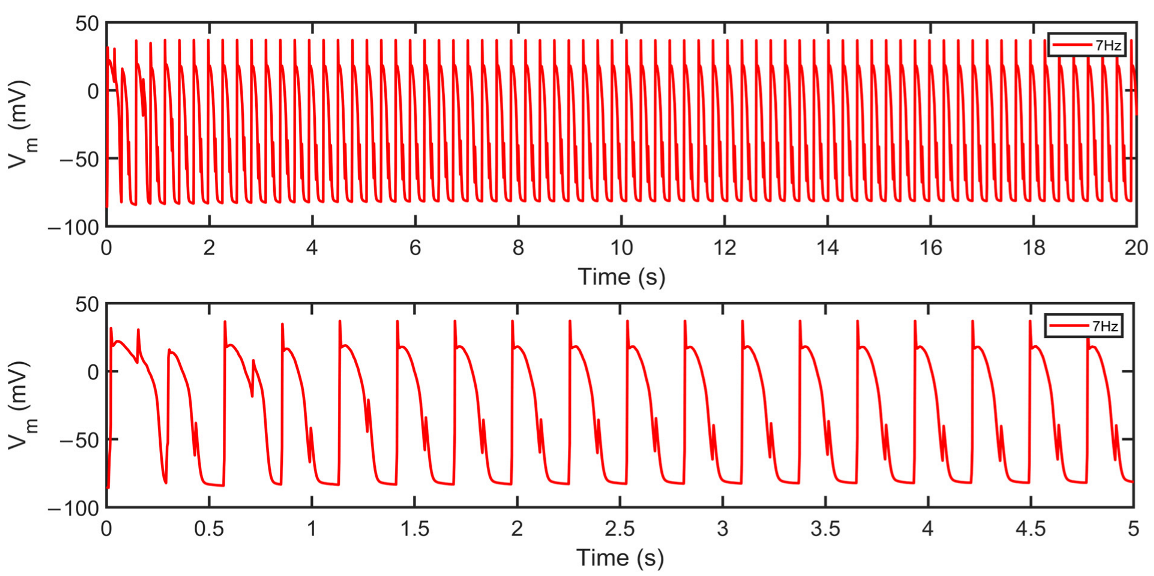
3.3. APD Rate Dependence and Mechanisms Involved at Higher Pacing Rates
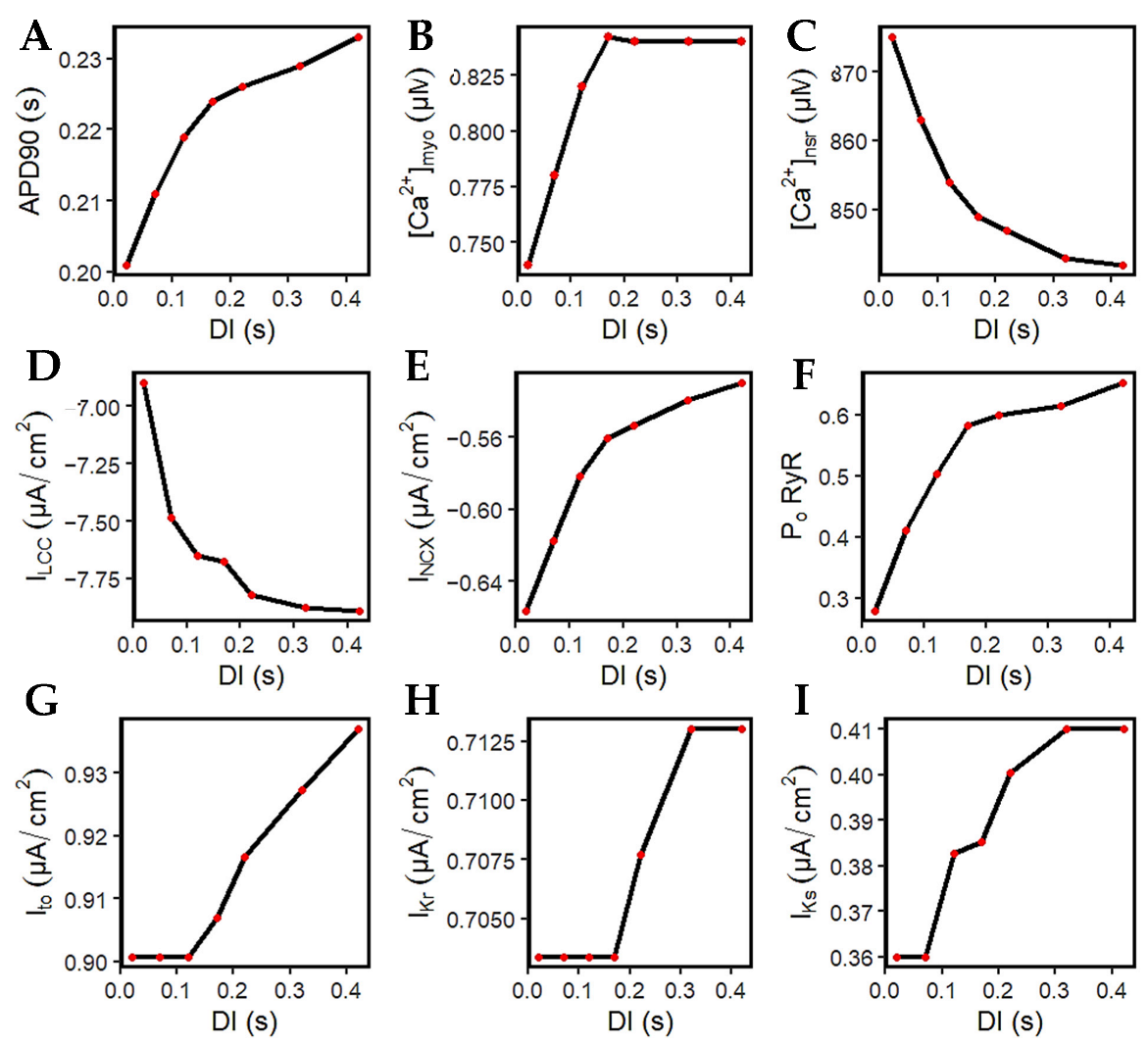
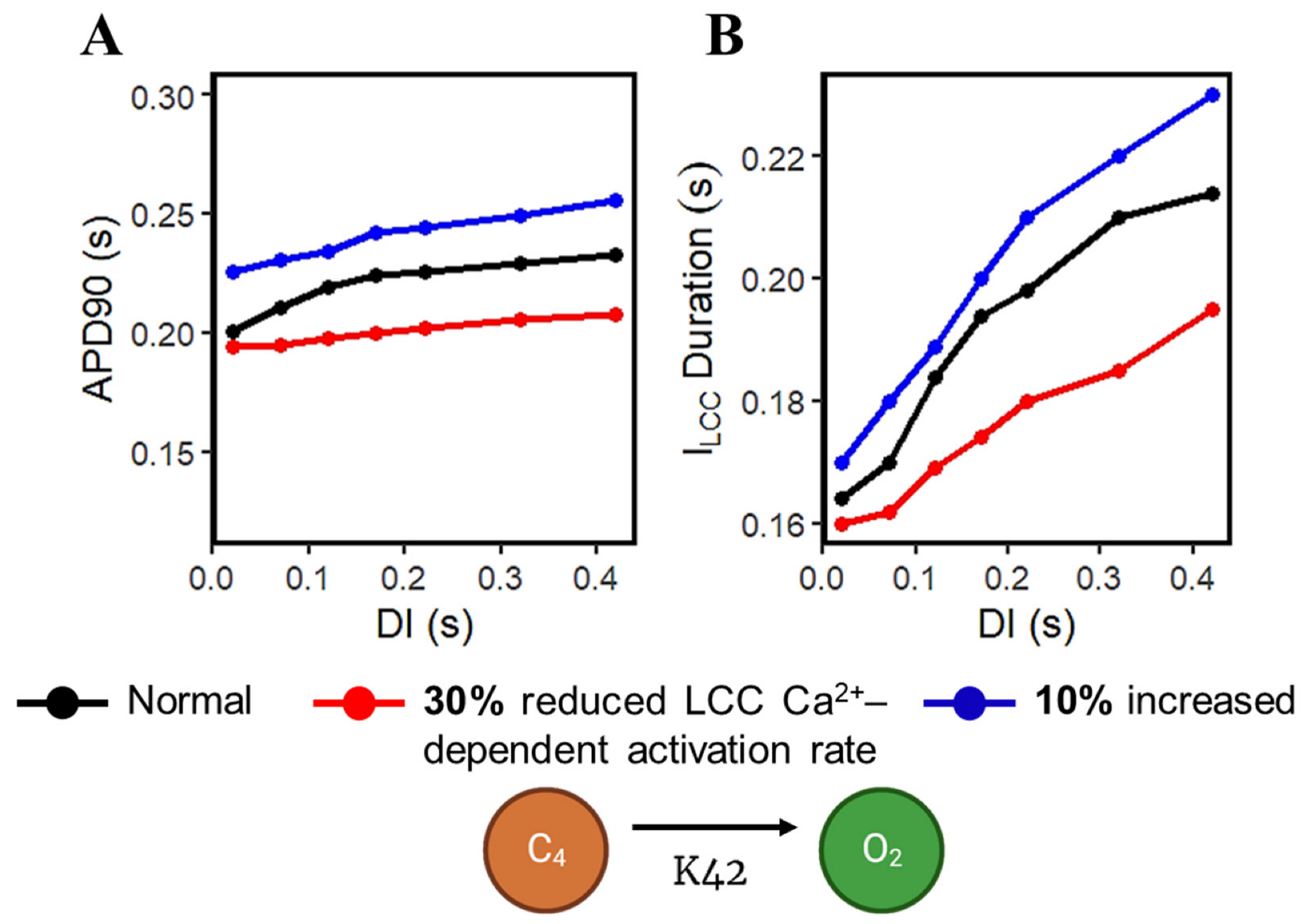
3.4. Restitution Analyses through S1S2 Protocol
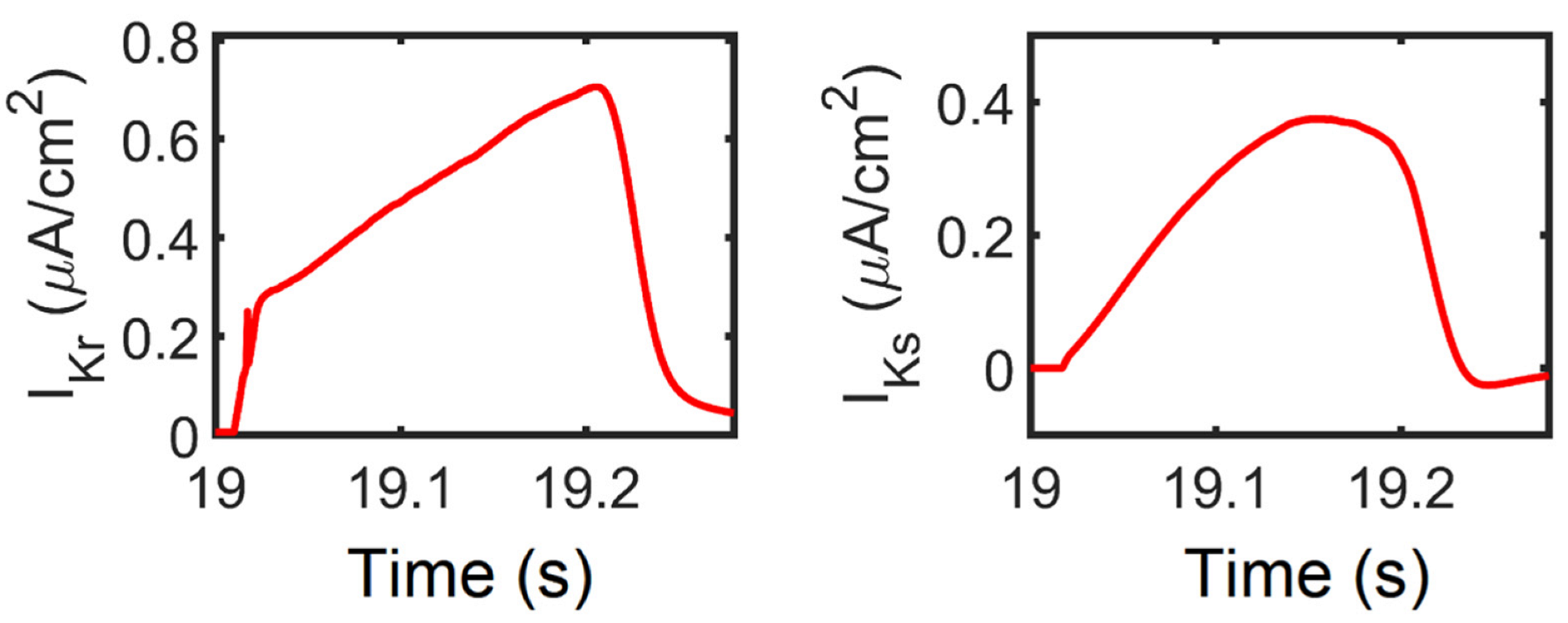
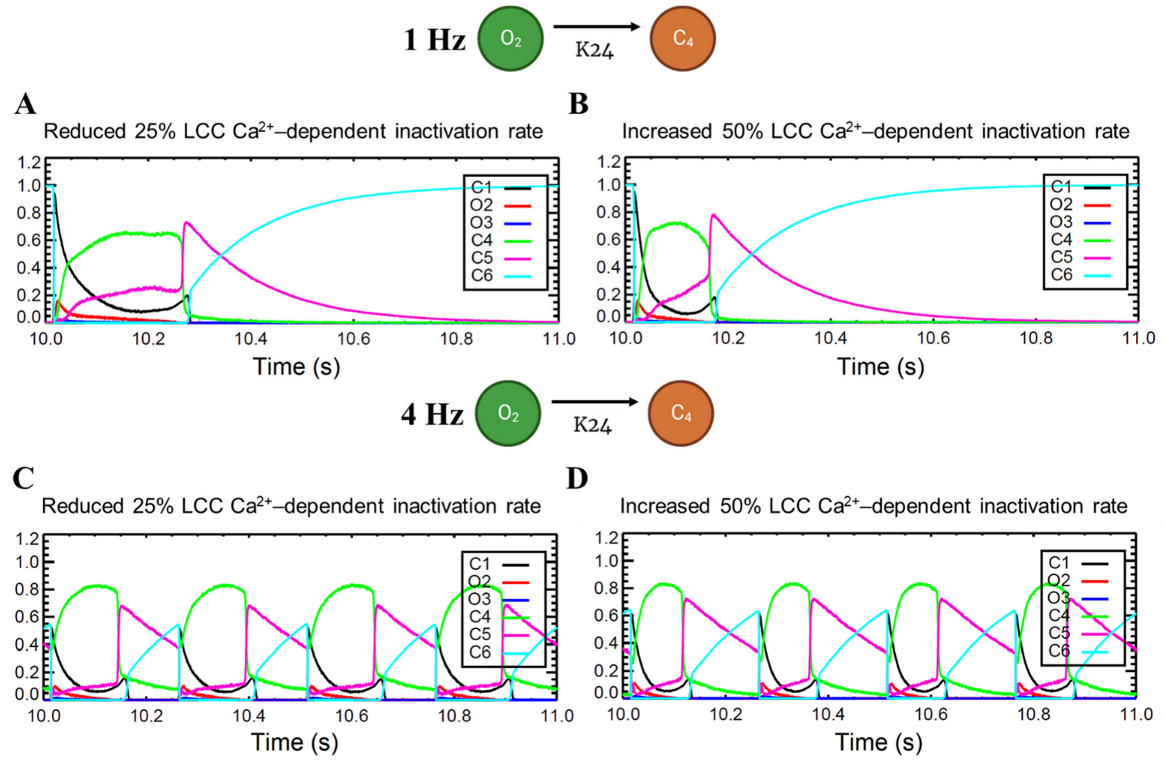
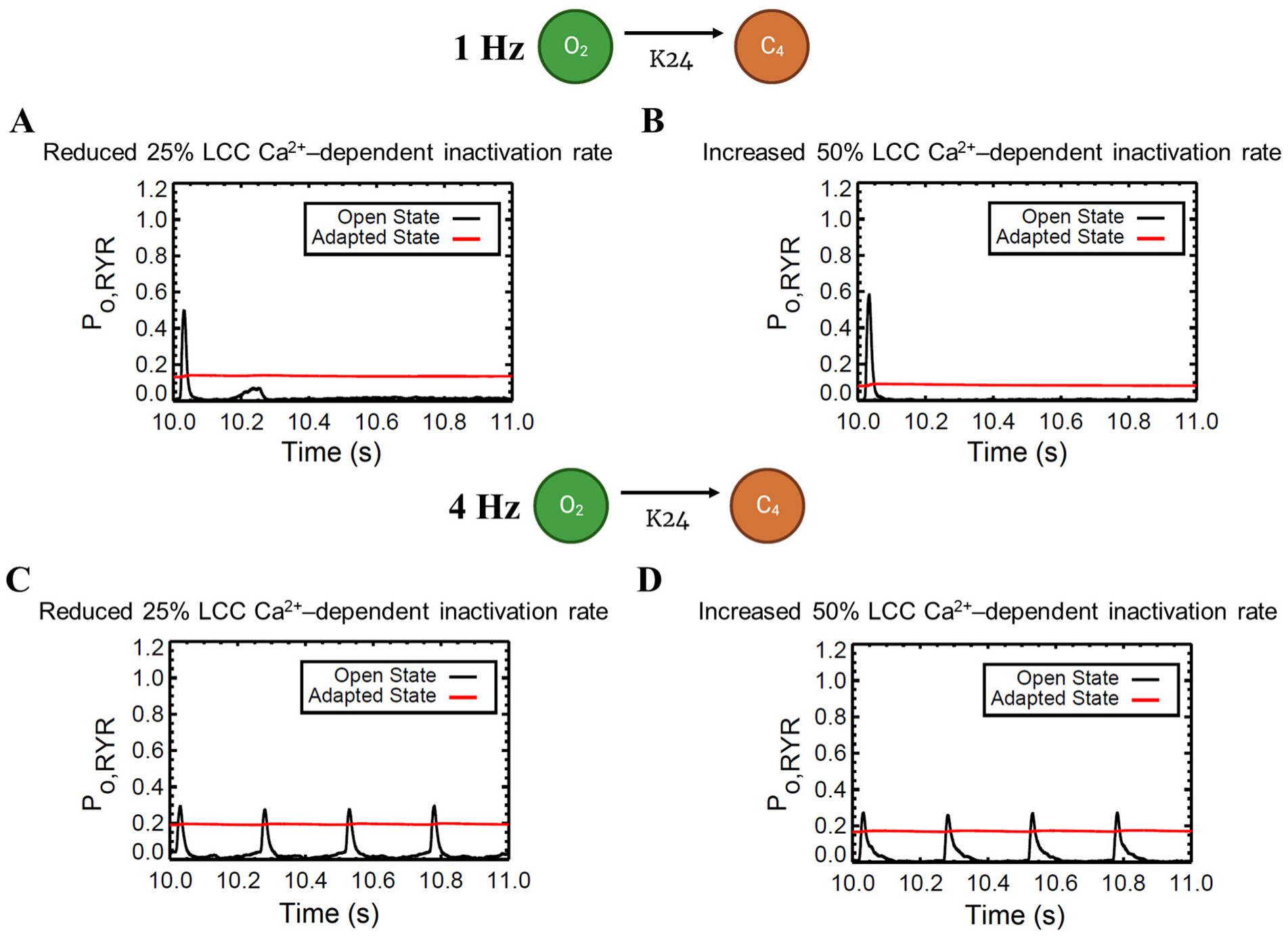
3.5. APD Restitution Mechanisms and Determinants: L-Type Calcium Channel, RyR Open Probability, and IK Currents
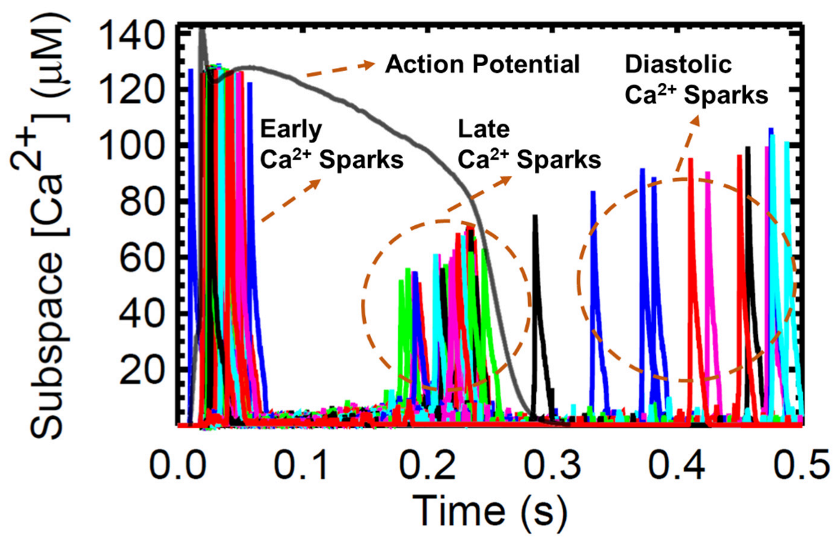
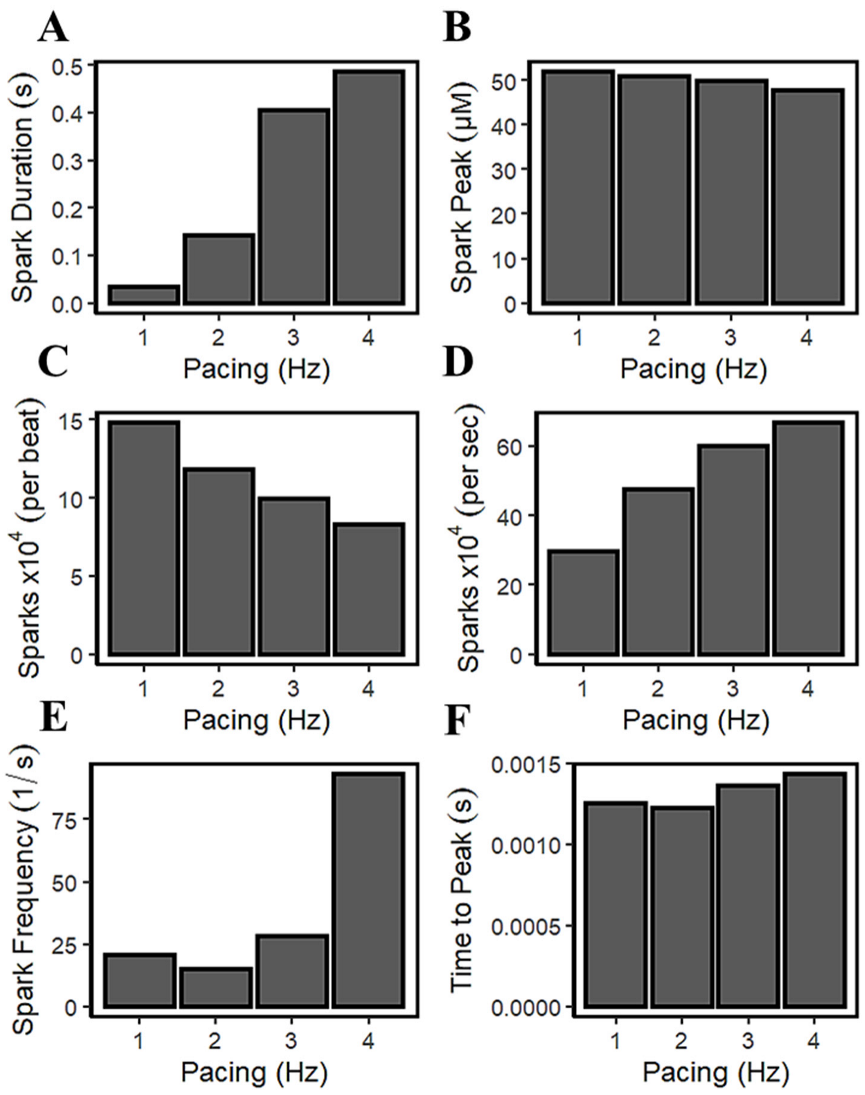
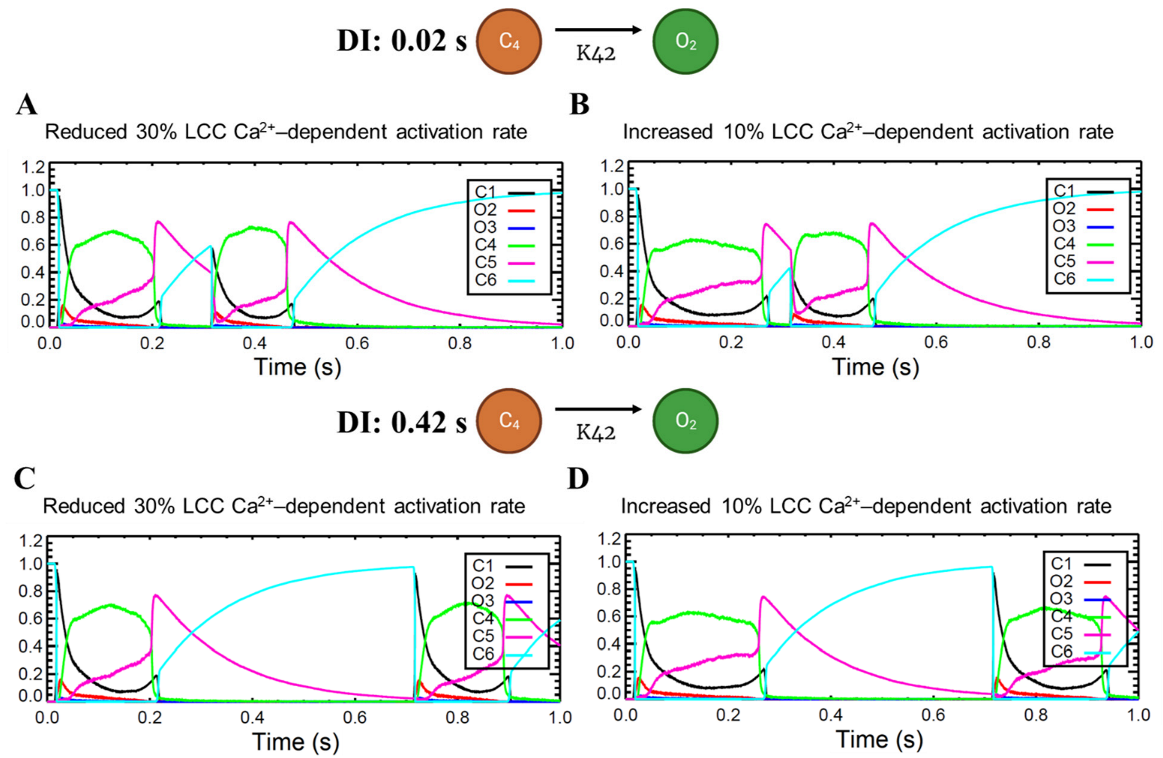
3.6. Frequency-Dependent Calcium Spark Behaviors and Characteristics
4. Discussion
4.1. Advantages of the Model versus Early Studies
4.2. Interval-Force Relations Depend on RyR Dynamics
4.3. Increased Predicted Force Is Accompanied by Reduced SR Ca2+ Fractional Release during Rapid Pacing
4.4. Rate-Dependent Changes and Mechanisms in APD Shortening
4.5. S1S2 Restitution Mechanisms and Determinants
4.6. Calcium Spark Characteristics Change with Pacing
4.7. Implications of Frequency-Dependent Changes in Ca2+ Mishandling
4.8. Model Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A
Appendix A.1. Appendix Figures








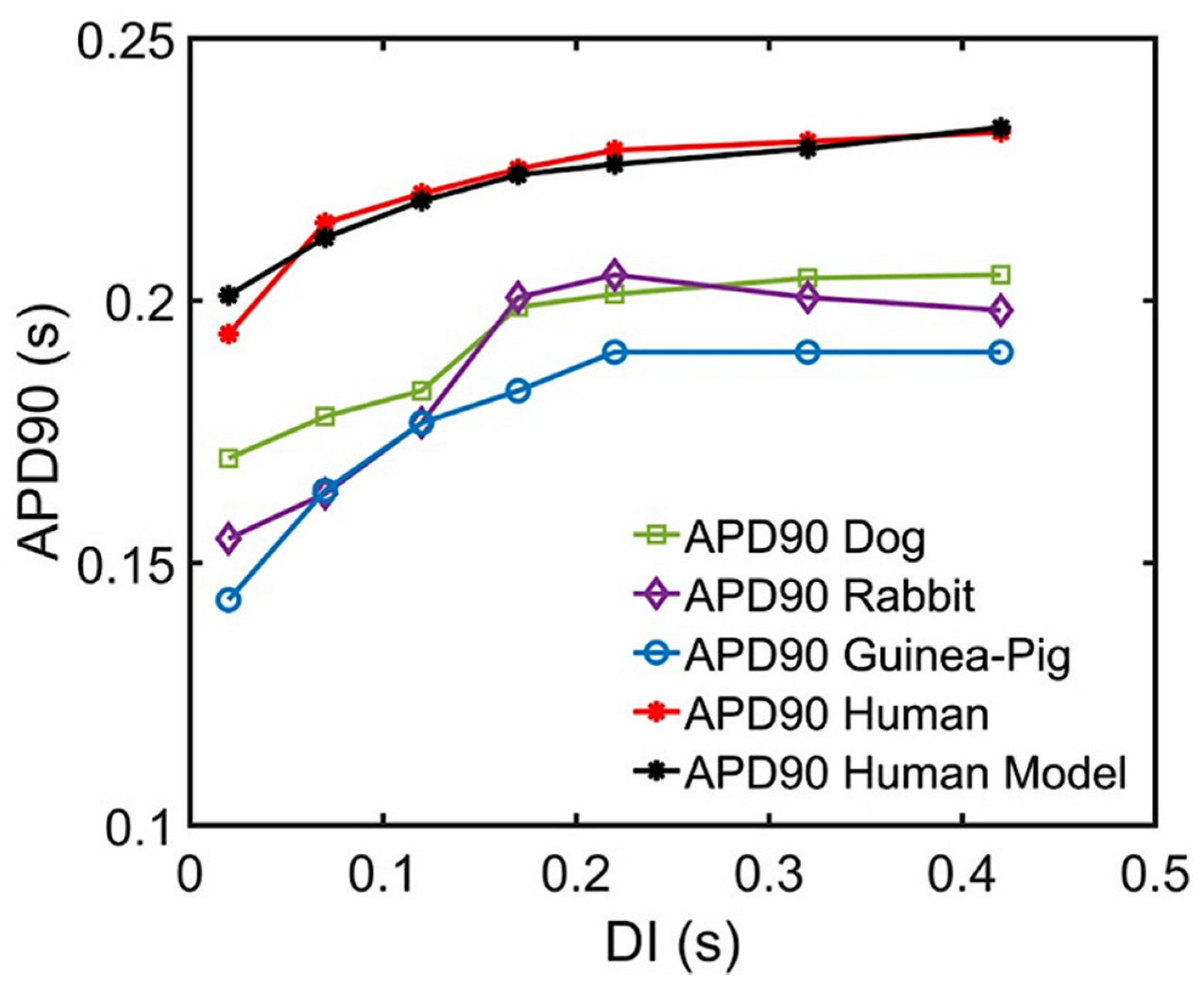
Appendix A.2. Interspecies APD Restitution and Slope



Appendix A.3. Appendix Table

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Time (s) | 14.0 | 15.0 | 15.4 | 15.8 | 16.2 | 16.6 | 17.0 | 17.4 | 17.8 | 18.2 | 18.6 | 19.0 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Peak Systolic [Ca2+]myo (µM) | 0.73 | 0.75 | 0.82 | 0.91 | 0.95 | 0.97 | 0.97 | 0.98 | 0.98 | 1.0 | 1.0 | 1.0 |
| Predicted Steady-State Force | 0.280 | 0.297 | 0.355 | 0.430 | 0.462 | 0.477 | 0.477 | 0.485 | 0.485 | 0.500 | 0.500 | 0.500 |
| Peak Diastolic [Ca2+]nsr (µM) | 980 | 960 | 970 | 985 | 1005 | 1020 | 1030 | 1030 | 1040 | 1040 | 1050 | 1050 |
| Peak Systolic [Ca2+]nsr (µM) | 810 | 840 | 870 | 880 | 890 | 900 | 910 | 910 | 920 | 920 | 920 | 920 |
| Fractional SR Release | 0.21 | 0.14 | 0.11 | 0.12 | 0.13 | 0.13 | 0.13 | 0.13 | 0.13 | 0.13 | 0.14 | 0.14 |
References
- Courtemanche, M.; Ramirez, R.J.; Nattel, S. Ionic mechanisms underlying human atrial action potential properties: Insights from a mathematical model. Am. J. Physiol. Heart Circ. Physiol. 1998, 275, H301–H321. [Google Scholar] [CrossRef]
- Nygren, A.; Fiset, C.; Firek, L.; Clark, J.W.; Lindblad, D.S.; Clark, R.B.; Giles, W.R. Mathematical Model of an Adult Human Atrial Cell. Circ. Res. 1998, 82, 63–81. [Google Scholar] [CrossRef] [PubMed]
- Grandi, E.; Pasqualini, F.S.; Bers, D.M. A novel computational model of the human ventricular action potential and Ca transient. J. Mol. Cell. Cardiol. 2010, 48, 112–121. [Google Scholar] [CrossRef] [PubMed]
- Cannell, M.B.; Cheng, H.; Lederer, W.J. Spatial non-uniformities in [Ca2+]i during excitation-contraction coupling in cardiac myocytes. Biophys. J. 1994, 67, 1942–1956. [Google Scholar] [CrossRef]
- Stern, M.D. Theory of excitation-contraction coupling in cardiac muscle. Biophys. J. 1992, 63, 497–517. [Google Scholar] [CrossRef]
- Rice, J.J.; Jafri, M.S.; Winslow, R.L. Modeling gain and gradedness of Ca2+ release in the functional unit of the cardiac diadic space. Biophys. J. 1999, 77, 1871–1884. [Google Scholar] [CrossRef]
- Greenstein, J.L.; Winslow, R.L. An integrative model of the cardiac ventricular myocyte incorporating local control of Ca2+ release. Biophys. J. 2002, 83, 2918–2945. [Google Scholar] [CrossRef]
- Himeno, Y.; Asakura, K.; Cha, C.Y.; Memida, H.; Powell, T.; Amano, A.; Noma, A. A human ventricular myocyte model with a refined representation of excitation-contraction coupling. Biophys. J. 2015, 109, 415–427. [Google Scholar] [CrossRef]
- Priebe, L.; Beuckelmann, D.J. Simulation study of cellular electric properties in heart failure. Circ. Res. 1998, 82, 1206–1223. [Google Scholar] [CrossRef]
- Luo, C.H.; Rudy, Y. A model of the ventricular cardiac action potential. Depolarization, repolarization, and their interaction. Circ. Res. 1991, 68, 1501–1526. [Google Scholar] [CrossRef]
- Luo, C.H.; Rudy, Y. A dynamic model of the cardiac ventricular action potential. I. Simulations of ionic currents and concentration changes. Circ. Res. 1994, 74, 1071–1096. [Google Scholar] [CrossRef]
- Nivala, M.; de Lange, E.; Rovetti, R.; Qu, Z. Computational Modeling and Numerical Methods for Spatiotemporal Calcium Cycling in Ventricular Myocytes. Front. Physiol. 2012, 3, 114. [Google Scholar] [CrossRef] [PubMed]
- Hoang-Trong, T.M.; Ullah, A.; Lederer, W.J.; Jafri, M.S. A Stochastic Spatiotemporal Model of Rat Ventricular Myocyte Calcium Dynamics Demonstrated Necessary Features for Calcium Wave Propagation. Membranes 2021, 11, 989. [Google Scholar] [CrossRef] [PubMed]
- Tomek, J.; Bueno-Orovio, A.; Passini, E.; Zhou, X.; Minchole, A.; Britton, O.; Bartolucci, C.; Severi, S.; Shrier, A.; Virag, L.; et al. Development, calibration, and validation of a novel human ventricular myocyte model in health, disease, and drug block. Elife 2019, 8, e48890. [Google Scholar] [CrossRef] [PubMed]
- Page, G.; Ratchada, P.; Miron, Y.; Steiner, G.; Ghetti, A.; Miller, P.E.; Reynolds, J.A.; Wang, K.; Greiter-Wilke, A.; Polonchuk, L.; et al. Human ex-vivo action potential model for pro-arrhythmia risk assessment. J. Pharmacol. Toxicol. Methods 2016, 81, 183–195. [Google Scholar] [CrossRef]
- O’Hara, T.; Virág, L.; Varró, A.; Rudy, Y. Simulation of the Undiseased Human Cardiac Ventricular Action Potential: Model Formulation and Experimental Validation. PLoS Comput. Biol. 2011, 7, e1002061. [Google Scholar] [CrossRef] [PubMed]
- Ramanathan, C.; Jia, P.; Ghanem, R.; Ryu, K.; Rudy, Y. Activation and repolarization of the normal human heart under complete physiological conditions. Proc. Natl. Acad. Sci. USA 2006, 103, 6309–6314. [Google Scholar] [CrossRef]
- Jafri, M.S.; Rice, J.J.; Winslow, R.L. Cardiac Ca2+ dynamics: The roles of ryanodine receptor adaptation and sarcoplasmic reticulum load. Biophys. J. 1998, 74, 1149–1168. [Google Scholar] [CrossRef]
- Williams, G.S.; Chikando, A.C.; Tuan, H.T.; Sobie, E.A.; Lederer, W.J.; Jafri, M.S. Dynamics of calcium sparks and calcium leak in the heart. Biophys. J. 2011, 101, 1287–1296. [Google Scholar] [CrossRef]
- Paudel, R. A Local-Control Model of the Guinea Pig Ventricular Myocyte Allows Understanding of Force-Interval Relations at the Calcium Spark Level. Biophys. J. 2019, 116, 113a. [Google Scholar] [CrossRef]
- Tusscher, K.H.W.J.t.; Noble, D.; Noble, P.J.; Panfilov, A.V. A model for human ventricular tissue. Am. J. Physiol.-Heart Circ. Physiol. 2004, 286, H1573–H1589. [Google Scholar] [CrossRef] [PubMed]
- Tran, K.; Smith, N.P.; Loiselle, D.S.; Crampin, E.J. A Thermodynamic Model of the Cardiac Sarcoplasmic/Endoplasmic Ca2+ (SERCA) Pump. Biophys. J. 2009, 96, 2029–2042. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Fan, J.S.; Clark, J.W.; Palade, P.T. A model of the L-type Ca2+ channel in rat ventricular myocytes: Ion selectivity and inactivation mechanisms. J. Physiol. 2000, 529 Pt 1, 139–158. [Google Scholar] [CrossRef]
- Wagner, E.; Lauterbach, M.A.; Kohl, T.; Westphal, V.; Williams, G.S.B.; Steinbrecher, J.H.; Streich, J.-H.; Korff, B.; Tuan, H.-T.M.; Hagen, B.; et al. Stimulated Emission Depletion Live-Cell Super-Resolution Imaging Shows Proliferative Remodeling of T-Tubule Membrane Structures After Myocardial Infarction. Circ. Res. 2012, 111, 402–414. [Google Scholar] [CrossRef]
- Bers, D.M.; Stiffel, V.M. Ratio of ryanodine to dihydropyridine receptors in cardiac and skeletal muscle and implications for E-C coupling. Am. J. Physiol.-Cell Physiol. 1993, 264, C1587–C1593. [Google Scholar] [CrossRef] [PubMed]
- Goldman, D.E. Potential, Impedance, and rectification in membranes. J. Gen. Physiol. 1943, 27, 37–60. [Google Scholar] [CrossRef] [PubMed]
- Pitzer, K.S.; Mayorga, G. Thermodynamics of electrolytes. II. Activity and osmotic coefficients for strong electrolytes with one or both ions univalent. J. Phys. Chem. 1973, 77, 2300–2308. [Google Scholar] [CrossRef]
- Lee, K.S.; Tsien, R.W. High selectivity of calcium channels in single dialysed heart cells of the guinea-pig. J. Physiol. 1984, 354, 253–272. [Google Scholar] [CrossRef]
- Hess, P.; Lansman, J.B.; Tsien, R.W. Calcium channel selectivity for divalent and monovalent cations. Voltage and concentration dependence of single channel current in ventricular heart cells. J. Gen. Physiol. 1986, 88, 293–319. [Google Scholar] [CrossRef]
- Clark, R.B.; Giles, W.R.; Imaizumi, Y. Properties of the transient outward current in rabbit atrial cells. J. Physiol. 1988, 405, 147–168. [Google Scholar] [CrossRef]
- Escande, D.; Coulombe, A.; Faivre, J.F.; Deroubaix, E.; Coraboeuf, E. Two types of transient outward currents in adult human atrial cells. Am. J. Physiol.-Heart Circ. Physiol. 1987, 252, H142–H148. [Google Scholar] [CrossRef] [PubMed]
- Giles, W.R.; van Ginneken, A.C. A transient outward current in isolated cells from the crista terminalis of rabbit heart. J. Physiol. 1985, 368, 243–264. [Google Scholar] [CrossRef] [PubMed]
- Shibata, E.F.; Drury, T.; Refsum, H.; Aldrete, V.; Giles, W. Contributions of a transient outward current to repolarization in human atrium. Am. J. Physiol.-Heart Circ. Physiol. 1989, 257, H1773–H1781. [Google Scholar] [CrossRef] [PubMed]
- Fermini, B.; Wang, Z.; Duan, D.; Nattel, S. Differences in rate dependence of transient outward current in rabbit and human atrium. Am. J. Physiol.-Heart Circ. Physiol. 1992, 263, H1747–H1754. [Google Scholar] [CrossRef]
- Jafri, M.S.; Hoang-Trong, T.M.; Williams, G.S.B. Method and System for Utilizing Markov Chain Monte Carlo Simulations. Google Patents US9009095B1, 14 April 2015. [Google Scholar]
- Afshar, Y.; Schmid, F.; Pishevar, A.; Worley, S. Exploiting seeding of random number generators for efficient domain decomposition parallelization of dissipative particle dynamics. Comput. Phys. Commun. 2013, 184, 1119–1128. [Google Scholar] [CrossRef]
- Ullah, A.; Hoang-Trong, M.T.; Lederer, W.J.; Winslow, R.L.; Jafri, M.S. Critical Requirements for the Initiation of a Cardiac Arrhythmia in Rat Ventricle: How Many Myocytes? Cells 2022, 11, 1878. [Google Scholar] [CrossRef]
- Smith, G.D. Modeling the stochastic gating of ion channels. In Computational Cell Biology; Springer: Berlin/Heidelberg, Germany, 2002; pp. 285–319. [Google Scholar]
- Groff, J.R.; DeRemigio, H.; Smith, G.D. Markov chain models of ion channels and calcium release sites. In Stochastic Methods in Neuroscience; Oxford Academic: Oxford, UK, 2009; pp. 29–64. [Google Scholar] [CrossRef]
- Hoang-Trong, M.T.; Ullah, A.; Lederer, W.J.; Jafri, M.S. Cardiac Alternans Occurs through the Synergy of Voltage- and Calcium-Dependent Mechanisms. Membranes 2021, 11, 794. [Google Scholar] [CrossRef]
- Sun, Y.B.; Irving, M. The molecular basis of the steep force-calcium relation in heart muscle. J. Mol. Cell. Cardiol. 2010, 48, 859–865. [Google Scholar] [CrossRef]
- Sun, Y.-B.; Lou, F.; Irving, M. Calcium- and myosin-dependent changes in troponin structure during activation of heart muscle. J. Physiol. 2009, 587, 155–163. [Google Scholar] [CrossRef]
- Li, G.-R.; Yang, B.; Feng, J.; Bosch, R.F.; Carrier, M.; Nattel, S. TransmembraneI Ca contributes to rate-dependent changes of action potentials in human ventricular myocytes. Am. J. Physiol.-Heart Circ. Physiol. 1999, 276, H98–H106. [Google Scholar] [CrossRef]
- Näbauer, M.; Beuckelmann, D.J.; Überfuhr, P.; Steinbeck, G. Regional Differences in Current Density and Rate-Dependent Properties of the Transient Outward Current in Subepicardial and Subendocardial Myocytes of Human Left Ventricle. Circulation 1996, 93, 168–177. [Google Scholar] [CrossRef]
- Iost, N.; Virág, L.; Opincariu, M.; Szécsi, J.; Varró, A.; Papp, J.G. Delayed rectifier potassium current in undiseased human ventricular myocytes. Cardiovasc. Res. 1998, 40, 508–515. [Google Scholar] [CrossRef] [PubMed]
- Virág, L.; Iost, N.; Opincariu, M.; Szolnoky, J.; Szécsi, J.; Bogáts, G.; Szenohradszky, P.; Varró, A.; Papp, J.G. The slow component of the delayed rectifier potassium current in undiseased human ventricular myocytes. Cardiovasc. Res. 2001, 49, 790–797. [Google Scholar] [CrossRef]
- Fülöp, L.; Bányász, T.; Magyar, J.; Szentandrássy, N.; Varró, A.; Nánási, P.P. Reopening of L-type calcium channels in human ventricular myocytes during applied epicardial action potentials. Acta Physiol. Scand. 2004, 180, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Faber, G.M.; Silva, J.; Livshitz, L.; Rudy, Y. Kinetic properties of the cardiac L-type Ca2+ channel and its role in myocyte electrophysiology: A theoretical investigation. Biophys. J. 2007, 92, 1522–1543. [Google Scholar] [CrossRef]
- Fill, M.; Zahradníková, A.; Villalba-Galea, C.A.; Zahradník, I.; Escobar, A.L.; Györke, S. Ryanodine receptor adaptation. J. Gen. Physiol. 2000, 116, 873–882. [Google Scholar] [CrossRef] [PubMed]
- Árpádffy-Lovas, T.; Baczkó, I.; Baláti, B.; Bitay, M.; Jost, N.; Lengyel, C.; Nagy, N.; Takács, J.; Varró, A.; Virág, L. Electrical Restitution and Its Modifications by Antiarrhythmic Drugs in Undiseased Human Ventricular Muscle. Front. Pharmacol. 2020, 11, 479. [Google Scholar] [CrossRef]
- Bárándi, L.; Virág, L.; Jost, N.; Horváth, Z.; Koncz, I.; Papp, R.; Harmati, G.; Horváth, B.; Szentandrássy, N.; Bányász, T.; et al. Reverse rate-dependent changes are determined by baseline action potential duration in mammalian and human ventricular preparations. Basic Res. Cardiol. 2010, 105, 315–323. [Google Scholar] [CrossRef]
- Van Petegem, F. Ryanodine receptors: Structure and function. J. Biol. Chem. 2012, 287, 31624–31632. [Google Scholar] [CrossRef]
- Bers, D.M. Cardiac excitation–contraction coupling. Nature 2002, 415, 198–205. [Google Scholar] [CrossRef]
- Singh, B. Amiodarone: Historical development and pharmacologic profile. Am. Heart J. 1983, 106, 788–797. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, Y.; Peters, W.; Khan, S.S.; Mandel, W.J.; Karagueuzian, H.S. Cellular mechanisms of differential action potential duration restitution in canine ventricular muscle cells during single versus double premature stimuli. Circulation 1992, 86, 955–967. [Google Scholar] [CrossRef] [PubMed]
- Bjørnstad, H.; Tande, P.M.; Lathrop, D.A.; Refsum, H. Effects of temperature on cycle length dependent changes and restitution of action potential duration in guinea pig ventricular muscle. Cardiovasc. Res. 1993, 27, 946–950. [Google Scholar] [CrossRef]
- Sanguinetti, M.C.; Jurkiewicz, N.K. Two components of cardiac delayed rectifier K+ current. Differential sensitivity to block by class III antiarrhythmic agents. J. Gen. Physiol. 1990, 96, 195–215. [Google Scholar] [CrossRef] [PubMed]
- Matsuura, H.; Ehara, T.; Imoto, Y. An analysis of the delayed outward current in single ventricular cells of the guinea-pig. Pflügers Arch. 1987, 410, 596–603. [Google Scholar] [CrossRef] [PubMed]
- Martin, C.L.; Chinn, K. Contribution of delayed rectifier and inward rectifier to repolarization of the action potential: Pharmacologic separation. J. Cardiovasc. Pharmacol. 1992, 19, 830–837. [Google Scholar] [CrossRef] [PubMed]
- Giles, W.R.; Imaizumi, Y. Comparison of potassium currents in rabbit atrial and ventricular cells. J. Physiol. 1988, 405, 123–145. [Google Scholar] [CrossRef]
- Chinn, K. Two delayed rectifiers in guinea pig ventricular myocytes distinguished by tail current kinetics. J. Pharmacol. Exp. Ther. 1993, 264, 553–560. [Google Scholar]
- Johnson, E.K.; Springer, S.J.; Wang, W.; Dranoff, E.J.; Zhang, Y.; Kanter, E.M.; Yamada, K.A.; Nerbonne, J.M. Differential Expression and Remodeling of Transient Outward Potassium Currents in Human Left Ventricles. Circ. Arrhythmia Electrophysiol. 2018, 11, e005914. [Google Scholar] [CrossRef]
- Yu, H.; McKinnon, D.; Dixon, J.E.; Gao, J.; Wymore, R.; Cohen, I.S.; Danilo, P.; Shvilkin, A.; Anyukhovsky, E.P.; Sosunov, E.A.; et al. Transient Outward Current, Ito1, Is Altered in Cardiac Memory. Circulation 1999, 99, 1898–1905. [Google Scholar] [CrossRef]
- Wei, X.; Yohannan, S.; Richards, J.R. Physiology, Cardiac Repolarization Dispersion and Reserve. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Chiamvimonvat, N.; Chen-Izu, Y.; Clancy, C.E.; Deschenes, I.; Dobrev, D.; Heijman, J.; Izu, L.; Qu, Z.; Ripplinger, C.M.; Vandenberg, J.I.; et al. Potassium currents in the heart: Functional roles in repolarization, arrhythmia and therapeutics. J. Physiol. 2017, 595, 2229–2252. [Google Scholar] [CrossRef] [PubMed]
- Zeng, J.; Laurita, K.R.; Rosenbaum, D.S.; Rudy, Y. Two Components of the Delayed Rectifier K+ Current in Ventricular Myocytes of the Guinea Pig Type. Circ. Res. 1995, 77, 140–152. [Google Scholar] [CrossRef] [PubMed]
- Hadley, R.W.; Hume, J.R. An intrinsic potential-dependent inactivation mechanism associated with calcium channels in guinea-pig myocytes. J. Physiol. 1987, 389, 205–222. [Google Scholar] [CrossRef] [PubMed]
- Shirokov, R.; Levis, R.; Shirokova, N.; Ríos, E. Ca(2+)-dependent inactivation of cardiac L-type Ca2+ channels does not affect their voltage sensor. J. Gen. Physiol. 1993, 102, 1005–1030. [Google Scholar] [CrossRef] [PubMed]
- Kubalová, Z. Inactivation of L-type calcium channels in cardiomyocytes. Experimental and theoretical approaches. Gen. Physiol. Biophys. 2003, 22, 441–454. [Google Scholar] [PubMed]
- Bers, D.M. Calcium and Cardiac Rhythms. Circ. Res. 2002, 90, 14–17. [Google Scholar] [CrossRef]
- Armoundas, A.A.; Hobai, I.A.; Tomaselli, G.F.; Winslow, R.L.; O’Rourke, B. Role of sodium-calcium exchanger in modulating the action potential of ventricular myocytes from normal and failing hearts. Circ. Res. 2003, 93, 46–53. [Google Scholar] [CrossRef]
- Taylor, D.G.; Parilak, L.D.; LeWinter, M.M.; Knot, H.J. Quantification of the rat left ventricle force and Ca2+ frequency relationships: Similarities to dog and human. Cardiovasc. Res. 2004, 61, 77–86. [Google Scholar] [CrossRef]
- Hardy, M.E.L.; Pervolaraki, E.; Bernus, O.; White, E. Dynamic Action Potential Restitution Contributes to Mechanical Restitution in Right Ventricular Myocytes From Pulmonary Hypertensive Rats. Front. Physiol. 2018, 9, 205. [Google Scholar] [CrossRef]
- Tse, G.; Wong, S.T.; Tse, V.; Lee, Y.T.; Lin, H.Y.; Yeo, J.M. Cardiac dynamics: Alternans and arrhythmogenesis. J. Arrhythm. 2016, 32, 411–417. [Google Scholar] [CrossRef]
- Banyasz, T.; Horvath, B.; Jian, Z.; Izu, L.T.; Chen-Izu, Y. Profile of L-type Ca2+ current and Na+/Ca2+ exchange current during cardiac action potential in ventricular myocytes. Heart Rhythm. 2012, 9, 134–142. [Google Scholar] [CrossRef] [PubMed]
- Qu, Z.; Weiss, J.N.; Garfinkel, A. Cardiac electrical restitution properties and stability of reentrant spiral waves: A simulation study. Am. J. Physiol. Heart Circ. Physiol. 1999, 276, H269–H283. [Google Scholar] [CrossRef]
- Qu, Z.; Garfinkel, A.; Chen, P.-S.; Weiss, J.N. Mechanisms of Discordant Alternans and Induction of Reentry in Simulated Cardiac Tissue. Circulation 2000, 102, 1664–1670. [Google Scholar] [CrossRef] [PubMed]
- Guo, T.; Gillespie, D.; Fill, M. Ryanodine Receptor Current Amplitude Controls Ca2+ Sparks in Cardiac Muscle. Circ. Res. 2012, 111, 28–36. [Google Scholar] [CrossRef]
- Cheng, H.; Lederer, W.J.; Cannell, M.B. Calcium sparks: Elementary events underlying excitation-contraction coupling in heart muscle. Science 1993, 262, 740–744. [Google Scholar] [CrossRef] [PubMed]
- Lukyanenko, V.; Subramanian, S.; Gyorke, I.; Wiesner, T.F.; Gyorke, S. The role of luminal Ca2+ in the generation of Ca2+ waves in rat ventricular myocytes. J. Physiol. 1999, 518, 173–186. [Google Scholar] [CrossRef]
- Satoh, H.; Blatter, L.A.; Bers, D.M. Effects of [Ca2+]i, SR Ca2+ load, and rest on Ca2+ spark frequency in ventricular myocytes. Am. J. Physiol.-Heart Circ. Physiol. 1997, 272, H657–H668. [Google Scholar] [CrossRef]
- Lindner, M.; Brandt, M.C.; Sauer, H.; Hescheler, J.; Böhle, T.; Beuckelmann, D.J. Calcium sparks in human ventricular cardiomyocytes from patients with terminal heart failure. Cell Calcium 2002, 31, 175–182. [Google Scholar] [CrossRef]
- Usman, A.; Gandhi, J.; Gupta, G. Physiology, Bowditch Effect. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Cannon, W.B. Henry Pickering Bowditch, Physiologist. Science 1938, 87, 471–474. [Google Scholar] [CrossRef]
- Maier, L.S.; Barckhausen, P.; Weisser, J.; Aleksic, I.; Baryalei, M.; Pieske, B. Ca2+ handling in isolated human atrial myocardium. Am. J. Physiol.-Heart Circ. Physiol. 2000, 279, H952–H958. [Google Scholar] [CrossRef] [PubMed]
- Pieske, B.; Maier, L.S.; Bers, D.M.; Hasenfuss, G. Ca2+ handling and sarcoplasmic reticulum Ca2+ content in isolated failing and nonfailing human myocardium. Circ. Res. 1999, 85, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Györke, S.; Fill, M. Ryanodine Receptor Adaptation: Control Mechanism of Ca2+-Induced Ca2+ Release in Heart. Science 1993, 260, 807–809. [Google Scholar] [CrossRef] [PubMed]
- Shannon, T.R.; Ginsburg, K.S.; Bers, D.M. Potentiation of fractional sarcoplasmic reticulum calcium release by total and free intra-sarcoplasmic reticulum calcium concentration. Biophys. J. 2000, 78, 334–343. [Google Scholar] [CrossRef] [PubMed]
- Eisner, D.A.; Caldwell, J.L.; Kistamás, K.; Trafford, A.W. Calcium and Excitation-Contraction Coupling in the Heart. Circ. Res. 2017, 121, 181–195. [Google Scholar] [CrossRef]
- Bassani, J.W.; Yuan, W.; Bers, D.M. Fractional SR Ca release is regulated by trigger Ca and SR Ca content in cardiac myocytes. Am. J. Physiol. 1995, 268, C1313–C1319. [Google Scholar] [CrossRef]
- Berlin, J.R.; Bassani, J.W.; Bers, D.M. Intrinsic cytosolic calcium buffering properties of single rat cardiac myocytes. Biophys. J. 1994, 67, 1775–1787. [Google Scholar] [CrossRef]
- Wang, L.; Myles, R.C.; Jesus, N.M.D.; Ohlendorf, A.K.P.; Bers, D.M.; Ripplinger, C.M. Optical Mapping of Sarcoplasmic Reticulum Ca2+ in the Intact Heart. Circ. Res. 2014, 114, 1410–1421. [Google Scholar] [CrossRef]
- Meissner, G.; Darling, E.; Eveleth, J. Kinetics of rapid Ca2+ release by sarcoplasmic reticulum. Effects of Ca2+, Mg2+, and adenine nucleotides. Biochemistry 1986, 25, 236–244. [Google Scholar] [CrossRef]
- Gettes, L.; Reuter, H. Slow recovery from inactivation of inward currents in mammalian myocardial fibres. J. Physiol. 1974, 240, 703–724. [Google Scholar] [CrossRef]
- Attwell, D.; Cohen, I.; Eisner, D.A. The effects of heart rate on the action potential of guinea-pig and human ventricular muscle. J. Physiol. 1981, 313, 439–461. [Google Scholar] [CrossRef]
- Xie, F.; Qu, Z.; Garfinkel, A.; Weiss, J.N. Electrical refractory period restitution and spiral wave reentry in simulated cardiac tissue. Am. J. Physiol.-Heart Circ. Physiol. 2002, 283, H448–H460. [Google Scholar] [CrossRef] [PubMed]
- Taggart, P.; Lab, M. Cardiac mechano-electric feedback and electrical restitution in humans. Prog. Biophys. Mol. Biol. 2008, 97, 452–460. [Google Scholar] [CrossRef] [PubMed]
- Nash, M.P.; Bradley, C.P.; Sutton, P.M.; Clayton, R.H.; Kallis, P.; Hayward, M.P.; Paterson, D.J.; Taggart, P. Whole heart action potential duration restitution properties in cardiac patients: A combined clinical and modelling study. Exp. Physiol. 2006, 91, 339–354. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Lederer, W.J. Calcium Sparks. Physiol. Rev. 2008, 88, 1491–1545. [Google Scholar] [CrossRef]
- Hoang-Trong, T.M.; Ullah, A.; Jafri, M.S. Calcium Sparks in the Heart: Dynamics and Regulation. Res. Rep. Biol. 2015, 6, 203–214. [Google Scholar] [CrossRef]
- Alpert, N.R.; Mulieri, L.A.; Warshaw, D. The failing human heart. Cardiovasc. Res. 2002, 54, 1–10. [Google Scholar] [CrossRef]
- Shannon, T.R.; Guo, T.; Bers, D.M. Ca2+ Scraps: Local Depletions of Free [Ca2+] in Cardiac Sarcoplasmic Reticulum during Contractions Leave Substantial Ca2+ Reserve. Circ. Res. 2003, 93, 40–45. [Google Scholar] [CrossRef]
- Greensmith, D.J.; Galli, G.L.J.; Trafford, A.W.; Eisner, D.A. Direct measurements of SR free Ca reveal the mechanism underlying the transient effects of RyR potentiation under physiological conditions. Cardiovasc. Res. 2014, 103, 554–563. [Google Scholar] [CrossRef]
- Lawson, C.S.; Downey, J.M. Preconditioning: State of the art myocardial protection. Cardiovasc. Res. 1993, 27, 542–550. [Google Scholar] [CrossRef]
- Koning, M.M.G.; Gho, B.C.G.; Klaarwater, E.v.; Opstal, R.L.J.; Duncker, D.J.; Verdouw, P.D. Rapid Ventricular Pacing Produces Myocardial Protection by Nonischemic Activation of KATP+ Channels. Circulation 1996, 93, 178–186. [Google Scholar] [CrossRef]
- Daehnert, I.; Rotzsch, C.; Wiener, M.; Schneider, P. Rapid right ventricular pacing is an alternative to adenosine in catheter interventional procedures for congenital heart disease. Heart 2004, 90, 1047–1050. [Google Scholar] [CrossRef] [PubMed]
- Fefer, P.; Bogdan, A.; Grossman, Y.; Berkovitch, A.; Brodov, Y.; Kuperstein, R.; Segev, A.; Guetta, V.; Barbash, I.M. Impact of Rapid Ventricular Pacing on Outcome After Transcatheter Aortic Valve Replacement. J. Am. Heart Assoc. 2018, 7, e009038. [Google Scholar] [CrossRef]
- Grabert, J.; Huber-Petersen, S.; Lampmann, T.; Eichhorn, L.; Vatter, H.; Coburn, M.; Velten, M.; Güresir, E. Rapid Ventricular Pacing as a Safe Procedure for Clipping of Complex Ruptured and Unruptured Intracranial Aneurysms. J. Clin. Med. 2021, 10, 5406. [Google Scholar] [CrossRef] [PubMed]
- Livshitz, L.M.; Rudy, Y. Regulation of Ca2+ and electrical alternans in cardiac myocytes: Role of CAMKII and repolarizing currents. Am. J. Physiol.-Heart Circ. Physiol. 2007, 292, H2854–H2866. [Google Scholar] [CrossRef] [PubMed]
- Hund, T.J.; Rudy, Y. Rate dependence and regulation of action potential and calcium transient in a canine cardiac ventricular cell model. Circulation 2004, 110, 3168–3174. [Google Scholar] [CrossRef] [PubMed]
- Ullah, A.; Hoang-Trong, T.M.; Williams, G.S.; Lederer, J.W.; Jafri, M.S. A small number of cells is sufficient to trigger a cardiac arrhythmia: Stochastic computational studies. Biophys. J. 2014, 106, 112a. [Google Scholar] [CrossRef]
- Sutanto, H.; Heijman, J. Integrative Computational Modeling of Cardiomyocyte Calcium Handling and Cardiac Arrhythmias: Current Status and Future Challenges. Cells 2022, 11, 1090. [Google Scholar] [CrossRef]
- Fu, Y.; Zhang, G.-Q.; Hao, X.-M.; Wu, C.-H.; Chai, Z.; Wang, S.-Q. Temperature dependence and thermodynamic properties of Ca2+ sparks in rat cardiomyocytes. Biophys. J. 2005, 89, 2533–2541. [Google Scholar] [CrossRef]
- O’Hara, T.; Rudy, Y. Quantitative comparison of cardiac ventricular myocyte electrophysiology and response to drugs in human and nonhuman species. Am. J. Physiol.-Heart Circ. Physiol. 2012, 302, H1023–H1030. [Google Scholar] [CrossRef]
- HDF-Group. 2014. Available online: http://www.hdfgroup.org/HDF5/ (accessed on 2 July 2023).
- Yang, Y.; Pascarel, C.; Steele, D.S.; Komukai, K.; Brette, F.; Orchard, C.H. Na+-Ca2+ exchange activity is localized in the T-tubules of rat ventricular myocytes. Circ. Res. 2002, 91, 315–322. [Google Scholar] [CrossRef]
- Sobie, E.A.; Dilly, K.W.; dos Santos Cruz, J.; Lederer, W.J.; Jafri, M.S. Termination of Cardiac Ca2+ Sparks: An Investigative Mathematical Model of Calcium-Induced Calcium Release. Biophys. J. 2002, 83, 59–78. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.D.; Keizer, J.E.; Stern, M.D.; Lederer, W.J.; Cheng, H. A simple numerical model of calcium spark formation and detection in cardiac myocytes. Biophys. J. 1998, 75, 15–32. [Google Scholar] [CrossRef]
- Groff, J.R.; Smith, G.D. Ryanodine receptor allosteric coupling and the dynamics of calcium sparks. Biophys. J. 2008, 95, 135–154. [Google Scholar] [CrossRef] [PubMed]
- Ehlers, M.D.; Augustine, G.J.; Field, R.O. Calmodulin at the channel gate. Nature 1999, 399, 105–107. [Google Scholar] [CrossRef] [PubMed]
- Fallon, J.L.; Baker, M.R.; Xiong, L.; Loy, R.E.; Yang, G.; Dirksen, R.T.; Hamilton, S.L.; Quiocho, F.A. Crystal structure of dimeric cardiac L-type calcium channel regulatory domains bridged by Ca2+* calmodulins. Proc. Natl. Acad. Sci. USA 2009, 106, 5135–5140. [Google Scholar] [CrossRef]
- Pitt, G.S.; Zühlke, R.D.; Hudmon, A.; Schulman, H.; Reuter, H.; Tsien, R.W. Molecular basis of calmodulin tethering and Ca2+-dependent inactivation of L-type Ca2+ channels. J. Biol. Chem. 2001, 276, 30794–30802. [Google Scholar] [CrossRef]
- Mori, M.X.; Erickson, M.G.; Yue, D.T. Functional stoichiometry and local enrichment of calmodulin interacting with Ca2+ channels. Science 2004, 304, 432–435. [Google Scholar] [CrossRef]
- Hodgkin, A.L.; Huxley, A.F. A quantiative description of membrane current and its application to conduction and excitation in nerve. J. Physiol. 1952, 117, 25–71. [Google Scholar] [CrossRef]
- Bondarenko, V.E.; Szigeti, G.P.; Bett, G.C.L.; Kim, S.-J.; Rasmusson, R.L. Computer model of action potential of mouse ventricular myocytes. Am. J. Physiol. Heart Circ. Physiol. 2004, 287, H1378–H1403. [Google Scholar] [CrossRef]
- Ramay, H.R.; Liu, O.Z.; Sobie, E.A. Recovery of cardiac calcium release is controlled by sarcoplasmic reticulum refilling and ryanodine receptor sensitivity. Cardiovasc. Res. 2011, 91, 598–605. [Google Scholar] [CrossRef]
- Li, G.-R.; Feng, J.; Yue, L.; Carrier, M.; Nattel, S. Evidence for Two Components of Delayed Rectifier K+ Current in Human Ventricular Myocytes. Circ. Res. 1996, 78, 689–696. [Google Scholar] [CrossRef] [PubMed]
- Roden, D.M. Long-QT Syndrome. N. Engl. J. Med. 2008, 358, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Jost, N.; Virág, L.; Bitay, M.; Takács, J.; Lengyel, C.; Biliczki, P.; Nagy, Z.; Bogáts, G.; Lathrop, D.A.; Papp, J.G.; et al. Restricting Excessive Cardiac Action Potential and QT Prolongation. Circulation 2005, 112, 1392–1399. [Google Scholar] [CrossRef]
- Taggart, P.; Sutton, P.; Chalabi, Z.; Boyett, M.R.; Simon, R.; Elliott, D.; Gill, J.S. Effect of Adrenergic Stimulation on Action Potential Duration Restitution in Humans. Circulation 2003, 107, 285–289. [Google Scholar] [CrossRef] [PubMed]
- Elharrar, V.; Surawicz, B. Cycle length effect on restitution of action potential duration in dog cardiac fibers. Am. J. Physiol.-Heart Circ. Physiol. 1983, 244, H782–H792. [Google Scholar] [CrossRef] [PubMed]
- Osadchii, O.E. Effects of ventricular pacing protocol on electrical restitution assessments in guinea-pig heart. Exp. Physiol. 2012, 97, 807–821. [Google Scholar] [CrossRef]
- Shattock, M.J.; Park, K.C.; Yang, H.-Y.; Lee, A.W.C.; Niederer, S.; MacLeod, K.T.; Winter, J. Restitution slope is principally determined by steady-state action potential duration. Cardiovasc. Res. 2017, 113, 817–828. [Google Scholar] [CrossRef]
- Coraboeuf, E.; Coulombe, A.; Deroubaix, E.; Hatem, S.; Mercadier, J.J. Transient outward potassium current and repolarization of cardiac cells. Bull. Acad. Natl. Med. 1998, 182, 325–333. [Google Scholar]














Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alvarez, J.A.E.; Jafri, M.S.; Ullah, A. Local Control Model of a Human Ventricular Myocyte: An Exploration of Frequency-Dependent Changes and Calcium Sparks. Biomolecules 2023, 13, 1259. https://doi.org/10.3390/biom13081259
Alvarez JAE, Jafri MS, Ullah A. Local Control Model of a Human Ventricular Myocyte: An Exploration of Frequency-Dependent Changes and Calcium Sparks. Biomolecules. 2023; 13(8):1259. https://doi.org/10.3390/biom13081259
Chicago/Turabian StyleAlvarez, Jerome Anthony E., M. Saleet Jafri, and Aman Ullah. 2023. "Local Control Model of a Human Ventricular Myocyte: An Exploration of Frequency-Dependent Changes and Calcium Sparks" Biomolecules 13, no. 8: 1259. https://doi.org/10.3390/biom13081259
APA StyleAlvarez, J. A. E., Jafri, M. S., & Ullah, A. (2023). Local Control Model of a Human Ventricular Myocyte: An Exploration of Frequency-Dependent Changes and Calcium Sparks. Biomolecules, 13(8), 1259. https://doi.org/10.3390/biom13081259