
_Kwok.png)
IgG Fc-Binding Peptide-Conjugated Pan-CoV Fusion Inhibitor Exhibits Extended In Vivo Half-Life and Synergistic Antiviral Effect When Combined with Neutralizing Antibodies




Abstract
:1. Introduction
2. Methods
2.1. Cell Lines, Viruses, Peptides, and Plasmids
2.2. Inhibition of HCoV S-Mediated Cell–Cell Fusion
2.3. Inhibition of Pseudotyped HCoV Infection
2.4. Inhibition of Live HCoV Replication
2.5. In Vivo Protective Study against Live HCoV-OC43 Infection
2.6. Circular Dichroism Spectroscopic Analysis
2.7. Expression and Purification of IgG/Antibody
2.8. Measurement of Peptide Binding to IgG
2.9. Evaluation of Tolerance to Proteolytic Enzymes
2.10. Cytotoxicity Assay
2.11. In Vivo Safety Assay
2.12. Pharmacokinetic Studies in Rhesus Monkeys
2.13. Detection of Synergistic Antiviral Effect of IBP-EK1 in Combination with Neutralizing Antibodies
2.14. Detection of Ex Vivo Synergistic Effect of IBP-EK1 in Combination with Neutralizing Antibodies
2.15. Statistical Analyses
3. Results
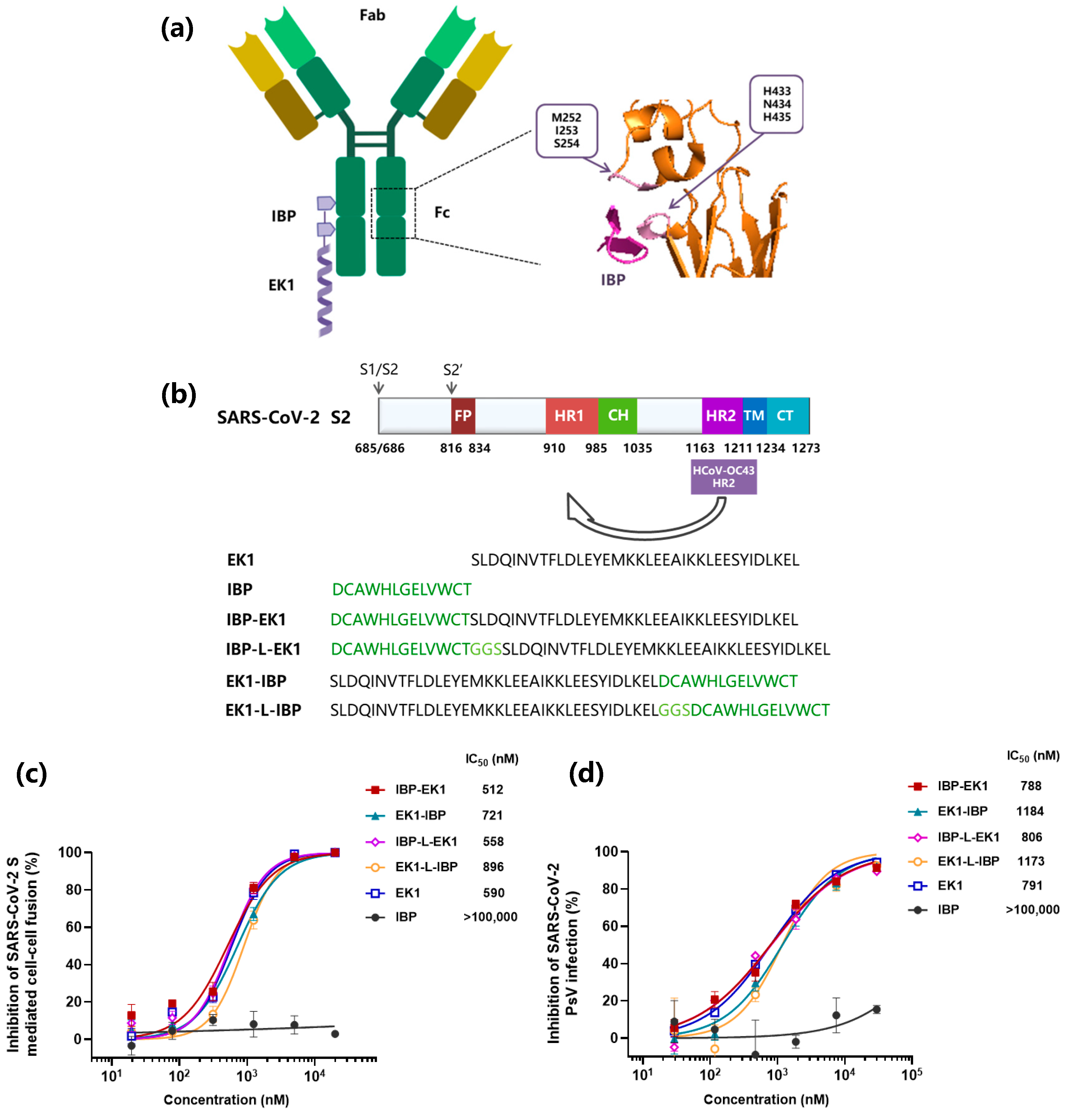
3.1. Design and Identification of IBP-Conjugated Peptides
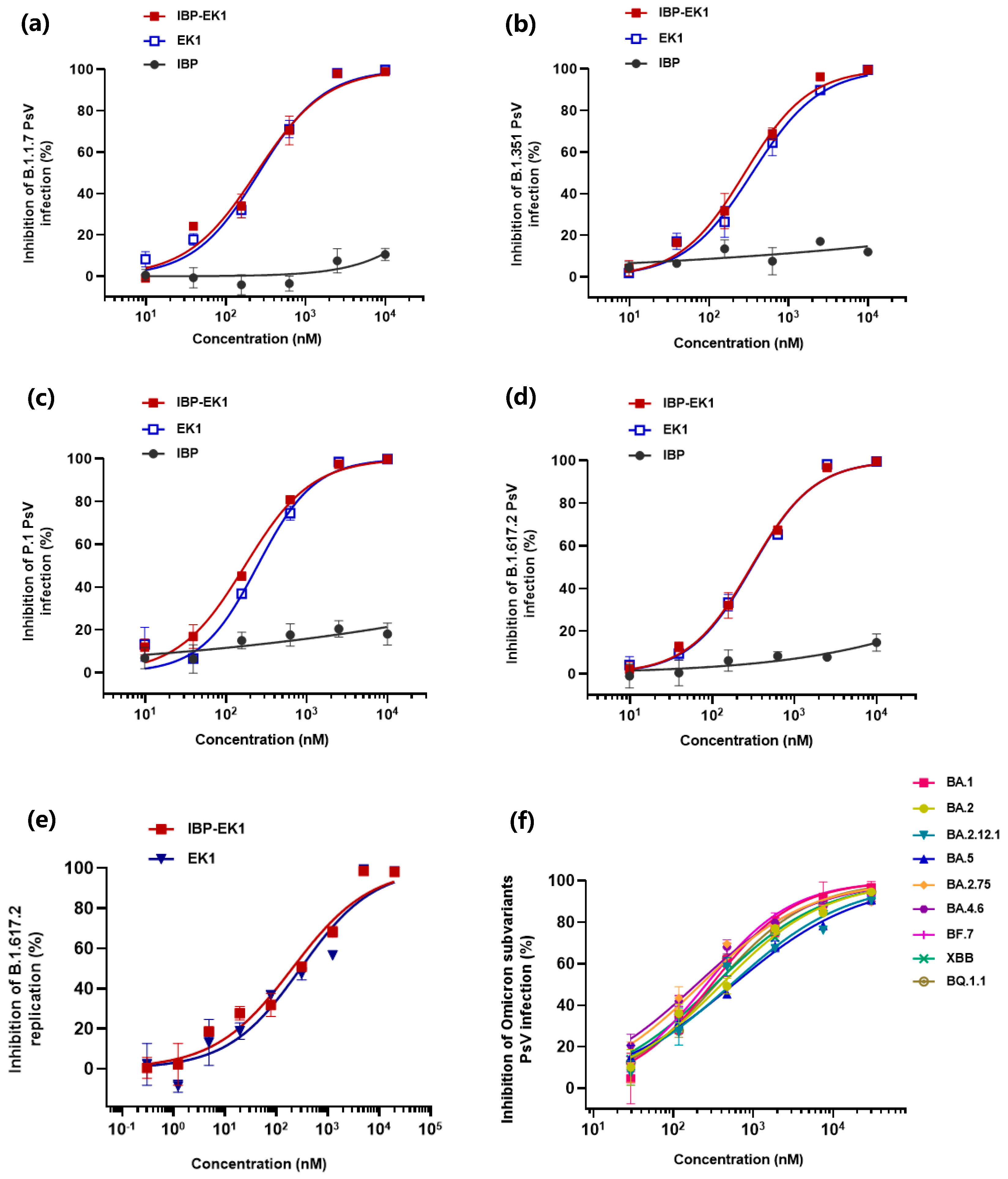
3.2. IBP-EK1 Showed Potent Inhibitory Activities against SARS-CoV-2 Variants
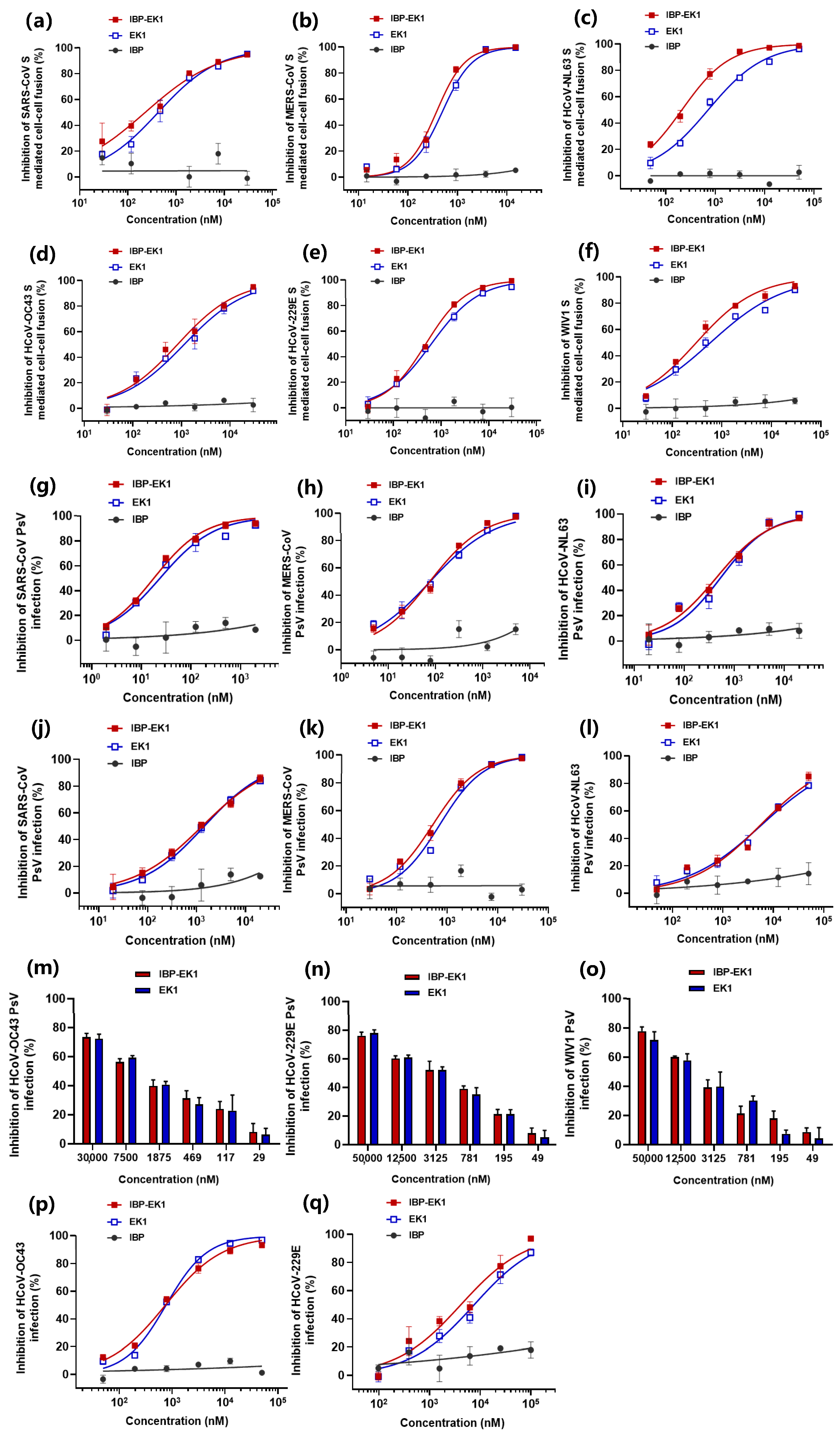
3.3. IBP-EK1 Exhibited Broadly Inhibitory Activities against HCoVs In Vitro
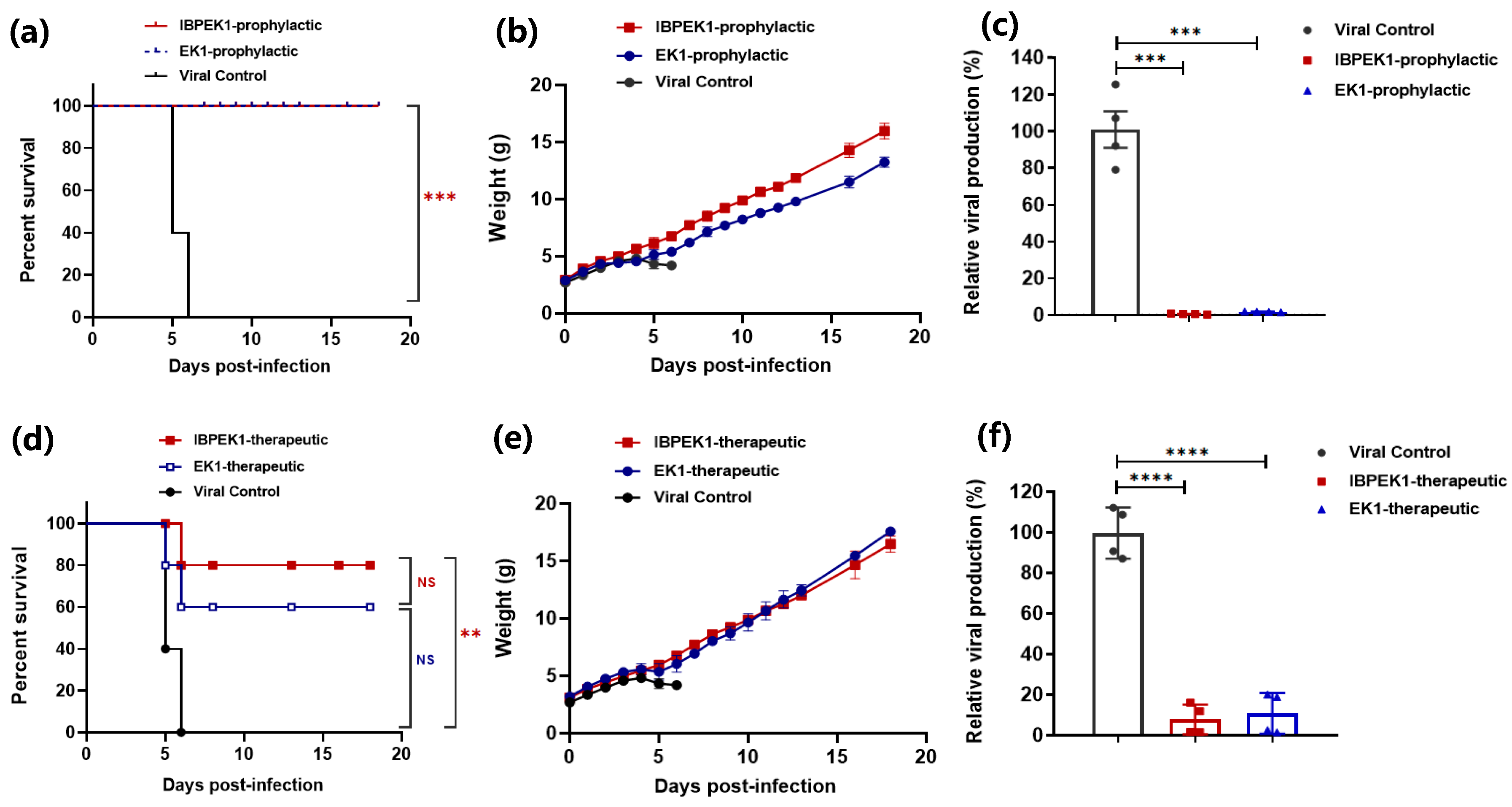
3.4. Intranasal Administration of IBP-EK1 Strongly Protected Mice against HCoV-OC43 Challenge
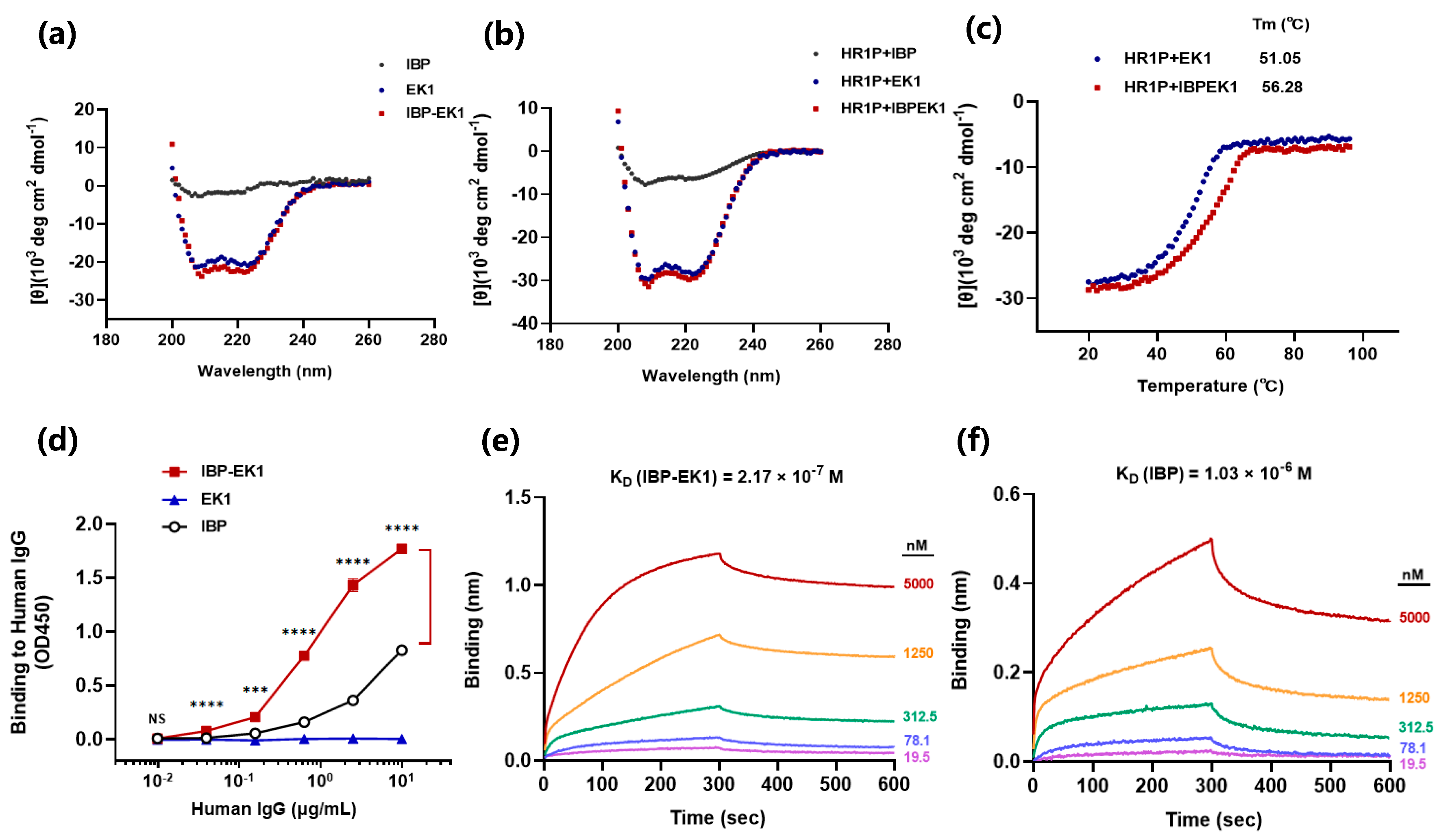
3.5. Influences of IBP Conjugation on the Original Properties of Peptides
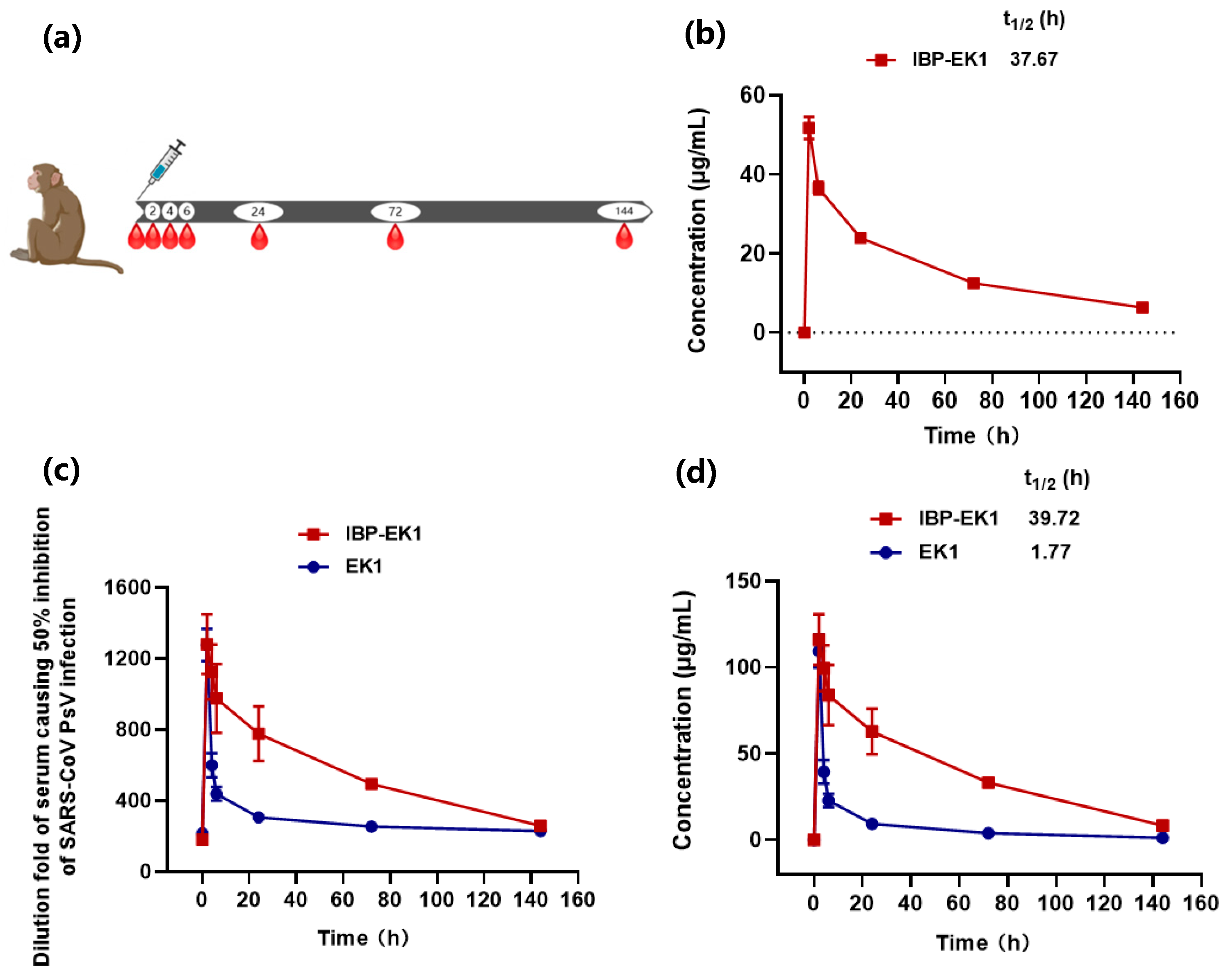
3.6. IBP-EK1 Displayed Extended In Vivo Half-Life
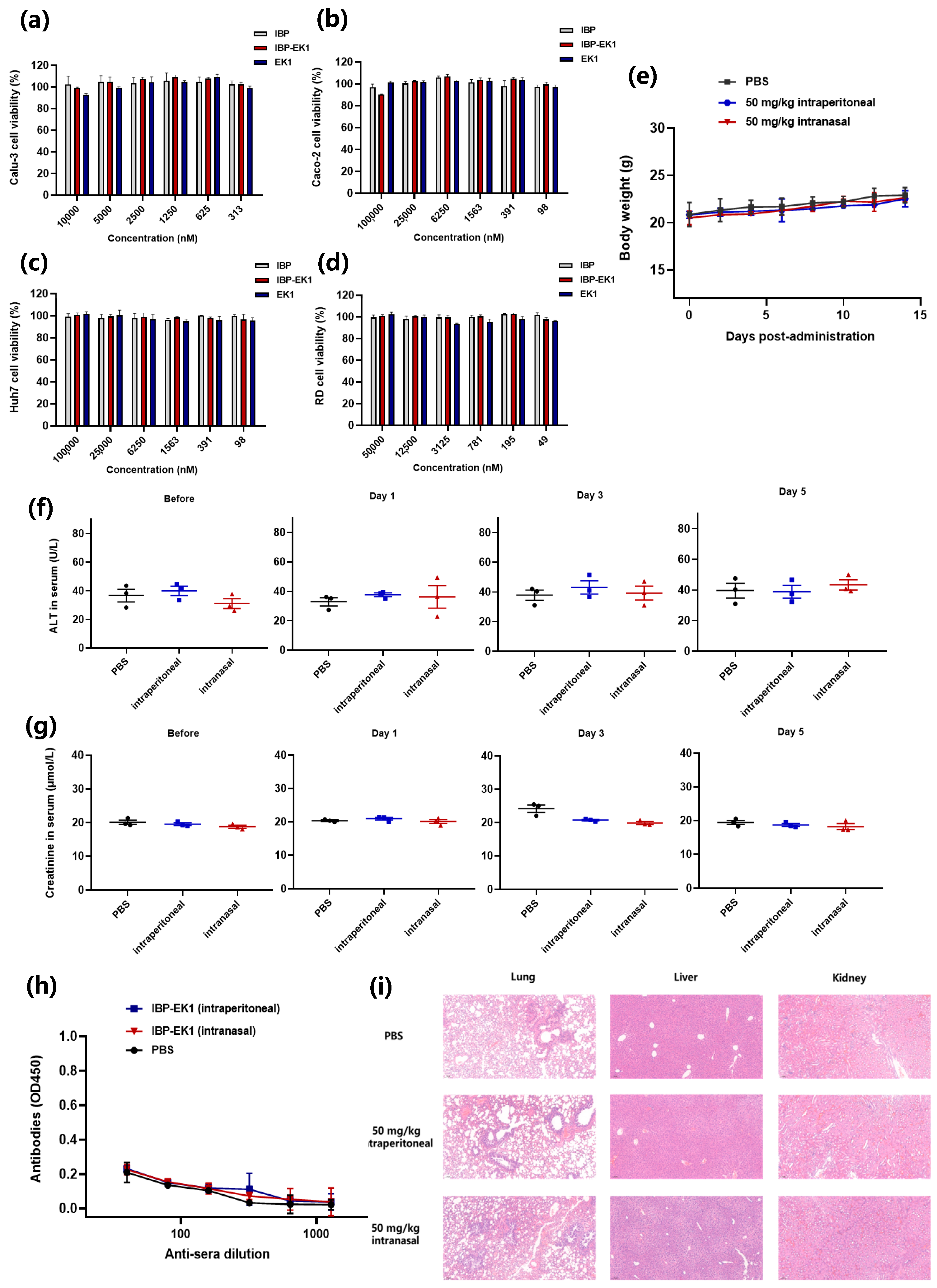
3.7. IBP-EK1 Showed Good In Vivo Safety
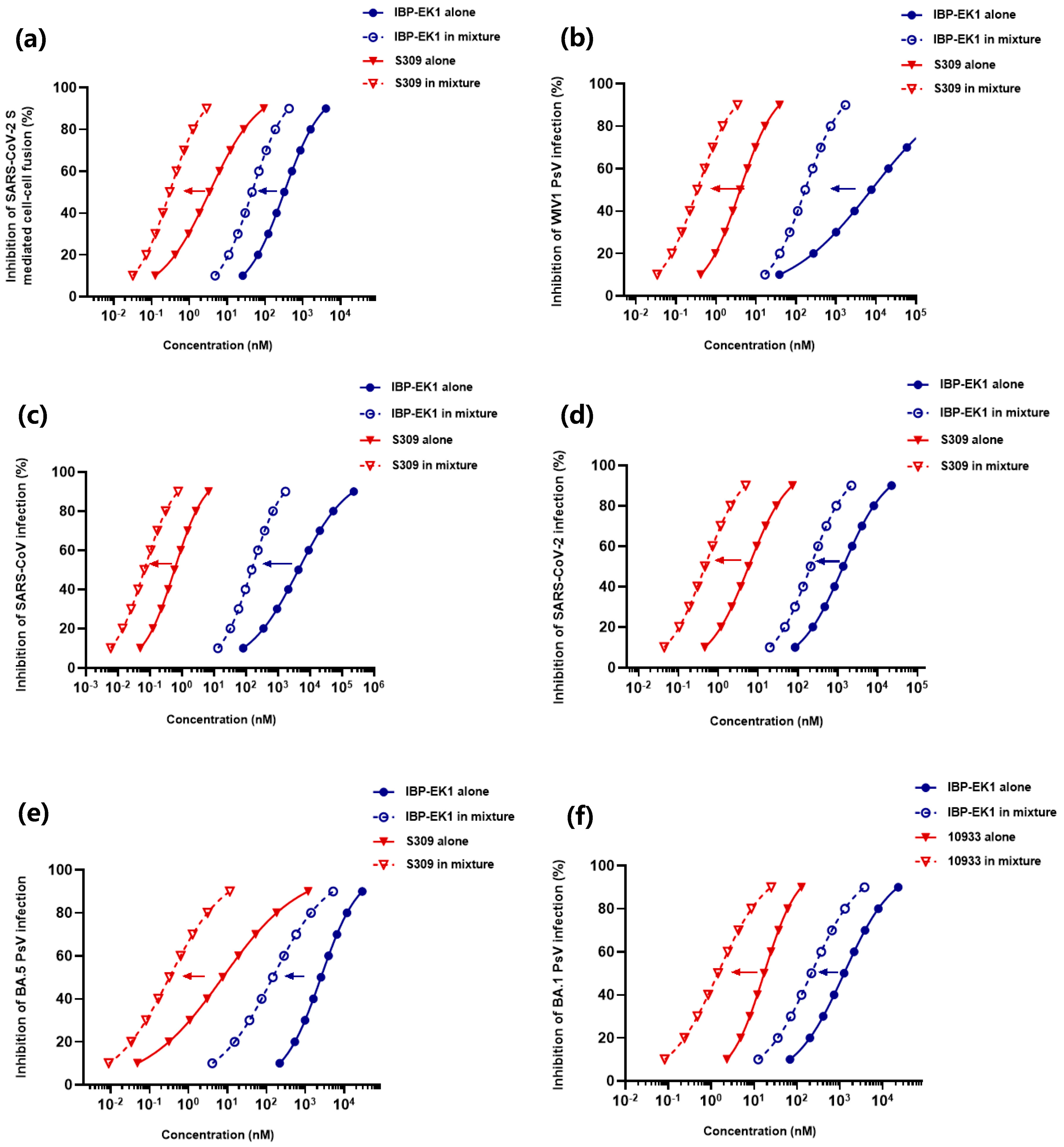
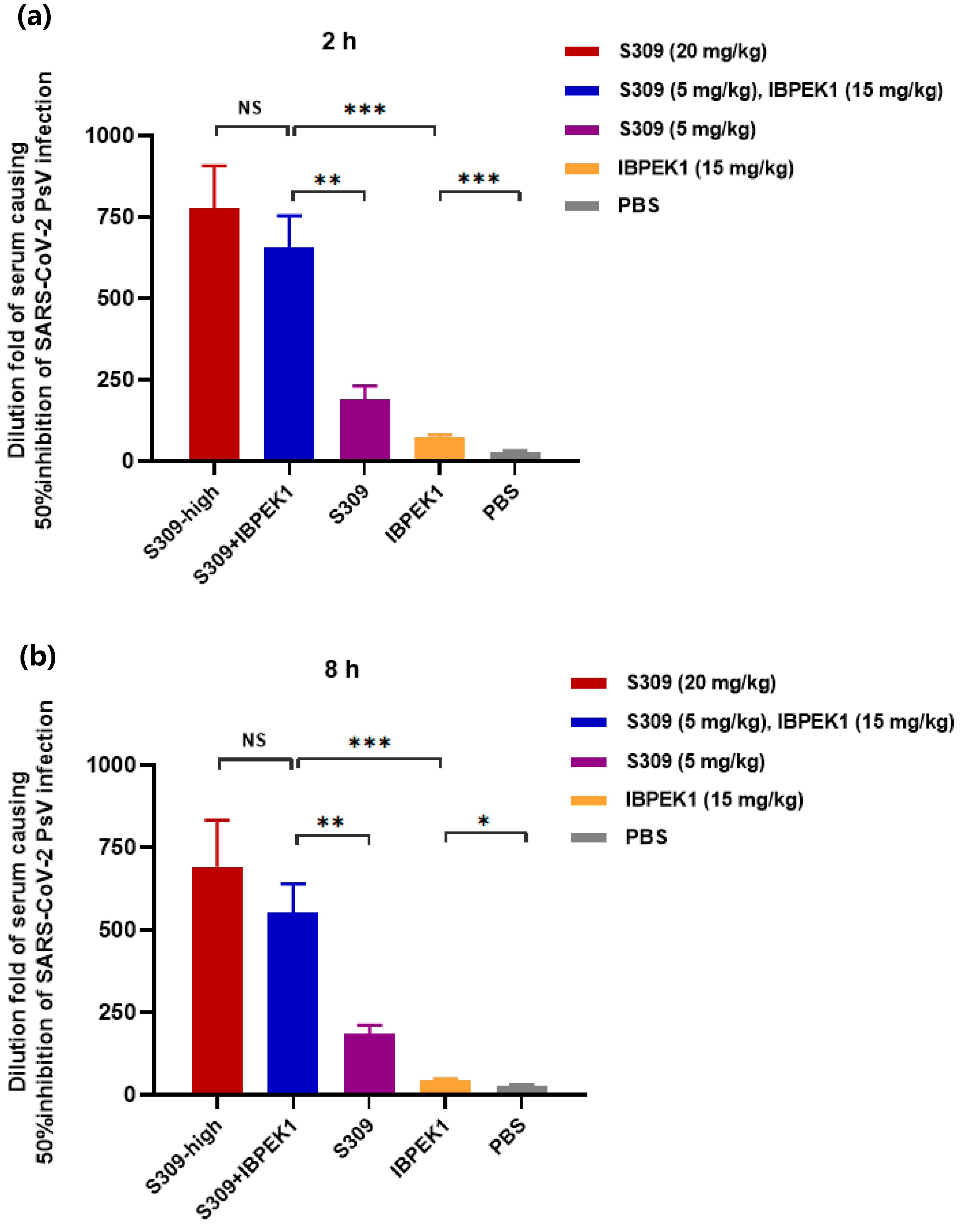
3.8. IBP-EK1 Synergized with Neutralizing Antibodies Targeting RBD to Robustly Inhibit CoVs In Vitro and Ex Vivo
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Adams, M.J.; Carstens, E.B. Ratification vote on taxonomic proposals to the International Committee on Taxonomy of Viruses (2012). Arch. Virol. 2012, 157, 1411–1422. [Google Scholar] [CrossRef]
- Cui, J.; Li, F.; Shi, Z.L. Origin and evolution of pathogenic coronaviruses. Nat. Rev. Microbiol. 2019, 17, 181–192. [Google Scholar] [CrossRef]
- Forni, D.; Cagliani, R.; Clerici, M.; Sironi, M. Molecular Evolution of Human Coronavirus Genomes. Trends Microbiol. 2017, 25, 35–48. [Google Scholar] [CrossRef] [PubMed]
- Su, S.; Wong, G.; Shi, W.; Liu, J.; Lai, A.C.K.; Zhou, J.; Liu, W.; Bi, Y.; Gao, G.F. Epidemiology, Genetic Recombination, and Pathogenesis of Coronaviruses. Trends Microbiol. 2016, 24, 490–502. [Google Scholar] [CrossRef] [PubMed]
- Drosten, C.; Gunther, S.; Preiser, W.; van der Werf, S.; Brodt, H.R.; Becker, S.; Rabenau, H.; Panning, M.; Kolesnikova, L.; Fouchier, R.A.; et al. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N. Engl. J. Med. 2003, 348, 1967–1976. [Google Scholar] [CrossRef] [PubMed]
- Zaki, A.M.; van Boheemen, S.; Bestebroer, T.M.; Osterhaus, A.D.; Fouchier, R.A. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N. Engl. J. Med. 2012, 367, 1814–1820. [Google Scholar] [CrossRef]
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed]
- Vlasova, A.N.; Diaz, A.; Damtie, D.; Xiu, L.; Toh, T.H.; Lee, J.S.; Saif, L.J.; Gray, G.C. Novel Canine Coronavirus Isolated from a Hospitalized Patient With Pneumonia in East Malaysia. Clin. Infect. Dis. 2022, 74, 446–454. [Google Scholar] [CrossRef]
- Lednicky, J.A.; Tagliamonte, M.S.; White, S.K.; Blohm, G.M.; Alam, M.M.; Iovine, N.M.; Salemi, M.; Mavian, C.; Morris, J.G. Isolation of a Novel Recombinant Canine Coronavirus From a Visitor to Haiti: Further Evidence of Transmission of Coronaviruses of Zoonotic Origin to Humans. Clin. Infect. Dis. 2022, 75, e1184–e1187. [Google Scholar] [CrossRef]
- Lednicky, J.A.; Tagliamonte, M.S.; White, S.K.; Elbadry, M.A.; Alam, M.M.; Stephenson, C.J.; Bonny, T.S.; Loeb, J.C.; Telisma, T.; Chavannes, S.; et al. Independent infections of porcine deltacoronavirus among Haitian children. Nature 2021, 600, 133–137. [Google Scholar] [CrossRef]
- Li, W.; Shi, Z.; Yu, M.; Ren, W.; Smith, C.; Epstein, J.H.; Wang, H.; Crameri, G.; Hu, Z.; Zhang, H.; et al. Bats are natural reservoirs of SARS-like coronaviruses. Science 2005, 310, 676–679. [Google Scholar] [CrossRef] [PubMed]
- Ge, X.Y.; Li, J.L.; Yang, X.L.; Chmura, A.A.; Zhu, G.; Epstein, J.H.; Mazet, J.K.; Hu, B.; Zhang, W.; Peng, C.; et al. Isolation and characterization of a bat SARS-like coronavirus that uses the ACE2 receptor. Nature 2013, 503, 535–538. [Google Scholar] [CrossRef] [PubMed]
- Menachery, V.D.; Yount, B.L., Jr.; Sims, A.C.; Debbink, K.; Agnihothram, S.S.; Gralinski, L.E.; Graham, R.L.; Scobey, T.; Plante, J.A.; Royal, S.R.; et al. SARS-like WIV1-CoV poised for human emergence. Proc. Natl. Acad. Sci. USA 2016, 113, 3048–3053. [Google Scholar] [CrossRef] [PubMed]
- Boni, M.F.; Lemey, P.; Jiang, X.; Lam, T.T.; Perry, B.W.; Castoe, T.A.; Rambaut, A.; Robertson, D.L. Evolutionary origins of the SARS-CoV-2 sarbecovirus lineage responsible for the COVID-19 pandemic. Nat. Microbiol. 2020, 5, 1408–1417. [Google Scholar] [CrossRef] [PubMed]
- Menachery, V.D.; Yount, B.L., Jr.; Debbink, K.; Agnihothram, S.; Gralinski, L.E.; Plante, J.A.; Graham, R.L.; Scobey, T.; Ge, X.Y.; Donaldson, E.F.; et al. A SARS-like cluster of circulating bat coronaviruses shows potential for human emergence. Nat. Med. 2015, 21, 1508–1513. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Mendelsohn, E.; Zong, C.; Zhang, W.; Hagan, E.; Wang, N.; Li, S.; Yan, H.; Huang, H.; Zhu, G.; et al. Human-animal interactions and bat coronavirus spillover potential among rural residents in Southern China. Biosaf. Health 2019, 1, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Hu, B.; Hu, C.; Zhu, F.; Liu, X.; Zhang, J.; Wang, B.; Xiang, H.; Cheng, Z.; Xiong, Y.; et al. Clinical Characteristics of 138 Hospitalized Patients With 2019 Novel Coronavirus-Infected Pneumonia in Wuhan, China. JAMA 2020, 323, 1061–1069. [Google Scholar] [CrossRef]
- Zhao, F.; Zai, X.; Zhang, Z.; Xu, J.; Chen, W. Challenges and developments in universal vaccine design against SARS-CoV-2 variants. NPJ Vaccines 2022, 7, 167. [Google Scholar] [CrossRef]
- McCallum, M.; Czudnochowski, N.; Rosen, L.E.; Zepeda, S.K.; Bowen, J.E.; Walls, A.C.; Hauser, K.; Joshi, A.; Stewart, C.; Dillen, J.R.; et al. Structural basis of SARS-CoV-2 Omicron immune evasion and receptor engagement. Science 2022, 375, 864–868. [Google Scholar] [CrossRef]
- Dyer, O. Covid-19: Omicron is causing more infections but fewer hospital admissions than delta, South African data show. BMJ 2021, 375, n3104. [Google Scholar] [CrossRef]
- Fan, Y.; Li, X.; Zhang, L.; Wan, S.; Zhang, L.; Zhou, F. SARS-CoV-2 Omicron variant: Recent progress and future perspectives. Signal Transduct. Target. Ther. 2022, 7, 141. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Jian, F.; Wang, J.; Yu, Y.; Song, W.; Yisimayi, A.; Wang, J.; An, R.; Chen, X.; Zhang, N.; et al. Imprinted SARS-CoV-2 humoral immunity induces convergent Omicron RBD evolution. Nature 2022, 614, 521–529. [Google Scholar] [CrossRef] [PubMed]
- Xia, S.; Xu, W.; Wang, Q.; Wang, C.; Hua, C.; Li, W.; Lu, L.; Jiang, S. Peptide-Based Membrane Fusion Inhibitors Targeting HCoV-229E Spike Protein HR1 and HR2 Domains. Int. J. Mol. Sci. 2018, 19, 487. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Xiao, G.; Chen, Y.; He, Y.; Niu, J.; Escalante, C.R.; Xiong, H.; Farmar, J.; Debnath, A.K.; Tien, P.; et al. Interaction between heptad repeat 1 and 2 regions in spike protein of SARS-associated coronavirus: Implications for virus fusogenic mechanism and identification of fusion inhibitors. Lancet 2004, 363, 938–947. [Google Scholar] [CrossRef] [PubMed]
- Bosch, B.J.; Martina, B.E.; Van Der Zee, R.; Lepault, J.; Haijema, B.J.; Versluis, C.; Heck, A.J.; De Groot, R.; Osterhaus, A.D.; Rottier, P.J. Severe acute respiratory syndrome coronavirus (SARS-CoV) infection inhibition using spike protein heptad repeat-derived peptides. Proc. Natl. Acad. Sci. USA 2004, 101, 8455–8460. [Google Scholar] [CrossRef]
- Sun, Y.; Zhang, H.; Shi, J.; Zhang, Z.; Gong, R. Identification of a Novel Inhibitor against Middle East Respiratory Syndrome Coronavirus. Viruses 2017, 9, 255. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Liu, Q.; Zhu, Y.; Chan, K.H.; Qin, L.; Li, Y.; Wang, Q.; Chan, J.F.; Du, L.; Yu, F.; et al. Structure-based discovery of Middle East respiratory syndrome coronavirus fusion inhibitor. Nat. Commun. 2014, 5, 3067. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Lu, G.; Qi, J.; Li, Y.; Wu, Y.; Deng, Y.; Geng, H.; Li, H.; Wang, Q.; Xiao, H.; et al. Structure of the fusion core and inhibition of fusion by a heptad repeat peptide derived from the S protein of Middle East respiratory syndrome coronavirus. J. Virol. 2013, 87, 13134–13140. [Google Scholar] [CrossRef]
- Xia, S.; Yan, L.; Xu, W.; Agrawal, A.S.; Algaissi, A.; Tseng, C.K.; Wang, Q.; Du, L.; Tan, W.; Wilson, I.A.; et al. A pan-coronavirus fusion inhibitor targeting the HR1 domain of human coronavirus spike. Sci. Adv. 2019, 5, eaav4580. [Google Scholar] [CrossRef]
- Sarin, H. Physiologic upper limits of pore size of different blood capillary types and another perspective on the dual pore theory of microvascular permeability. J. Angiogenes Res. 2010, 2, 14. [Google Scholar] [CrossRef]
- Haraldsson, B.; Nystrom, J.; Deen, W.M. Properties of the glomerular barrier and mechanisms of proteinuria. Physiol. Rev. 2008, 88, 451–487. [Google Scholar] [CrossRef] [PubMed]
- Su, S.; Rasquinha, G.; Du, L.; Wang, Q.; Xu, W.; Li, W.; Lu, L.; Jiang, S. A Peptide-Based HIV-1 Fusion Inhibitor with Two Tail-Anchors and Palmitic Acid Exhibits Substantially Improved In Vitro and Ex Vivo Anti-HIV-1 Activity and Prolonged In Vivo Half-Life. Molecules 2019, 24, 1134. [Google Scholar] [CrossRef] [PubMed]
- Datta-Mannan, A. Mechanisms Influencing the Pharmacokinetics and Disposition of Monoclonal Antibodies and Peptides. Drug Metab. Dispos. 2019, 47, 1100–1110. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.; Cholkar, K.; Mitra, A.K. Recent developments in protein and peptide parenteral delivery approaches. Ther. Deliv. 2014, 5, 337–365. [Google Scholar] [CrossRef] [PubMed]
- Strohl, W.R. Fusion Proteins for Half-Life Extension of Biologics as a Strategy to Make Biobetters. BioDrugs 2015, 29, 215–239. [Google Scholar] [CrossRef] [PubMed]
- Schellekens, H.; Hennink, W.E.; Brinks, V. The immunogenicity of polyethylene glycol: Facts and fiction. Pharm. Res. 2013, 30, 1729–1734. [Google Scholar] [CrossRef] [PubMed]
- Garay, R.P.; El-Gewely, R.; Armstrong, J.K.; Garratty, G.; Richette, P. Antibodies against polyethylene glycol in healthy subjects and in patients treated with PEG-conjugated agents. Expert. Opin. Drug Deliv. 2012, 9, 1319–1323. [Google Scholar] [CrossRef] [PubMed]
- Webster, R.; Didier, E.; Harris, P.; Siegel, N.; Stadler, J.; Tilbury, L.; Smith, D. PEGylated proteins: Evaluation of their safety in the absence of definitive metabolism studies. Drug Metab. Dispos. 2007, 35, 9–16. [Google Scholar] [CrossRef]
- Vallee, S.; Rakhe, S.; Reidy, T.; Walker, S.; Lu, Q.; Sakorafas, P.; Low, S.; Bitonti, A. Pulmonary administration of interferon Beta-1a-fc fusion protein in non-human primates using an immunoglobulin transport pathway. J. Interferon Cytokine Res. 2012, 32, 178–184. [Google Scholar] [CrossRef]
- Sleep, D.; Cameron, J.; Evans, L.R. Albumin as a versatile platform for drug half-life extension. Biochim. Biophys. Acta 2013, 1830, 5526–5534. [Google Scholar] [CrossRef]
- Choe, W.; Durgannavar, T.A.; Chung, S.J. Fc-Binding Ligands of Immunoglobulin G: An Overview of High Affinity Proteins and Peptides. Materials 2016, 9, 994. [Google Scholar] [CrossRef] [PubMed]
- Vukovic, N.; van Elsas, A.; Verbeek, J.S.; Zaiss, D.M.W. Isotype selection for antibody-based cancer therapy. Clin. Exp. Immunol. 2021, 203, 351–365. [Google Scholar] [CrossRef] [PubMed]
- DeLano, W.L.; Ultsch, M.H.; de Vos, A.M.; Wells, J.A. Convergent solutions to binding at a protein-protein interface. Science 2000, 287, 1279–1283. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Xu, W.; Liu, Z.; Wang, C.; Xia, S.; Lan, Q.; Cai, Y.; Su, S.; Pu, J.; Xing, L.; et al. A highly potent and stable pan-coronavirus fusion inhibitor as a candidate prophylactic and therapeutic for COVID-19 and other coronavirus diseases. Acta Pharm. Sin. B 2022, 12, 1652–1661. [Google Scholar] [CrossRef] [PubMed]
- Xia, S.; Liu, M.; Wang, C.; Xu, W.; Lan, Q.; Feng, S.; Qi, F.; Bao, L.; Du, L.; Liu, S.; et al. Inhibition of SARS-CoV-2 (previously 2019-nCoV) infection by a highly potent pan-coronavirus fusion inhibitor targeting its spike protein that harbors a high capacity to mediate membrane fusion. Cell Res. 2020, 30, 343–355. [Google Scholar] [CrossRef] [PubMed]
- Duan, Q.; Xia, S.; Jiao, F.; Wang, Q.; Wang, R.; Lu, L.; Jiang, S.; Xu, W. A Modified Fibronectin Type III Domain-Conjugated, Long-Acting Pan-Coronavirus Fusion Inhibitor with Extended Half-Life. Viruses 2022, 14, 655. [Google Scholar] [CrossRef]
- Jacomy, H.; Talbot, P.J. Vacuolating encephalitis in mice infected by human coronavirus OC43. Virology 2003, 315, 20–33. [Google Scholar] [CrossRef] [PubMed]
- Bi, W.; Xu, W.; Cheng, L.; Xue, J.; Wang, Q.; Yu, F.; Xia, S.; Wang, Q.; Li, G.; Qin, C.; et al. IgG Fc-binding motif-conjugated HIV-1 fusion inhibitor exhibits improved potency and in vivo half-life: Potential application in combination with broad neutralizing antibodies. PLoS Pathog. 2019, 15, e1008082. [Google Scholar] [CrossRef]
- Chong, H.; Xue, J.; Zhu, Y.; Cong, Z.; Chen, T.; Wei, Q.; Qin, C.; He, Y. Monotherapy with a low-dose lipopeptide HIV fusion inhibitor maintains long-term viral suppression in rhesus macaques. PLoS Pathog. 2019, 15, e1007552. [Google Scholar] [CrossRef]
- Allen, G.D. MODFIT: A pharmacokinetics computer program. Biopharm. Drug Dispos. 1990, 11, 477–498. [Google Scholar] [CrossRef]
- Chou, T.C. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol. Rev. 2006, 58, 621–681. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.C.; Talalay, P. Quantitative analysis of dose-effect relationships: The combined effects of multiple drugs or enzyme inhibitors. Adv. Enzyme Regul. 1984, 22, 27–55. [Google Scholar] [CrossRef]
- Chong, H.; Xue, J.; Zhu, Y.; Cong, Z.; Chen, T.; Guo, Y.; Wei, Q.; Zhou, Y.; Qin, C.; He, Y. Design of Novel HIV-1/2 Fusion Inhibitors with High Therapeutic Efficacy in Rhesus Monkey Models. J. Virol. 2018, 92, e00775-18. [Google Scholar] [CrossRef]
- Tang, T.; Bidon, M.; Jaimes, J.A.; Whittaker, G.R.; Daniel, S. Coronavirus membrane fusion mechanism offers a potential target for antiviral development. Antiviral Res. 2020, 178, 104792. [Google Scholar] [CrossRef] [PubMed]
- Jacomy, H.; St-Jean, J.R.; Brison, E.; Marceau, G.; Desforges, M.; Talbot, P.J. Mutations in the spike glycoprotein of human coronavirus OC43 modulate disease in BALB/c mice from encephalitis to flaccid paralysis and demyelination. J. Neurovirol. 2010, 16, 279–293. [Google Scholar] [CrossRef] [PubMed]
- Tolbert, W.D.; Gohain, N.; Kremer, P.G.; Hederman, A.P.; Nguyen, D.N.; Van, V.; Sherburn, R.; Lewis, G.K.; Finzi, A.; Pollara, J.; et al. Decoding human-macaque interspecies differences in Fc-effector functions: The structural basis for CD16-dependent effector function in Rhesus macaques. Front. Immunol. 2022, 13, 960411. [Google Scholar] [CrossRef] [PubMed]
- Tolbert, W.D.; Subedi, G.P.; Gohain, N.; Lewis, G.K.; Patel, K.R.; Barb, A.W.; Pazgier, M. From Rhesus macaque to human: Structural evolutionary pathways for immunoglobulin G subclasses. MAbs 2019, 11, 709–724. [Google Scholar] [CrossRef]
- Chen, Y.; Zhao, X.; Zhou, H.; Zhu, H.; Jiang, S.; Wang, P. Broadly neutralizing antibodies to SARS-CoV-2 and other human coronaviruses. Nat. Rev. Immunol. 2022, 23, 189–199. [Google Scholar] [CrossRef]
- Barnes, C.O.; Jette, C.A.; Abernathy, M.E.; Dam, K.A.; Esswein, S.R.; Gristick, H.B.; Malyutin, A.G.; Sharaf, N.G.; Huey-Tubman, K.E.; Lee, Y.E.; et al. SARS-CoV-2 neutralizing antibody structures inform therapeutic strategies. Nature 2020, 588, 682–687. [Google Scholar] [CrossRef]
- Hansen, J.; Baum, A.; Pascal, K.E.; Russo, V.; Giordano, S.; Wloga, E.; Fulton, B.O.; Yan, Y.; Koon, K.; Patel, K.; et al. Studies in humanized mice and convalescent humans yield a SARS-CoV-2 antibody cocktail. Science 2020, 369, 1010–1014. [Google Scholar] [CrossRef]
- Pinto, D.; Park, Y.J.; Beltramello, M.; Walls, A.C.; Tortorici, M.A.; Bianchi, S.; Jaconi, S.; Culap, K.; Zatta, F.; De Marco, A.; et al. Cross-neutralization of SARS-CoV-2 by a human monoclonal SARS-CoV antibody. Nature 2020, 583, 290–295. [Google Scholar] [CrossRef] [PubMed]
- Lempp, F.A.; Soriaga, L.B.; Montiel-Ruiz, M.; Benigni, F.; Noack, J.; Park, Y.J.; Bianchi, S.; Walls, A.C.; Bowen, J.E.; Zhou, J.; et al. Lectins enhance SARS-CoV-2 infection and influence neutralizing antibodies. Nature 2021, 598, 342–347. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; He, B.; Zhang, H.; Wang, X.; Zhang, Q.; Dai, W. IgG Fc Affinity Ligands and Their Applications in Antibody-Involved Drug Delivery: A Brief Review. Pharmaceutics 2023, 15, 187. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Gurgel, P.V.; Williams, D.K., Jr.; Bobay, B.G.; Cavanagh, J.; Muddiman, D.C.; Carbonell, R.G. Binding site on human immunoglobulin G for the affinity ligand HWRGWV. J. Mol. Recognit. 2010, 23, 271–282. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Gurgel, P.V.; Carbonell, R.G. Purification of human immunoglobulin G via Fc-specific small peptide ligand affinity chromatography. J. Chromatogr. A 2009, 1216, 910–918. [Google Scholar] [CrossRef] [PubMed]
- Menegatti, S.; Bobay, B.G.; Ward, K.L.; Islam, T.; Kish, W.S.; Naik, A.D.; Carbonell, R.G. Design of protease-resistant peptide ligands for the purification of antibodies from human plasma. J. Chromatogr. A 2016, 1445, 93–104. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.W.; Shi, Q.H.; Sun, Y. FYWHCLDE-based affinity chromatography of IgG: Effect of ligand density and purifications of human IgG and monoclonal antibody. J. Chromatogr. A 2014, 1355, 107–114. [Google Scholar] [CrossRef]
- Zhao, W.W.; Liu, F.F.; Shi, Q.H.; Sun, Y. Octapeptide-based affinity chromatography of human immunoglobulin G: Comparisons of three different ligands. J. Chromatogr. A 2014, 1359, 100–111. [Google Scholar] [CrossRef]
- Dias, R.L.; Fasan, R.; Moehle, K.; Renard, A.; Obrecht, D.; Robinson, J.A. Protein ligand design: From phage display to synthetic protein epitope mimetics in human antibody Fc-binding peptidomimetics. J. Am. Chem. Soc. 2006, 128, 2726–2732. [Google Scholar] [CrossRef]
- Gong, Y.; Zhang, L.; Li, J.; Feng, S.; Deng, H. Development of the Double Cyclic Peptide Ligand for Antibody Purification and Protein Detection. Bioconjug. Chem. 2016, 27, 1569–1573. [Google Scholar] [CrossRef]
- Pujadas, E.; Chaudhry, F.; McBride, R.; Richter, F.; Zhao, S.; Wajnberg, A.; Nadkarni, G.; Glicksberg, B.S.; Houldsworth, J.; Cordon-Cardo, C. SARS-CoV-2 viral load predicts COVID-19 mortality. Lancet Respir. Med. 2020, 8, e70. [Google Scholar] [CrossRef] [PubMed]
- Wolfel, R.; Corman, V.M.; Guggemos, W.; Seilmaier, M.; Zange, S.; Muller, M.A.; Niemeyer, D.; Jones, T.C.; Vollmar, P.; Rothe, C.; et al. Virological assessment of hospitalized patients with COVID-2019. Nature 2020, 581, 465–469. [Google Scholar] [CrossRef] [PubMed]
- Lau, J.L.; Dunn, M.K. Therapeutic peptides: Historical perspectives, current development trends, and future directions. Bioorg. Med. Chem. 2018, 26, 2700–2707. [Google Scholar] [CrossRef] [PubMed]
- Di, L. Strategic approaches to optimizing peptide ADME properties. AAPS J. 2015, 17, 134–143. [Google Scholar] [CrossRef] [PubMed]
- Stein, S.R.; Ramelli, S.C.; Grazioli, A.; Chung, J.Y.; Singh, M.; Yinda, C.K.; Winkler, C.W.; Sun, J.; Dickey, J.M.; Ylaya, K.; et al. SARS-CoV-2 infection and persistence in the human body and brain at autopsy. Nature 2022, 612, 758–763. [Google Scholar] [CrossRef] [PubMed]
- Taylor, P.C.; Adams, A.C.; Hufford, M.M.; de la Torre, I.; Winthrop, K.; Gottlieb, R.L. Neutralizing monoclonal antibodies for treatment of COVID-19. Nat. Rev. Immunol. 2021, 21, 382–393. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Zhang, W.; Hu, Y.; Tong, X.; Zheng, S.; Yang, J.; Kong, Y.; Ren, L.; Wei, Q.; Mei, H.; et al. Effect of Convalescent Plasma Therapy on Time to Clinical Improvement in Patients With Severe and Life-threatening COVID-19: A Randomized Clinical Trial. JAMA 2020, 324, 460–470. [Google Scholar] [CrossRef]
- Shen, C.; Wang, Z.; Zhao, F.; Yang, Y.; Li, J.; Yuan, J.; Wang, F.; Li, D.; Yang, M.; Xing, L.; et al. Treatment of 5 Critically Ill Patients With COVID-19 With Convalescent Plasma. JAMA 2020, 323, 1582–1589. [Google Scholar] [CrossRef]
- Liu, R.; Oldham, R.J.; Teal, E.; Beers, S.A.; Cragg, M.S. Fc-Engineering for Modulated Effector Functions-Improving Antibodies for Cancer Treatment. Antibodies 2020, 9, 64. [Google Scholar] [CrossRef]
- Xia, S.; Wang, L.; Jiao, F.; Yu, X.; Xu, W.; Huang, Z.; Li, X.; Wang, Q.; Zhu, Y.; Man, Q.; et al. SARS-CoV-2 Omicron subvariants exhibit distinct fusogenicity, but similar sensitivity, to pan-CoV fusion inhibitors. Emerg. Microbes Infect. 2023, 12, 2178241. [Google Scholar] [CrossRef]
- Youngnak, P.; Kozono, Y.; Kozono, H.; Iwai, H.; Otsuki, N.; Jin, H.; Omura, K.; Yagita, H.; Pardoll, D.M.; Chen, L.; et al. Differential binding properties of B7-H1 and B7-DC to programmed death-1. Biochem. Biophys. Res. Commun. 2003, 307, 672–677. [Google Scholar] [CrossRef]
- Pinto, D.; Sauer, M.M.; Czudnochowski, N.; Low, J.S.; Tortorici, M.A.; Housley, M.P.; Noack, J.; Walls, A.C.; Bowen, J.E.; Guarino, B.; et al. Broad betacoronavirus neutralization by a stem helix-specific human antibody. Science 2021, 373, 1109–1116. [Google Scholar] [CrossRef]
- Sun, X.; Yi, C.; Zhu, Y.; Ding, L.; Xia, S.; Chen, X.; Liu, M.; Gu, C.; Lu, X.; Fu, Y.; et al. Neutralization mechanism of a human antibody with pan-coronavirus reactivity including SARS-CoV-2. Nat. Microbiol. 2022, 7, 1063–1074. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SARS-CoV-2 Variants | IC50 (nM) against Pseudotyped SARS-CoV-2 Variants on Caco-2 Cells | IC50 (nM) against Pseudotyped SARS-CoV-2 Variants on Calu-3 Cells | ||||
|---|---|---|---|---|---|---|
| IBP | IBP-EK1 | EK1 | IBP | IBP-EK1 | EK1 | |
| N501Y | >50,000 | 718 | 737 | >50,000 | 266 | 347 |
| B.1.1.7 | >50,000 | 457 | 477 | >50,000 | 241 | 259 |
| B.1.351 | >50,000 | 677 | 719 | >50,000 | 281 | 349 |
| P.1 | >50,000 | 338 | 428 | >50,000 | 171 | 246 |
| B.1.617.2 | >50,000 | 389 | 527 | >50,000 | 296 | 303 |
| C.37 | >50,000 | 722 | 791 | >50,000 | 289 | 357 |
| BA.1 | >50,000 | 301 | 334 | NA | ||
| BA.2 | >50,000 | 399 | 544 | |||
| BA.2.12.1 | >50,000 | 527 | 592 | |||
| BA.2.75 | >50,000 | 221 | 248 | |||
| BA.5 | >50,000 | 564 | 690 | |||
| BF.7 | >50,000 | 257 | 260 | |||
| BA.4.6 | >50,000 | 195 | 201 | |||
| XBB | >50,000 | 330 | 366 | |||
| BQ.1.1 | >50,000 | 342 | 378 | |||
| Coronaviruses | Genus | Receptor (Reported) | IC50 (nM) against Pseudotyped CoVs on Caco-2 Cells | IC50 (nM) against Pseudotyped CoVs on Calu-3 Cells | IC50 (nM) against Live CoV Infection | IC50 (nM) against Cell-Cell Fusion Mediated by S Proteins of CoVs | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| IBP | IBP-EK1 | EK1 | IBP | IBP-EK1 | EK1 | IBP | IBP-EK1 | EK1 | IBP | IBP-EK1 | EK1 | |||
| SARS-CoV | β | ACE2 | >50,000 | 1301 | 1441 | >50,000 | 18 | 24 | ND | >50,000 | 229 | 426 | ||
| MERS-CoV | β | DPP4 | >50,000 | 522 | 885 | >50,000 | 76 | 77 | ND | >50,000 | 376 | 492 | ||
| SARS-CoV-2 | β | ACE2 | >50,000 | 512 | 590 | >50,000 | 180 | 253 | >50,000 | 204 | 296 | >50,000 | 512 | 590 |
| WIV1-CoV | β | ACE2 | >50,000 | 6047 | 6985 | NA | ND | >50,000 | 306 | 588 | ||||
| HCoV-OC43 | β | 9-O-Ac-Sias | >50,000 | 3574 | 3611 | NA | >50,000 | 744 | 764 | >50,000 | 829 | 1161 | ||
| HCoV-NL63 | α | ACE2 | >50,000 | 5641 | 5934 | >50,000 | 440 | 537 | ND | >50,000 | 212 | 721 | ||
| HCoV-229E | α | hAPN | >50,000 | 3344 | 3475 | NA | >50,000 | 4139 | 7592 | >50,000 | 478 | 623 | ||
| Parameter | IBP-EK1 (UPLC-Q-TOF-MS) | IBP-EK1 (Estimated) |
|---|---|---|
| t1/2 (h) | 37.67 | 39.72 |
| Tmax (h) | 2 | 2 |
| Cmax (μg/mL) | 51.73 | 116.09 |
| AUC (μg/mL × h) | 2207.03 | 5228.98 |
| IBP-EK1 Combination with Antibody against Sarbecoviruses | CI | Antibody | IBP-EK1 | ||||
|---|---|---|---|---|---|---|---|
| IC50 (nM) | Dose Reduction (n-Fold) | IC50 (nM) | Dose Reduction (n-Fold) | ||||
| Alone | In Mixture | Alone | In Mixture | ||||
| 10,933 | |||||||
| B.1.351 (Beta) | 0.32 | 0.51 | 0.11 | 4.5 | 1673 | 168.8 | 9.9 |
| P.1 (Gamma) | 0.38 | 0.46 | 0.13 | 3.6 | 1556 | 154.9 | 10.1 |
| BA.1 | 0.26 | 17.22 | 1.44 | 11.9 | 1256 | 216.6 | 5.8 |
| S309 | |||||||
| SARS-CoV-2 | 0.23 | 5.81 | 0.46 | 12.6 | 1381 | 208.4 | 6.6 |
| B.1.117 (Alpha) | 0.22 | 3.10 | 0.24 | 12.8 | 801.0 | 109.2 | 7.3 |
| B.1.351 (Beta) | 0.23 | 1.20 | 0.15 | 7.9 | 670.6 | 68.3 | 9.8 |
| P.1 (Gamma) | 0.24 | 4.01 | 0.26 | 15.6 | 647.6 | 115.7 | 5.6 |
| B.1.617.2 (Delta) | 0.14 | 1.20 | 0.12 | 10.2 | 1533 | 70.5 | 21.7 |
| BA.1 | 0.19 | 3.28 | 0.30 | 11.0 | 1416 | 134.7 | 10.5 |
| BA.5 | 0.10 | 7.62 | 0.32 | 23.5 | 2520 | 145.8 | 17.3 |
| XBB | 0.14 | 5.28 | 0.43 | 12.2 | 1217 | 70.8 | 17.2 |
| BQ.1.1 | 0.20 | 52.98 | 3.11 | 17.0 | 1123 | 155.7 | 7.2 |
| SARS-CoV | 0.15 | 0.59 | 0.07 | 8.8 | 4358 | 150.3 | 29.0 |
| WIV1-CoV | 0.11 | 4.03 | 0.34 | 11.8 | 7713 | 170.5 | 45.3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Su, X.; Huang, Z.; Xu, W.; Wang, Q.; Xing, L.; Lu, L.; Jiang, S.; Xia, S. IgG Fc-Binding Peptide-Conjugated Pan-CoV Fusion Inhibitor Exhibits Extended In Vivo Half-Life and Synergistic Antiviral Effect When Combined with Neutralizing Antibodies. Biomolecules 2023, 13, 1283. https://doi.org/10.3390/biom13091283
Su X, Huang Z, Xu W, Wang Q, Xing L, Lu L, Jiang S, Xia S. IgG Fc-Binding Peptide-Conjugated Pan-CoV Fusion Inhibitor Exhibits Extended In Vivo Half-Life and Synergistic Antiviral Effect When Combined with Neutralizing Antibodies. Biomolecules. 2023; 13(9):1283. https://doi.org/10.3390/biom13091283
Chicago/Turabian StyleSu, Xiaojie, Ziqi Huang, Wei Xu, Qian Wang, Lixiao Xing, Lu Lu, Shibo Jiang, and Shuai Xia. 2023. "IgG Fc-Binding Peptide-Conjugated Pan-CoV Fusion Inhibitor Exhibits Extended In Vivo Half-Life and Synergistic Antiviral Effect When Combined with Neutralizing Antibodies" Biomolecules 13, no. 9: 1283. https://doi.org/10.3390/biom13091283
APA StyleSu, X., Huang, Z., Xu, W., Wang, Q., Xing, L., Lu, L., Jiang, S., & Xia, S. (2023). IgG Fc-Binding Peptide-Conjugated Pan-CoV Fusion Inhibitor Exhibits Extended In Vivo Half-Life and Synergistic Antiviral Effect When Combined with Neutralizing Antibodies. Biomolecules, 13(9), 1283. https://doi.org/10.3390/biom13091283