
Targeting Breast Cancer: The Familiar, the Emerging, and the Uncharted Territories



Abstract
:1. Introduction
2. Where We Stand

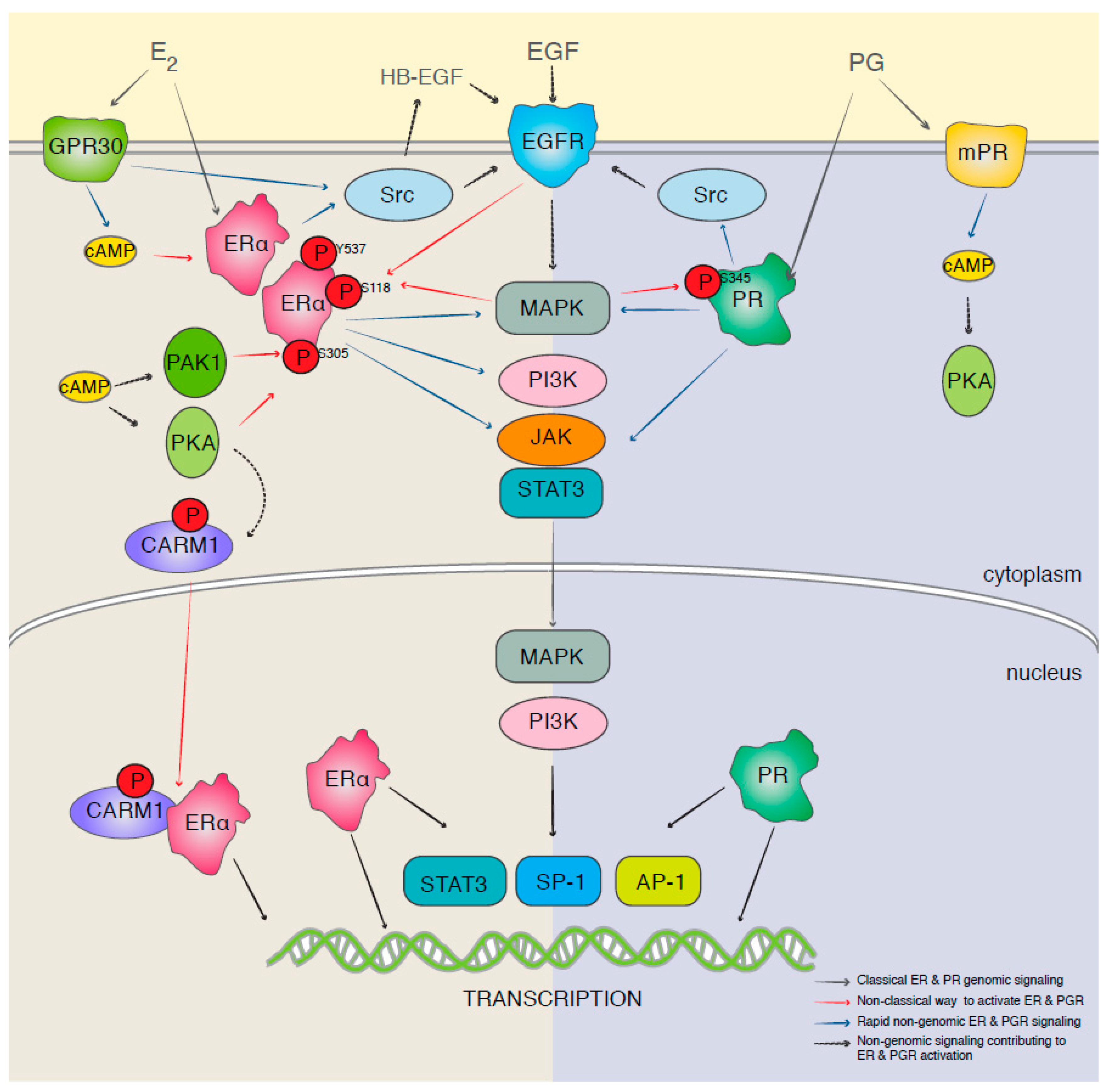{kind=link}
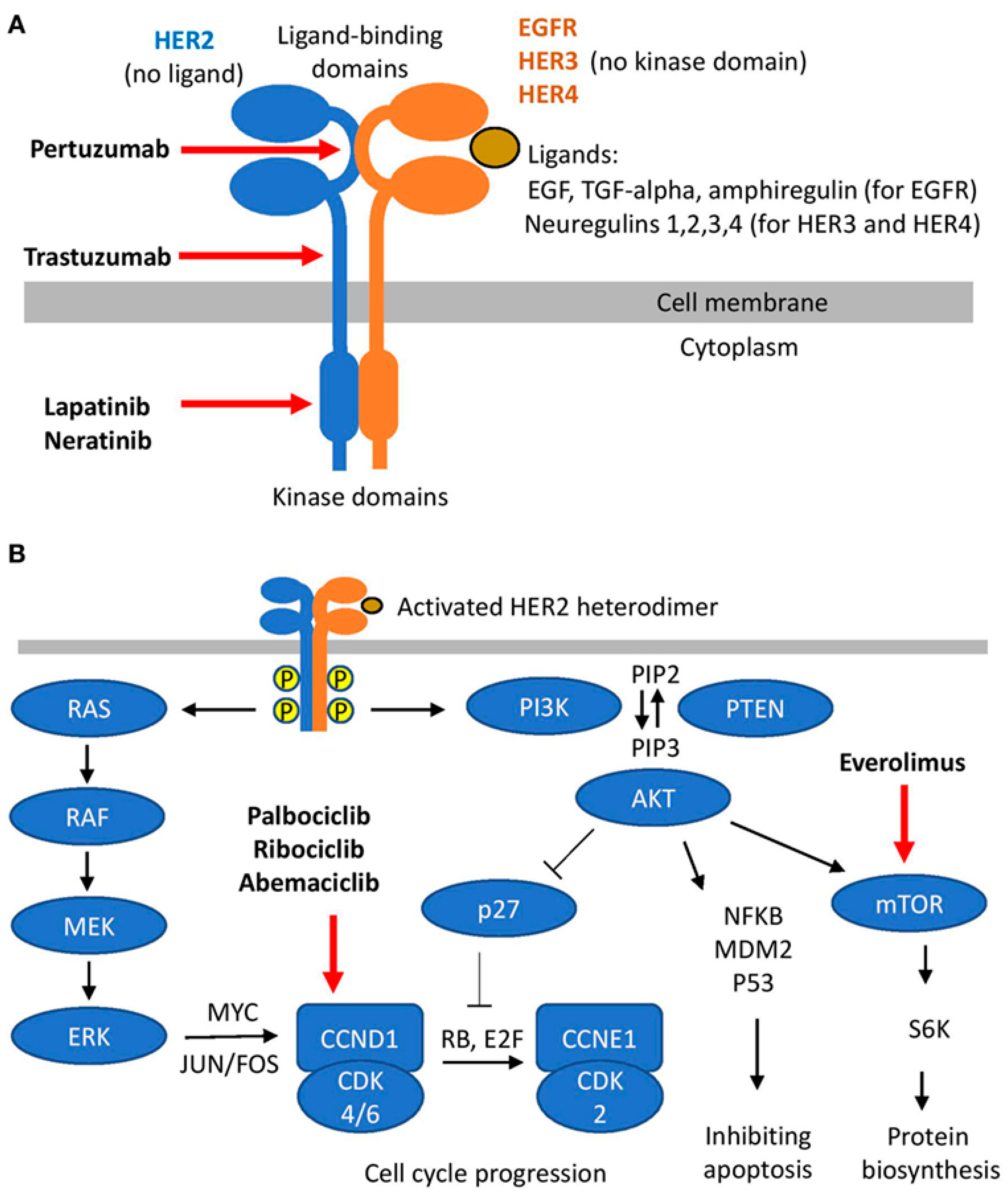{kind=link}
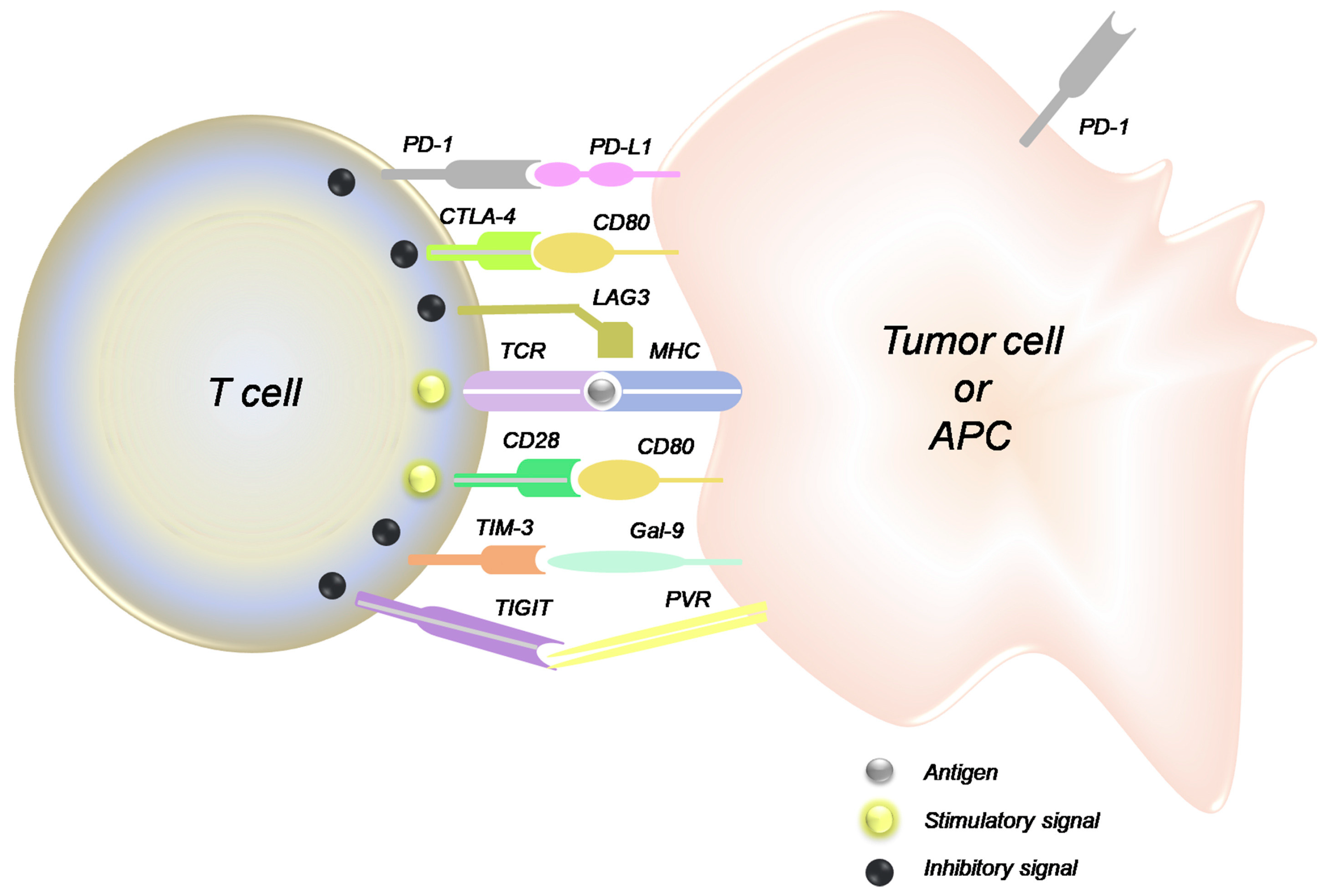{kind=link}
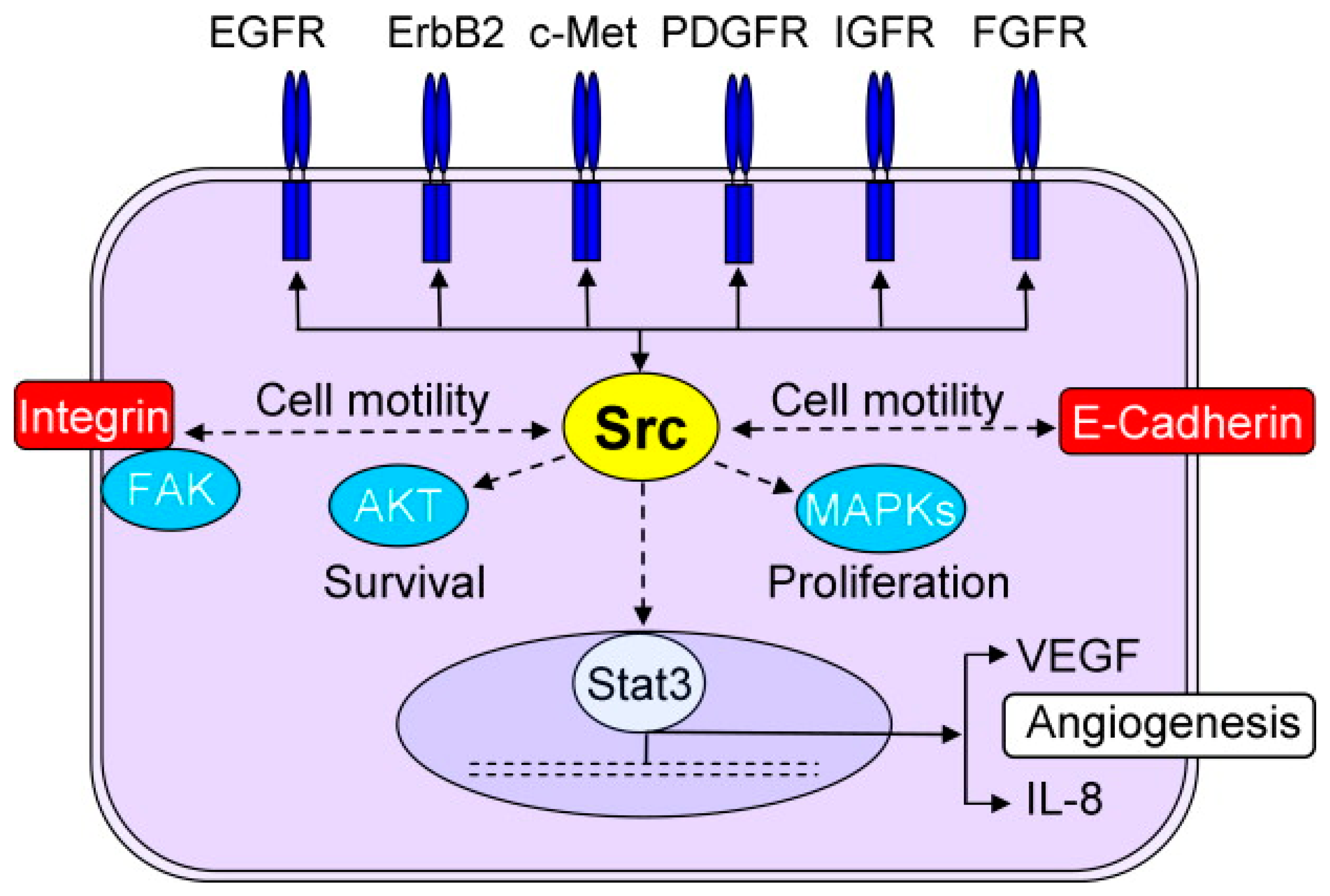{kind=link}
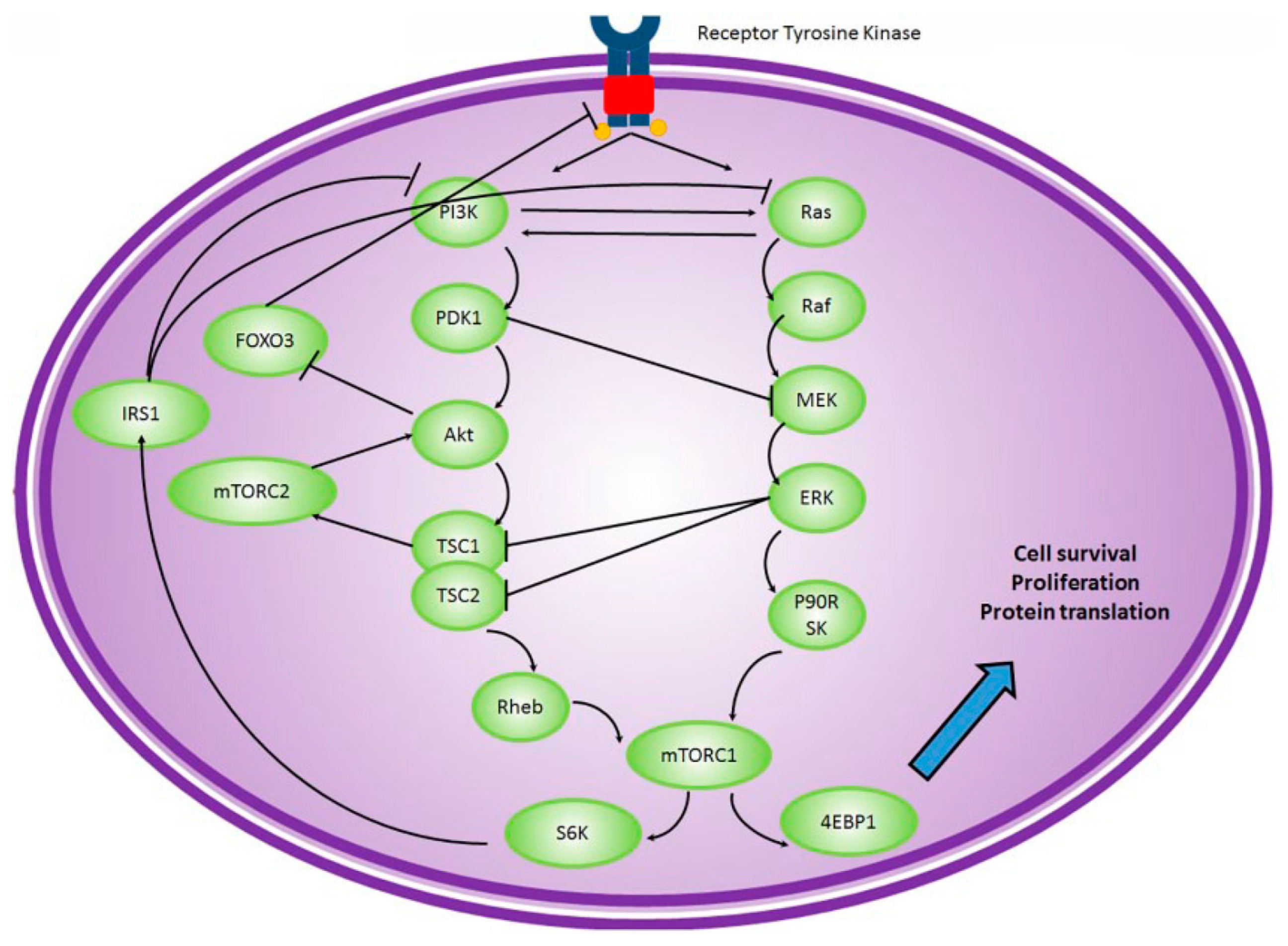{kind=link}
| Drug | Targeted Protein | Indication | Used in Combination | Selected Combinations in Clinical Trials |
|---|---|---|---|---|
| Abemaciclib | CDK4/6 | HR-Positive, HER2-negative | Fulvestrant, aromatase inhibitors, Tamoxifen | Phase Ib/II (NCT04791384) with elacestrant; Phase II (NCT04432454) with lasofoxifene (ERA); Phase I (NCT05095207) with bicalutamide (AA); Phase I (NCT05464173) with Fulvestrant and chidamide (HDACI); Phase I (NCT03846583) with tucatinib and trastuzumab |
| Alpelisib | PI3K (predominantly against PI3K catalyst subunit α) | HR-Positive, HER2-negative with PIK3CA mutation | Fulvestrant | Phase I (NCT05143229) with Sacituzumab govitecan; Phase II (NCT05625087) with ribociclib; Phase I (NCT05230810) with tucatinib |
| Anastrozole | Aromatase | HR-Positive | - | Several with fulvestrant, goserelin, tamoxifen, gefitinib (EGFRI), etc. |
| Atezolizumab | PD-L1 | Advanced or metastatic TNBC | It is only approved in combination with albumin-bound paclitaxel † | Phase II (NCT04759248) with trastuzumab and vinorelbine (chemotherapy); Phase I/II (NCT02708680) with entinostat (HDACI); Phase II (NCT04690855) with Talazoparib; Phase I (NCT04584112) with tiragolumab (TIGIT blocker) |
| Elacestrant | Estrogen receptor α | ER-positive and HER2-negative with ESR1 mutation | - | Phase III (NCT05512364) with tamoxifen, letrozole, anastrozole, and exemestane; Phase Ib/II (NCT04791384) with abemaciclib; Phase I (NCT05618613) with onapristone (AP) |
| Everolimus | mTOR | HR-Positive, HER2-negative | Exemestane | Several with letrozole, fulvestrant, goserelin, erlotinib, trastuzumab, etc. |
| Exemestane | Aromatase | ER-positive | - | Several with Dasatinib (TKI), chidamide (HDACI), raloxifene (ERAA), everolimus, fulvestrant, etc. |
| Fulvestrant | Estrogen receptor | HR-Positive, HER2-negative | Ribociclib, palbociclib, or abemaciclib (in metastatic cases that got worse with hormone therapy alone) | Several with enzalutamide (second generation AA), neratinib or pyrotinib (TKIs; in HR-positive, HER2-positive cases), goserelin, anastrozole, etc. |
| Goserelin | LHRH receptor | Advanced breast cancer | - | Phase II (NCT02072512) with fulvestrant and anastrozole; Phase II (NCT00010010) with exemestane; Phase II (NCT00217659) with anastrozole; Phase I (NCT02586675) with tamoxifen and ribociclib |
| Lapatinib | EGFR/HER2 | Advanced HER2-positive | Capecitabine (chemotherapy), or with letrozole (in HR-positive, HER2-positive cases) | Phase I (NCT00424164) with tamoxifen; Phase II/III (NCT03085368) with trastuzumab; Phase II (NCT01272141) with everolimus; Phase II (NCT01275859) with letrozole; and several trials with a variety of chemotherapeutic agents |
| Letrozole | Aromatase | Early stage or metastatic HR-positive | - | Several with nintedanib (TKI), lenvatinib (TKI), pembrolizumab, palbociclib, anastrozole, ribociclib, tucatinib, dalpiciclib (CDK4/6 inhibitor), bevacizumab (VEGF inhibitor), etc. |
| Margetuximab | HER2 | Metastatic HER2-positive | - | Phase II (NCT04262804) and Phase III (NCT02492711) with trastuzumab; Phase II (NCT04425018) with pertuzumab; Phase I (NCT03219268) with tebotelimab (PD-1 and LAG-3 blocker) |
| Neratinib | EGFR, HER2, HER4 | HER2-positive | Capecitabin (chemotherapy) in advanced or metastatic cases | Phase II (NCT03289039) with Fulvestrant; Phase II (NCT01111825) with temsirolimus (mTOR inhibitor); Phase II (NCT00915018) with trastuzumab) |
| Olaparib | PARP enzymes | HER2-negative with mutations in BRCA1 or BRCA2 | - | Phase II (NCT03025035) with pembrolizumab; Phase II (NCT03594396) with durvalumab (PD-L1 antibody); Phase II (NCT05536128) with fulvestrant; Phase II (NCT02849496) with atezolizumab |
| Palbociclib | CDK4/6 | HR-Positive, HER2-negative | Fulvestrant, aromatase inhibitors | Several with letrozole, avelumab (PD-1 antibody), erdafitinib (FGFR inhibitor), inavolisib (PI3Kα inhibitor), etc. |
| Pembrolizumab | PD-1 | TNBC | Chemotherapy | Several with tamoxifen, mifepristone (PRA), fulvestrant, lenvatinib (TKI), olaparib, exemestane, trastuzumab, etc. |
| Pertuzumab | HER2 (and EGFR, HER3 and HER4) | HER2-positive | Trastuzumab | Phase II (NCT05574881) with dalpiciclib (CDK4/6 inhibitor) and fulvestrant; Phase II (NCT03988036) with pembrolizumab; Phase III (NCT05132582) with tucatinib; Phase II (NCT03820141) with durvalumab (PD-L1 antibody) |
| Ribociclib | CDK4/6 | HR-Positive, HER2-negative | Fulvestrant, aromatase inhibitors | Phase Ib/II (NCT02657343) with trastuzumab; Phase II (NCT05625087) with Alpelisib; Phase I (NCT02586675) with tamoxifen; Phase I (NCT01857193) with everolimus/exemestane |
| Talazoparib | PARP enzymes | HER2-negative with mutations in BRCA1 or BRCA2 | - | Phase I/II (NCT03964532) with avelumab (PD-1 antibody); Phase II (NCT04690855) with atezolizumab; Phase I/II (NCT05035745) with Selinexor (XPO1 inhibitor) |
| Tamoxifen | Estrogen receptor | Metastatic breast cancer | Surgery and radiotherapy | Several with pembrolizumab, gefitinib (EGFRI), raloxifene (ERAA), toremifene, anastrozole, etc. |
| Toremifene | Estrogen receptor | Metastatic breast cancer (ER-positive or unknown) | - | Phase IV (NCT02344940) with tamoxifen; Phase IV (NCT02089854) with anastrozole; Phase III (NCT02132390) with goserelin; Phase III (NCT00044291) with atamestane (AI) |
| Trastuzumab | HER2 | HER2-positive | Chemotherapy in hormone receptor negative, high-risk, or metastatic breast cancer | Several with pertuzumab, bevacizumab (VEGF inhibitor), everolimus, monalizumab (NKG2A inhibitor), fulvestrant/pertuzumab/dalpiciclib (CDK4/6 inhibitor), and several chemotherapies |
| Tucatinib | HER2 | HER2-positive | Trastuzumab and capecitabine †† | Phase I/II (NCT05230810) with alpelisib |
3. The Familiar
3.1. Hormone Therapy
3.2. HER Family of Receptors
3.3. CDK4/6
4. The Emerging
4.1. Immune Checkpoint Inhibitors
4.2. PARP Inhibitors
4.3. PI3K/AKT Pathway Inhibitors
5. The Uncharted
5.1. Src
5.2. RAS/RAF/MEK/ERK Pathway
5.3. JAK/STAT Pathway
5.4. Others
5.4.1. PPAR
5.4.2. Syndecans (SDCs)
5.4.3. RUNX2 and HDACs
5.4.4. Hyaluronic Acid
5.4.5. NDRG1
5.4.6. Chimeric Antigen Receptor (CAR) T-Cell Immunotherapy
6. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Arnold, M.; Morgan, E.; Rumgay, H.; Mafra, A.; Singh, D.; Laversanne, M.; Vignat, J.; Gralow, J.R.; Cardoso, F.; Siesling, S.; et al. Current and future burden of breast cancer: Global statistics for 2020 and 2040. Breast 2022, 66, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Cao, B.; Soerjomataram, I.; Bray, F. World Cancer Report; World Health Organization (WHO): Geneva, Switzerland, 2020. [Google Scholar]
- Sottoriva, A.; Kang, H.; Ma, Z.; Graham, T.A.; Salomon, M.P.; Zhao, J.; Marjoram, P.; Siegmund, K.; Press, M.F.; Shibata, D.; et al. A Big Bang model of human colorectal tumor growth. Nat. Genet. 2015, 47, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Amirel, A.D.; Davis, K.L.; Tadmor, M.D.; Simonds, E.F.; Levine, J.H.; Bendall, S.C.; Shenfeld, D.K.; Krishnaswamy, S.; Nolan, G.P.; Pe’er, D. viSNE enables visualization of high dimensional single-cell data and reveals phenotypic heterogeneity of leukemia. Nat. Biotechnol. 2013, 31, 545–552. [Google Scholar] [CrossRef] [PubMed]
- Alderton, G.K. Heterogeneity: Explosive beginnings. Nat. Rev. Cancer 2015, 15, 198–199. [Google Scholar] [CrossRef] [PubMed]
- Holzel, M.; Bovier, A.; Tuting, T. Plasticity of tumour and immune cells: A source of heterogeneity and a cause for therapy resistance? Nat. Rev. Cancer 2013, 13, 365–376. [Google Scholar] [CrossRef]
- Nikolaou, M.; Pavlopoulou, A.; Georgakilas, A.G.; Kyrodimos, E. The challenge of drug resistance in cancer treatment: A current overview. Clin. Exp. Metastasis 2018, 35, 309–318. [Google Scholar] [CrossRef]
- Tortora, G.; Bianco, R.; Daniele, G.; Ciardiello, F.; McCubrey, J.A.; Ricciardi, M.R.; Ciuffreda, L.; Cognetti, F.; Tafuri, A.; Milella, M. Overcoming resistance to molecularly targeted anticancer therapies: Rational drug combinations based on EGFR and MAPK inhibition for solid tumours and haematologic malignancies. Drug Resist. Updat. 2007, 10, 81–100. [Google Scholar] [CrossRef]
- Asao, T.; Takahashi, F.; Takahashi, K. Resistance to molecularly targeted therapy in non-small-cell lung cancer. Respir. Investig. 2019, 57, 20–26. [Google Scholar] [CrossRef]
- Shien, K.; Papadimitrakopoulou, V.A.; Ruder, D.; Behrens, C.; Shen, L.; Kalhor, N.; Song, J.; Lee, J.J.; Wang, J.; Tang, X.; et al. JAK1/STAT3 Activation through a Proinflammatory Cytokine Pathway Leads to Resistance to Molecularly Targeted Therapy in Non-Small Cell Lung Cancer. Mol. Cancer Ther. 2017, 16, 2234–2245. [Google Scholar] [CrossRef]
- Whitesell, L.; Santagata, S.; Mendillo, M.L.; Lin, N.U.; Proia, D.A.; Lindquist, S. HSP90 empowers evolution of resistance to hormonal therapy in human breast cancer models. Proc. Natl. Acad. Sci. USA 2014, 111, 18297–18302. [Google Scholar] [CrossRef]
- Rosenzweig, S.A. Acquired resistance to drugs targeting receptor tyrosine kinases. Biochem. Pharmacol. 2012, 83, 1041–1048. [Google Scholar] [CrossRef] [PubMed]
- Rosenzweig, S.A. Acquired Resistance to Drugs Targeting Tyrosine Kinases. Adv. Cancer Res. 2018, 138, 71–98. [Google Scholar] [CrossRef] [PubMed]
- Qi, J.; McTigue, M.A.; Rogers, A.; Lifshits, E.; Christensen, J.G.; Janne, P.A.; Engelman, J.A. Multiple mutations and bypass mechanisms can contribute to development of acquired resistance to MET inhibitors. Cancer Res. 2011, 71, 1081–1091. [Google Scholar] [CrossRef]
- Ordog, T.; Zornig, M.; Hayashi, Y. Targeting Disease Persistence in Gastrointestinal Stromal Tumors. Stem Cells Transl. Med. 2015, 4, 702–707. [Google Scholar] [CrossRef]
- Kosaka, T.; Yamaki, E.; Mogi, A.; Kuwano, H. Mechanisms of resistance to EGFR TKIs and development of a new generation of drugs in non-small-cell lung cancer. J. Biomed. Biotechnol. 2011, 2011, 165214. [Google Scholar] [CrossRef]
- Walz, C.; Sattler, M. Novel targeted therapies to overcome imatinib mesylate resistance in chronic myeloid leukemia (CML). Crit. Rev. Oncol. Hematol. 2006, 57, 145–164. [Google Scholar] [CrossRef] [PubMed]
- Singleton, K.R.; Earley, K.T.; Heasley, L.E. Analysis of Drug Resistance Using Kinome-Wide Functional Screens. Methods Mol. Biol. 2017, 1636, 163–177. [Google Scholar] [CrossRef]
- Al-Lazikani, B.; Banerji, U.; Workman, P. Combinatorial drug therapy for cancer in the post-genomic era. Nat. Biotechnol. 2012, 30, 679–692. [Google Scholar] [CrossRef]
- Kelly, C.M.; Buzdar, A.U. Using multiple targeted therapies in oncology: Considerations for use, and progress to date in breast cancer. Drugs 2013, 73, 505–515. [Google Scholar] [CrossRef]
- Cheung, K.L. Treatment Strategies and Survival Outcomes in Breast Cancer. Cancers 2020, 12, 735. [Google Scholar] [CrossRef] [PubMed]
- Plesca, M.; Bordea, C.; El Houcheimi, B.; Ichim, E.; Blidaru, A. Evolution of radical mastectomy for breast cancer. J. Med. Life 2016, 9, 183–186. [Google Scholar] [PubMed]
- Alghamdi, M.A.A.; Esam Mahmood, S. Role of Surgery in Metastatic Breast Cancer: Insights from a Narrative Review. Breast Cancer 2023, 15, 349–358. [Google Scholar] [CrossRef]
- Upadhyay, R.; Bazan, J.G. Advances in Radiotherapy for Breast Cancer. Surg. Oncol. Clin. N. Am. 2023, 32, 515–536. [Google Scholar] [CrossRef] [PubMed]
- Goel, S.; DeCristo, M.J.; McAllister, S.S.; Zhao, J.J. CDK4/6 Inhibition in Cancer: Beyond Cell Cycle Arrest. Trends Cell Biol. 2018, 28, 911–925. [Google Scholar] [CrossRef] [PubMed]
- Chen, A. PARP inhibitors: Its role in treatment of cancer. Chin. J. Cancer 2011, 30, 463–471. [Google Scholar] [CrossRef]
- DePolo, J. Genentech Withdraws Breast Cancer Indication From Tecentriq. Available online: https://www.breastcancer.org/research-news/genentech-withdraws-breast-cancer-indication-from-tecentriq (accessed on 23 February 2023).
- Quirke, V.M. Tamoxifen from Failed Contraceptive Pill to Best-Selling Breast Cancer Medicine: A Case-Study in Pharmaceutical Innovation. Front. Pharmacol. 2017, 8, 620. [Google Scholar] [CrossRef]
- Sleightholm, R.; Neilsen, B.K.; Elkhatib, S.; Flores, L.; Dukkipati, S.; Zhao, R.; Choudhury, S.; Gardner, B.; Carmichael, J.; Smith, L.; et al. Percentage of Hormone Receptor Positivity in Breast Cancer Provides Prognostic Value: A Single-Institute Study. J. Clin. Med. Res. 2021, 13, 9–19. [Google Scholar] [CrossRef]
- Breast Cancer Hormone Receptor Status. Available online: https://www.breastcancer.org/pathology-report/hormone-receptor-status (accessed on 12 April 2023).
- Toss, A.; Venturelli, M.; Peterle, C.; Piacentini, F.; Cascinu, S.; Cortesi, L. Molecular Biomarkers for Prediction of Targeted Therapy Response in Metastatic Breast Cancer: Trick or Treat? Int. J. Mol. Sci. 2017, 18, 85. [Google Scholar] [CrossRef]
- Kulkoyluoglu, E.; Madak-Erdogan, Z. Nuclear and extranuclear-initiated estrogen receptor signaling crosstalk and endocrine resistance in breast cancer. Steroids 2016, 114, 41–47. [Google Scholar] [CrossRef]
- Oza, A.; Ma, C.X. New Insights in Estrogen Receptor (ER) Biology and Implications for Treatment. Curr. Breast Vancer Rep. 2017, 9, 13–25. [Google Scholar] [CrossRef]
- Kundu, N.; Brekman, A.; Kim, J.Y.; Xiao, G.; Gao, C.; Bargonetti, J. Estrogen-activated MDM2 disrupts mammary tissue architecture through a p53-independent pathway. Oncotarget 2017, 8, 47916–47930. [Google Scholar] [CrossRef] [PubMed]
- Tanos, T.; Rojo, L.; Echeverria, P.; Brisken, C. ER and PR signaling nodes during mammary gland development. Breast Cancer Res. 2012, 14, 210. [Google Scholar] [CrossRef] [PubMed]
- Filardo, E.J. Epidermal growth factor receptor (EGFR) transactivation by estrogen via the G-protein-coupled receptor, GPR30: A novel signaling pathway with potential significance for breast cancer. J. Steroid Biochem. Mol. Biol. 2002, 80, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Boonyaratanakornkit, V.; Scott, M.P.; Ribon, V.; Sherman, L.; Anderson, S.M.; Maller, J.L.; Miller, W.T.; Edwards, D.P. Progesterone receptor contains a proline-rich motif that directly interacts with SH3 domains and activates c-Src family tyrosine kinases. Mol. Cell 2001, 8, 269–280. [Google Scholar] [CrossRef]
- Burgess, D.J. Breast cancer: SRC hits the mark. Nat. Rev. Cancer 2011, 11, 314–315. [Google Scholar] [CrossRef]
- Bai, X.; Sun, P.; Wang, X.; Long, C.; Liao, S.; Dang, S.; Zhuang, S.; Du, Y.; Zhang, X.; Li, N.; et al. Structure and dynamics of the EGFR/HER2 heterodimer. Cell Discov. 2023, 9, 18. [Google Scholar] [CrossRef]
- Zaczek, A.; Brandt, B.; Bielawski, K.P. The diverse signaling network of EGFR, HER2, HER3 and HER4 tyrosine kinase receptors and the consequences for therapeutic approaches. Histol. Histopathol. 2005, 20, 1005–1015. [Google Scholar] [CrossRef]
- Larionov, A.A. Current Therapies for Human Epidermal Growth Factor Receptor 2-Positive Metastatic Breast Cancer Patients. Front. Oncol. 2018, 8, 89. [Google Scholar] [CrossRef]
- Paplomata, E.; O’Regan, R. New and emerging treatments for estrogen receptor-positive breast cancer: Focus on everolimus. Ther. Clin. Risk Manag. 2013, 9, 27–36. [Google Scholar] [CrossRef]
- Kavarthapu, R.; Anbazhagan, R.; Dufau, M.L. Crosstalk between PRLR and EGFR/HER2 Signaling Pathways in Breast Cancer. Cancers 2021, 13, 4685. [Google Scholar] [CrossRef] [PubMed]
- Sirkisoon, S.R.; Carpenter, R.L.; Rimkus, T.; Miller, L.; Metheny-Barlow, L.; Lo, H.W. EGFR and HER2 signaling in breast cancer brain metastasis. Front Biosci. (Elite Ed.) 2016, 8, 245–263. [Google Scholar] [CrossRef] [PubMed]
- Bousoik, E.; Montazeri Aliabadi, H. “Do We Know Jack” About JAK? A Closer Look at JAK/STAT Signaling Pathway. Front. Oncol. 2018, 8, 287. [Google Scholar] [CrossRef] [PubMed]
- Yeatman, T.J. A renaissance for SRC. Nat. Rev. Cancer 2004, 4, 470–480. [Google Scholar] [CrossRef]
- McBryan, J.; Theissen, S.M.; Byrne, C.; Hughes, E.; Cocchiglia, S.; Sande, S.; O’Hara, J.; Tibbitts, P.; Hill, A.D.; Young, L.S. Metastatic progression with resistance to aromatase inhibitors is driven by the steroid receptor coactivator SRC-1. Cancer Res. 2012, 72, 548–559. [Google Scholar] [CrossRef]
- Yue, W.; Fan, P.; Wang, J.; Li, Y.; Santen, R.J. Mechanisms of acquired resistance to endocrine therapy in hormone-dependent breast cancer cells. J. Steroid Biochem. Mol. Biol. 2007, 106, 102–110. [Google Scholar] [CrossRef]
- Zhang, S.; Huang, W.C.; Li, P.; Guo, H.; Poh, S.B.; Brady, S.W.; Xiong, Y.; Tseng, L.M.; Li, S.H.; Ding, Z.; et al. Combating trastuzumab resistance by targeting SRC, a common node downstream of multiple resistance pathways. Nat. Med. 2011, 17, 461–469. [Google Scholar] [CrossRef]
- Rimawi, M.F.; Schiff, R.; Osborne, C.K. Targeting HER2 for the treatment of breast cancer. Annu. Rev. Med. 2015, 66, 111–128. [Google Scholar] [CrossRef]
- Schlam, I.; Swain, S.M. HER2-positive breast cancer and tyrosine kinase inhibitors: The time is now. NPJ Breast Cancer 2021, 7, 56. [Google Scholar] [CrossRef]
- Wynn, C.S.; Tang, S.C. Anti-HER2 therapy in metastatic breast cancer: Many choices and future directions. Cancer Metastasis Rev. 2022, 41, 193–209. [Google Scholar] [CrossRef]
- Nebenfuehr, S.; Kollmann, K.; Sexl, V. The role of CDK6 in cancer. Int. J. Cancer 2020, 147, 2988–2995. [Google Scholar] [CrossRef] [PubMed]
- Goel, S.; Bergholz, J.S.; Zhao, J.J. Targeting CDK4 and CDK6 in cancer. Nat. Rev. Cancer 2022, 22, 356–372. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan, C.C.; Suman, V.J.; Goetz, M.P. The emerging role of CDK4/6i in HER2-positive breast cancer. Ther. Adv. Med. Oncol. 2019, 11, 1758835919887665. [Google Scholar] [CrossRef] [PubMed]
- Koirala, N.; Dey, N.; Aske, J.; De, P. Targeting Cell Cycle Progression in HER2+ Breast Cancer: An Emerging Treatment Opportunity. Int. J. Mol. Sci. 2022, 23, 6547. [Google Scholar] [CrossRef] [PubMed]
- Mughal, M.J.; Bhadresha, K.; Kwok, H.F. CDK inhibitors from past to present: A new wave of cancer therapy. Semin. Cancer Biol. 2023, 88, 106–122. [Google Scholar] [CrossRef]
- Ashai, N.; Swain, S.M. Post-CDK 4/6 Inhibitor Therapy: Current Agents and Novel Targets. Cancers 2023, 15, 1855. [Google Scholar] [CrossRef]
- Zhao, M.; Hanson, K.A.; Zhang, Y.; Zhou, A.; Cha-Silva, A.S. Place in Therapy of Cyclin-Dependent Kinase 4/6 Inhibitors in Breast Cancer: A Targeted Literature Review. Target. Oncol. 2023, 18, 327–358. [Google Scholar] [CrossRef]
- Zhou, F.H.; Downton, T.; Freelander, A.; Hurwitz, J.; Caldon, C.E.; Lim, E. CDK4/6 inhibitor resistance in estrogen receptor positive breast cancer, a 2023 perspective. Front Cell Dev. Biol. 2023, 11, 1148792. [Google Scholar] [CrossRef]
- Stanciu, I.M.; Parosanu, A.I.; Orlov-Slavu, C.; Iaciu, I.C.; Popa, A.M.; Olaru, C.M.; Pirlog, C.F.; Vrabie, R.C.; Nitipir, C. Mechanisms of Resistance to CDK4/6 Inhibitors and Predictive Biomarkers of Response in HR+/HER2-Metastatic Breast Cancer-A Review of the Literature. Diagnostics 2023, 13, 987. [Google Scholar] [CrossRef]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef]
- Debien, V.; De Caluwe, A.; Wang, X.; Piccart-Gebhart, M.; Tuohy, V.K.; Romano, E.; Buisseret, L. Immunotherapy in breast cancer: An overview of current strategies and perspectives. NPJ Breast Cancer 2023, 9, 7. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Aziz, A.K.; Saadeldin, M.K.; D’Amico, P.; Orecchioni, S.; Bertolini, F.; Curigliano, G.; Minucci, S. Preclinical models of breast cancer: Two-way shuttles for immune checkpoint inhibitors from and to patient bedside. Eur. J. Cancer 2019, 122, 22–41. [Google Scholar] [CrossRef]
- Patsoukis, N.; Wang, Q.; Strauss, L.; Boussiotis, V.A. Revisiting the PD-1 pathway. Sci. Adv. 2020, 6, eabd2712. [Google Scholar] [CrossRef]
- Brunet, J.F.; Denizot, F.; Luciani, M.F.; Roux-Dosseto, M.; Suzan, M.; Mattei, M.G.; Golstein, P. A new member of the immunoglobulin superfamily--CTLA-4. Nature 1987, 328, 267–270. [Google Scholar] [CrossRef] [PubMed]
- Leach, D.R.; Krummel, M.F.; Allison, J.P. Enhancement of antitumor immunity by CTLA-4 blockade. Science 1996, 271, 1734–1736. [Google Scholar] [CrossRef] [PubMed]
- Schoffski, P.; Tan, D.S.W.; Martin, M.; Ochoa-de-Olza, M.; Sarantopoulos, J.; Carvajal, R.D.; Kyi, C.; Esaki, T.; Prawira, A.; Akerley, W.; et al. Phase I/II study of the LAG-3 inhibitor ieramilimab (LAG525) +/- anti-PD-1 spartalizumab (PDR001) in patients with advanced malignancies. J. Immunother. Cancer 2022, 10, e003776. [Google Scholar] [CrossRef]
- Curigliano, G.; Gelderblom, H.; Mach, N.; Doi, T.; Tai, D.; Forde, P.M.; Sarantopoulos, J.; Bedard, P.L.; Lin, C.C.; Hodi, F.S.; et al. Phase I/Ib Clinical Trial of Sabatolimab, an Anti-TIM-3 Antibody, Alone and in Combination with Spartalizumab, an Anti-PD-1 Antibody, in Advanced Solid Tumors. Clin. Cancer Res. 2021, 27, 3620–3629. [Google Scholar] [CrossRef]
- Dirix, L.Y.; Takacs, I.; Jerusalem, G.; Nikolinakos, P.; Arkenau, H.T.; Forero-Torres, A.; Boccia, R.; Lippman, M.E.; Somer, R.; Smakal, M.; et al. Avelumab, an anti-PD-L1 antibody, in patients with locally advanced or metastatic breast cancer: A phase 1b JAVELIN Solid Tumor study. Breast Cancer Res. Treat. 2018, 167, 671–686. [Google Scholar] [CrossRef]
- Vicier, C.; Isambert, N.; Cropet, C.; Hamimed, M.; Osanno, L.; Legrand, F.; de La Motte Rouge, T.; Ciccolini, J.; Goncalves, A. MOVIE: A phase I, open-label, multicenter study to evaluate the safety and tolerability of metronomic vinorelbine combined with durvalumab plus tremelimumab in patients with advanced solid tumors. ESMO Open 2022, 7, 100646. [Google Scholar] [CrossRef]
- Fumet, J.D.; Limagne, E.; Thibaudin, M.; Truntzer, C.; Bertaut, A.; Rederstorff, E.; Ghiringhelli, F. Precision medicine phase II study evaluating the efficacy of a double immunotherapy by durvalumab and tremelimumab combined with olaparib in patients with solid cancers and carriers of homologous recombination repair genes mutation in response or stable after olaparib treatment. BMC Cancer 2020, 20, 748. [Google Scholar] [CrossRef]
- Pusztai, L.; Yau, C.; Wolf, D.M.; Han, H.S.; Du, L.; Wallace, A.M.; String-Reasor, E.; Boughey, J.C.; Chien, A.J.; Elias, A.D.; et al. Durvalumab with olaparib and paclitaxel for high-risk HER2-negative stage II/III breast cancer: Results from the adaptively randomized I-SPY2 trial. Cancer Cell 2021, 39, 989–998.e1–e5. [Google Scholar] [CrossRef] [PubMed]
- Domchek, S.M.; Postel-Vinay, S.; Im, S.A.; Park, Y.H.; Delord, J.P.; Italiano, A.; Alexandre, J.; You, B.; Bastian, S.; Krebs, M.G.; et al. Olaparib and durvalumab in patients with germline BRCA-mutated metastatic breast cancer (MEDIOLA): An open-label, multicentre, phase 1/2, basket study. Lancet Oncol. 2020, 21, 1155–1164. [Google Scholar] [CrossRef] [PubMed]
- Quintela-Fandino, M.; Holgado, E.; Manso, L.; Morales, S.; Bermejo, B.; Colomer, R.; Apala, J.V.; Blanco, R.; Munoz, M.; Caleiras, E.; et al. Immuno-priming durvalumab with bevacizumab in HER2-negative advanced breast cancer: A pilot clinical trial. Breast Cancer Res. 2020, 22, 124. [Google Scholar] [CrossRef]
- Lampert, E.J.; Zimmer, A.; Padget, M.; Cimino-Mathews, A.; Nair, J.R.; Liu, Y.; Swisher, E.M.; Hodge, J.W.; Nixon, A.B.; Nichols, E.; et al. Combination of PARP Inhibitor Olaparib, and PD-L1 Inhibitor Durvalumab, in Recurrent Ovarian Cancer: A Proof-of-Concept Phase II Study. Clin. Cancer Res. 2020, 26, 4268–4279. [Google Scholar] [CrossRef] [PubMed]
- Zimmer, A.S.; Nichols, E.; Cimino-Mathews, A.; Peer, C.; Cao, L.; Lee, M.J.; Kohn, E.C.; Annunziata, C.M.; Lipkowitz, S.; Trepel, J.B.; et al. A phase I study of the PD-L1 inhibitor, durvalumab, in combination with a PARP inhibitor, olaparib, and a VEGFR1-3 inhibitor, cediranib, in recurrent women’s cancers with biomarker analyses. J. Immunother. Cancer 2019, 7, 197. [Google Scholar] [CrossRef] [PubMed]
- Chia, S.; Bedard, P.L.; Hilton, J.; Amir, E.; Gelmon, K.; Goodwin, R.; Villa, D.; Cabanero, M.; Tu, D.; Tsao, M.; et al. A Phase Ib Trial of Durvalumab in Combination with Trastuzumab in HER2-Positive Metastatic Breast Cancer (CCTG IND.229). Oncologist 2019, 24, 1439–1445. [Google Scholar] [CrossRef]
- Hong, D.; Rasco, D.; Veeder, M.; Luke, J.J.; Chandler, J.; Balmanoukian, A.; George, T.J.; Munster, P.; Berlin, J.D.; Gutierrez, M.; et al. A Phase 1b/2 Study of the Bruton Tyrosine Kinase Inhibitor Ibrutinib and the PD-L1 Inhibitor Durvalumab in Patients with Pretreated Solid Tumors. Oncology 2019, 97, 102–111. [Google Scholar] [CrossRef]
- Liu, J.; Liu, Q.; Li, Y.; Li, Q.; Su, F.; Yao, H.; Su, S.; Wang, Q.; Jin, L.; Wang, Y.; et al. Efficacy and safety of camrelizumab combined with apatinib in advanced triple-negative breast cancer: An open-label phase II trial. J. Immunother. Cancer 2020, 8, e000696. [Google Scholar] [CrossRef]
- Liu, J.; Wang, Y.; Tian, Z.; Lin, Y.; Li, H.; Zhu, Z.; Liu, Q.; Su, S.; Zeng, Y.; Jia, W.; et al. Multicenter phase II trial of Camrelizumab combined with Apatinib and Eribulin in heavily pretreated patients with advanced triple-negative breast cancer. Nat. Commun. 2022, 13, 3011. [Google Scholar] [CrossRef]
- Zhang, Q.; Shao, B.; Tong, Z.; Ouyang, Q.; Wang, Y.; Xu, G.; Li, S.; Li, H. A phase Ib study of camrelizumab in combination with apatinib and fuzuloparib in patients with recurrent or metastatic triple-negative breast cancer. BMC Med. 2022, 20, 321. [Google Scholar] [CrossRef]
- Wu, S.Y.; Xu, Y.; Chen, L.; Fan, L.; Ma, X.Y.; Zhao, S.; Song, X.Q.; Hu, X.; Yang, W.T.; Chai, W.J.; et al. Combined angiogenesis and PD-1 inhibition for immunomodulatory TNBC: Concept exploration and biomarker analysis in the FUTURE-C-Plus trial. Mol. Cancer 2022, 21, 84. [Google Scholar] [CrossRef] [PubMed]
- Adams, S.; Othus, M.; Patel, S.P.; Miller, K.D.; Chugh, R.; Schuetze, S.M.; Chamberlin, M.D.; Haley, B.J.; Storniolo, A.M.V.; Reddy, M.P.; et al. A Multicenter Phase II Trial of Ipilimumab and Nivolumab in Unresectable or Metastatic Metaplastic Breast Cancer: Cohort 36 of Dual Anti-CTLA-4 and Anti-PD-1 Blockade in Rare Tumors (DART, SWOG S1609). Clin. Cancer Res. 2022, 28, 271–278. [Google Scholar] [CrossRef]
- Wagner, M.J.; Othus, M.; Patel, S.P.; Ryan, C.; Sangal, A.; Powers, B.; Budd, G.T.; Victor, A.I.; Hsueh, C.T.; Chugh, R.; et al. Multicenter phase II trial (SWOG S1609, cohort 51) of ipilimumab and nivolumab in metastatic or unresectable angiosarcoma: A substudy of dual anti-CTLA-4 and anti-PD-1 blockade in rare tumors (DART). J. Immunother. Cancer 2021, 9, e002990. [Google Scholar] [CrossRef] [PubMed]
- Kyte, J.A.; Andresen, N.K.; Russnes, H.G.; Fretland, S.O.; Falk, R.S.; Lingjaerde, O.C.; Naume, B. ICON: A randomized phase IIb study evaluating immunogenic chemotherapy combined with ipilimumab and nivolumab in patients with metastatic hormone receptor positive breast cancer. J. Transl. Med. 2020, 18, 269. [Google Scholar] [CrossRef] [PubMed]
- Ozaki, Y.; Tsurutani, J.; Mukohara, T.; Iwasa, T.; Takahashi, M.; Tanabe, Y.; Kawabata, H.; Masuda, N.; Futamura, M.; Minami, H.; et al. Safety and efficacy of nivolumab plus bevacizumab, paclitaxel for HER2-negative metastatic breast cancer: Primary results and biomarker data from a phase 2 trial (WJOG9917B). Eur. J. Cancer 2022, 171, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Yordanova, M.; Hubert, A.; Hassan, S. Expanding the Use of PARP Inhibitors as Monotherapy and in Combination in Triple-Negative Breast Cancer. Pharmaceuticals 2021, 14, 1270. [Google Scholar] [CrossRef]
- Roy, R.; Chun, J.; Powell, S.N. BRCA1 and BRCA2: Different roles in a common pathway of genome protection. Nat. Rev. Cancer 2011, 12, 68–78. [Google Scholar] [CrossRef]
- Morales, J.; Li, L.; Fattah, F.J.; Dong, Y.; Bey, E.A.; Patel, M.; Gao, J.; Boothman, D.A. Review of poly (ADP-ribose) polymerase (PARP) mechanisms of action and rationale for targeting in cancer and other diseases. Crit. Rev. Eukaryot. Gene Expr. 2014, 24, 15–28. [Google Scholar] [CrossRef]
- Layman, R.M.; Arun, B. PARP Inhibitors in Triple-Negative Breast Cancer Including Those With BRCA Mutations. Cancer J. 2021, 27, 67–75. [Google Scholar] [CrossRef]
- Gupta, T.; Vinayak, S.; Telli, M. Emerging strategies: PARP inhibitors in combination with immune checkpoint blockade in BRCA1 and BRCA2 mutation-associated and triple-negative breast cancer. Breast Cancer Res. Treat. 2023, 197, 51–56. [Google Scholar] [CrossRef]
- Barchiesi, G.; Roberto, M.; Verrico, M.; Vici, P.; Tomao, S.; Tomao, F. Emerging Role of PARP Inhibitors in Metastatic Triple Negative Breast Cancer. Current Scenario and Future Perspectives. Front. Oncol. 2021, 11, 769280. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.C.; Balmana, J.; Poncet, C.; Goulioti, T.; Tryfonidis, K.; Honkoop, A.H.; Zoppoli, G.; Razis, E.; Johannsson, O.T.; Colleoni, M.; et al. Niraparib for Advanced Breast Cancer with Germline BRCA1 and BRCA2 Mutations: The EORTC 1307-BCG/BIG5-13/TESARO PR-30-50-10-C BRAVO Study. Clin. Cancer Res. 2021, 27, 5482–5491. [Google Scholar] [CrossRef] [PubMed]
- O’Shaughnessy, J.; Osborne, C.; Pippen, J.E.; Yoffe, M.; Patt, D.; Rocha, C.; Koo, I.C.; Sherman, B.M.; Bradley, C. Iniparib plus chemotherapy in metastatic triple-negative breast cancer. N. Engl. J. Med. 2011, 364, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Manzo, J.; Puhalla, S.; Pahuja, S.; Ding, F.; Lin, Y.; Appleman, L.; Tawbi, H.; Stoller, R.; Lee, J.J.; Diergaarde, B.; et al. A phase 1 and pharmacodynamic study of chronically-dosed, single-agent veliparib (ABT-888) in patients with BRCA1- or BRCA2-mutated cancer or platinum-refractory ovarian or triple-negative breast cancer. Cancer Chemother. Pharmacol. 2022, 89, 721–735. [Google Scholar] [CrossRef] [PubMed]
- Ellis, H.; Ma, C.X. PI3K Inhibitors in Breast Cancer Therapy. Curr. Oncol. Rep. 2019, 21, 110. [Google Scholar] [CrossRef]
- Hoxhaj, G.; Manning, B.D. The PI3K-AKT network at the interface of oncogenic signalling and cancer metabolism. Nat. Rev. Cancer 2020, 20, 74–88. [Google Scholar] [CrossRef]
- Chai, C.; Wu, H.H.; Abuetabh, Y.; Sergi, C.; Leng, R. Regulation of the tumor suppressor PTEN in triple-negative breast cancer. Cancer Lett. 2022, 527, 41–48. [Google Scholar] [CrossRef]
- Sarbassov, D.D.; Guertin, D.A.; Ali, S.M.; Sabatini, D.M. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 2005, 307, 1098–1101. [Google Scholar] [CrossRef]
- Yuan, Y.; Long, H.; Zhou, Z.; Fu, Y.; Jiang, B. PI3K-AKT-Targeting Breast Cancer Treatments: Natural Products and Synthetic Compounds. Biomolecules 2023, 13, 93. [Google Scholar] [CrossRef]
- Dienstmann, R.; Rodon, J.; Serra, V.; Tabernero, J. Picking the point of inhibition: A comparative review of PI3K/AKT/mTOR pathway inhibitors. Mol. Cancer Ther. 2014, 13, 1021–1031. [Google Scholar] [CrossRef]
- Carvalho, S.; Schmitt, F. Potential role of PI3K inhibitors in the treatment of breast cancer. Future Oncol. 2010, 6, 1251–1263. [Google Scholar] [CrossRef] [PubMed]
- Presti, D.; Quaquarini, E. The PI3K/AKT/mTOR and CDK4/6 Pathways in Endocrine Resistant HR+/HER2- Metastatic Breast Cancer: Biological Mechanisms and New Treatments. Cancers 2019, 11, 1242. [Google Scholar] [CrossRef]
- Pavitra, E.; Kancharla, J.; Gupta, V.K.; Prasad, K.; Sung, J.Y.; Kim, J.; Tej, M.B.; Choi, R.; Lee, J.H.; Han, Y.K.; et al. The role of NF-kappaB in breast cancer initiation, growth, metastasis, and resistance to chemotherapy. Biomed. Pharmacother. 2023, 163, 114822. [Google Scholar] [CrossRef]
- Khongthong, P.; Roseweir, A.K.; Edwards, J. The NF-KB pathway and endocrine therapy resistance in breast cancer. Endocr. Relat. Cancer 2019, 26, R369–R380. [Google Scholar] [CrossRef]
- Eitsuka, T.; Tatewaki, N.; Nishida, H.; Nakagawa, K.; Miyazawa, T. Synergistic Anticancer Effect of Tocotrienol Combined with Chemotherapeutic Agents or Dietary Components: A Review. Int. J. Mol. Sci. 2016, 17, 1605. [Google Scholar] [CrossRef]
- Rascio, F.; Spadaccino, F.; Rocchetti, M.T.; Castellano, G.; Stallone, G.; Netti, G.S.; Ranieri, E. The Pathogenic Role of PI3K/AKT Pathway in Cancer Onset and Drug Resistance: An Updated Review. Cancers 2021, 13, 3949. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Chen, Y.; Liu, G.; Li, C.; Song, Y.; Cao, Z.; Li, W.; Hu, J.; Lu, C.; Liu, Y. PI3K/AKT pathway as a key link modulates the multidrug resistance of cancers. Cell Death Dis. 2020, 11, 797. [Google Scholar] [CrossRef] [PubMed]
- Massihnia, D.; Galvano, A.; Fanale, D.; Perez, A.; Castiglia, M.; Incorvaia, L.; Listi, A.; Rizzo, S.; Cicero, G.; Bazan, V.; et al. Triple negative breast cancer: Shedding light onto the role of pi3k/akt/mtor pathway. Oncotarget 2016, 7, 60712–60722. [Google Scholar] [CrossRef]
- Costa, R.L.B.; Han, H.S.; Gradishar, W.J. Targeting the PI3K/AKT/mTOR pathway in triple-negative breast cancer: A review. Breast Cancer Res. Treat. 2018, 169, 397–406. [Google Scholar] [CrossRef]
- Liu, H.; Murphy, C.J.; Karreth, F.A.; Emdal, K.B.; White, F.M.; Elemento, O.; Toker, A.; Wulf, G.M.; Cantley, L.C. Identifying and Targeting Sporadic Oncogenic Genetic Aberrations in Mouse Models of Triple-Negative Breast Cancer. Cancer Discov. 2018, 8, 354–369. [Google Scholar] [CrossRef]
- Pascual, J.; Turner, N.C. Targeting the PI3-kinase pathway in triple-negative breast cancer. Ann. Oncol. 2019, 30, 1051–1060. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.A.; Jain, V.K.; Rizwanullah, M.; Ahmad, J.; Jain, K. PI3K/AKT/mTOR pathway inhibitors in triple-negative breast cancer: A review on drug discovery and future challenges. Drug Discov. Today 2019, 24, 2181–2191. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.J.; Tan, T.J.Y.; Dent, R.A. Novel therapeutic avenues in triple-negative breast cancer: PI3K/AKT inhibition, androgen receptor blockade, and beyond. Ther. Adv. Med. Oncol. 2019, 11, 1758835919880429. [Google Scholar] [CrossRef] [PubMed]
- Pinilla, K.; Drewett, L.M.; Lucey, R.; Abraham, J.E. Precision Breast Cancer Medicine: Early Stage Triple Negative Breast Cancer-A Review of Molecular Characterisation, Therapeutic Targets and Future Trends. Front. Oncol. 2022, 12, 866889. [Google Scholar] [CrossRef]
- Tolcher, A.W.; Kurzrock, R.; Valero, V.; Gonzalez, R.; Heist, R.S.; Tan, A.R.; Means-Powell, J.; Werner, T.L.; Becerra, C.; Wang, C.; et al. Phase I dose-escalation trial of the oral AKT inhibitor uprosertib in combination with the oral MEK1/MEK2 inhibitor trametinib in patients with solid tumors. Cancer Chemother. Pharmacol. 2020, 85, 673–683. [Google Scholar] [CrossRef]
- Yang, K.; Tang, X.J.; Xu, F.F.; Liu, J.H.; Tan, Y.Q.; Gao, L.; Sun, Q.; Ding, X.; Liu, B.H.; Chen, Q.X. PI3K/mTORC1/2 inhibitor PQR309 inhibits proliferation and induces apoptosis in human glioblastoma cells. Oncol. Rep. 2020, 43, 773–782. [Google Scholar] [CrossRef]
- Mensah, F.A.; Blaize, J.P.; Bryan, L.J. Spotlight on copanlisib and its potential in the treatment of relapsed/refractory follicular lymphoma: Evidence to date. Onco Targets Ther. 2018, 11, 4817–4827. [Google Scholar] [CrossRef]
- Luo, J.; Zou, H.; Guo, Y.; Tong, T.; Ye, L.; Zhu, C.; Deng, L.; Wang, B.; Pan, Y.; Li, P. SRC kinase-mediated signaling pathways and targeted therapies in breast cancer. Breast Cancer Res. 2022, 24, 99. [Google Scholar] [CrossRef]
- Ishizawar, R.; Parsons, S.J. c-Src and cooperating partners in human cancer. Cancer Cell 2004, 6, 209–214. [Google Scholar] [CrossRef]
- Elshazly, A.M.; Gewirtz, D.A. An overview of resistance to Human epidermal growth factor receptor 2 (Her2) targeted therapies in breast cancer. Cancer Drug Resist. 2022, 5, 472–486. [Google Scholar] [CrossRef]
- Martin-Perez, J.; Garcia-Martinez, J.M.; Sanchez-Bailon, M.P.; Mayoral-Varo, V.; Calcabrini, A. Role of SRC family kinases in prolactin signaling. Adv. Exp. Med. Biol. 2015, 846, 163–188. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R., Jr. Src protein-tyrosine kinase structure, mechanism, and small molecule inhibitors. Pharmacol. Res. 2015, 94, 9–25. [Google Scholar] [CrossRef] [PubMed]
- Tatton, L.; Morley, G.M.; Chopra, R.; Khwaja, A. The Src-selective kinase inhibitor PP1 also inhibits Kit and Bcr-Abl tyrosine kinases. J. Biol. Chem. 2003, 278, 4847–4853. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Park, S.; Ryu, J.S.; Kang, J.; Kim, I.; Son, S.; Lee, B.S.; Kim, C.H.; Kim, Y.S. c-Src inhibitor PP2 inhibits head and neck cancer progression through regulation of the epithelial-mesenchymal transition. Exp. Biol. Med. (Maywood) 2022, 248, 492–500. [Google Scholar] [CrossRef]
- Bielefeld, E.C.; Wantuck, R.; Henderson, D. Postexposure treatment with a Src-PTK inhibitor in combination with N-l-acetyl cysteine to reduce noise-induced hearing loss. Noise Health 2011, 13, 292–298. [Google Scholar] [CrossRef]
- Gurbani, D.; Du, G.; Henning, N.J.; Rao, S.; Bera, A.K.; Zhang, T.; Gray, N.S.; Westover, K.D. Structure and Characterization of a Covalent Inhibitor of Src Kinase. Front. Mol. Biosci. 2020, 7, 81. [Google Scholar] [CrossRef]
- Lopez-Contreras, L.; Hernandez-Ramirez, V.I.; Herrera-Martinez, M.; Montano, S.; Constantino-Jonapa, L.A.; Chavez-Munguia, B.; Talamas-Rohana, P. Structural and functional characterization of the divergent Entamoeba Src using Src inhibitor-1. Parasit. Vectors 2017, 10, 500. [Google Scholar] [CrossRef]
- Pusztai, L.; Moulder, S.; Altan, M.; Kwiatkowski, D.; Valero, V.; Ueno, N.T.; Esteva, F.J.; Avritscher, R.; Qi, Y.; Strauss, L.; et al. Gene signature-guided dasatinib therapy in metastatic breast cancer. Clin. Cancer Res. 2014, 20, 5265–5271. [Google Scholar] [CrossRef]
- Rocca, A.; Braga, L.; Volpe, M.C.; Maiocchi, S.; Generali, D. The Predictive and Prognostic Role of RAS-RAF-MEK-ERK Pathway Alterations in Breast Cancer: Revision of the Literature and Comparison with the Analysis of Cancer Genomic Datasets. Cancers 2022, 14, 5306. [Google Scholar] [CrossRef]
- Ali, E.S.; Akter, S.; Ramproshad, S.; Mondal, B.; Riaz, T.A.; Islam, M.T.; Khan, I.N.; Docea, A.O.; Calina, D.; Sharifi-Rad, J.; et al. Targeting Ras-ERK cascade by bioactive natural products for potential treatment of cancer: An updated overview. Cancer Cell Int. 2022, 22, 246. [Google Scholar] [CrossRef]
- Song, Y.; Bi, Z.; Liu, Y.; Qin, F.; Wei, Y.; Wei, X. Targeting RAS-RAF-MEK-ERK signaling pathway in human cancer: Current status in clinical trials. Genes. Dis. 2023, 10, 76–88. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Wang, C.; Huang, M.; Tao, Z.; Yan, W.; Du, Y. Naturally Occurring Sesquiterpene Lactone-Santonin, Exerts Anticancer Effects in Multi-Drug Resistant Breast Cancer Cells by Inducing Mitochondrial Mediated Apoptosis, Caspase Activation, Cell Cycle Arrest, and by Targeting Ras/Raf/MEK/ERK Signaling Pathway. Med. Sci. Monit. 2019, 25, 3676–3682. [Google Scholar] [CrossRef] [PubMed]
- Kurzrock, R.; Bowles, D.W.; Kang, H.; Meric-Bernstam, F.; Hainsworth, J.; Spigel, D.R.; Bose, R.; Burris, H.; Sweeney, C.J.; Beattie, M.S.; et al. Targeted therapy for advanced salivary gland carcinoma based on molecular profiling: Results from MyPathway, a phase IIa multiple basket study. Ann. Oncol. 2020, 31, 412–421. [Google Scholar] [CrossRef] [PubMed]
- Ferrario, C.; Strepponi, I.; Esfahani, K.; Charamis, H.; Langleben, A.; Scarpi, E.; Nanni, O.; Miller, W.H., Jr.; Panasci, L.C. Phase I/II Trial of Sorafenib in Combination with Vinorelbine as First-Line Chemotherapy for Metastatic Breast Cancer. PLoS ONE 2016, 11, e0167906. [Google Scholar] [CrossRef]
- Decker, T.; Overkamp, F.; Rosel, S.; Nusch, A.; Gohler, T.; Indorf, M.; Sahlmann, J.; Trarbach, T. A randomized phase II study of paclitaxel alone versus paclitaxel plus sorafenib in second- and third-line treatment of patients with HER2-negative metastatic breast cancer (PASO). BMC Cancer 2017, 17, 499. [Google Scholar] [CrossRef]
- Mavratzas, A.; Baek, S.; Gerber, B.; Schmidt, M.; Moebus, V.; Foerster, F.; Grischke, E.M.; Fasching, P.; Strumberg, D.; Solomayer, E.; et al. Sorafenib in combination with docetaxel as first-line therapy for HER2-negative metastatic breast cancer: Final results of the randomized, double-blind, placebo-controlled phase II MADONNA study. Breast 2019, 45, 22–28. [Google Scholar] [CrossRef]
- Baselga, J.; Zamagni, C.; Gomez, P.; Bermejo, B.; Nagai, S.E.; Melichar, B.; Chan, A.; Mangel, L.; Bergh, J.; Costa, F.; et al. RESILIENCE: Phase III Randomized, Double-Blind Trial Comparing Sorafenib With Capecitabine Versus Placebo With Capecitabine in Locally Advanced or Metastatic HER2-Negative Breast Cancer. Clin. Breast Cancer 2017, 17, 585–594 e584. [Google Scholar] [CrossRef]
- Darvishi, B.; Farahmand, L.; Eslami, S.Z.; Majidzadeh, A.K. NF-kappaB as the main node of resistance to receptor tyrosine kinase inhibitors in triple-negative breast cancer. Tumour Biol. 2017, 39, 585–594.e4. [Google Scholar] [CrossRef]
- Infante, J.R.; Papadopoulos, K.P.; Bendell, J.C.; Patnaik, A.; Burris, H.A., 3rd; Rasco, D.; Jones, S.F.; Smith, L.; Cox, D.S.; Durante, M.; et al. A phase 1b study of trametinib, an oral Mitogen-activated protein kinase kinase (MEK) inhibitor, in combination with gemcitabine in advanced solid tumours. Eur. J. Cancer 2013, 49, 2077–2085. [Google Scholar] [CrossRef]
- Johnson, D.B.; Zhao, F.; Noel, M.; Riely, G.J.; Mitchell, E.P.; Wright, J.J.; Chen, H.X.; Gray, R.J.; Li, S.; McShane, L.M.; et al. Trametinib Activity in Patients with Solid Tumors and Lymphomas Harboring BRAF Non-V600 Mutations or Fusions: Results from NCI-MATCH (EAY131). Clin. Cancer Res. 2020, 26, 1812–1819. [Google Scholar] [CrossRef]
- Zaman, K.; Winterhalder, R.; Mamot, C.; Hasler-Strub, U.; Rochlitz, C.; Mueller, A.; Berset, C.; Wiliders, H.; Perey, L.; Rudolf, C.B.; et al. Fulvestrant with or without selumetinib, a MEK 1/2 inhibitor, in breast cancer progressing after aromatase inhibitor therapy: A multicentre randomised placebo-controlled double-blind phase II trial, SAKK 21/08. Eur. J. Cancer 2015, 51, 1212–1220. [Google Scholar] [CrossRef] [PubMed]
- Brufsky, A.; Kim, S.B.; Zvirbule, Z.; Eniu, A.; Mebis, J.; Sohn, J.H.; Wongchenko, M.; Chohan, S.; Amin, R.; Yan, Y.; et al. A phase II randomized trial of cobimetinib plus chemotherapy, with or without atezolizumab, as first-line treatment for patients with locally advanced or metastatic triple-negative breast cancer (COLET): Primary analysis. Ann. Oncol. 2021, 32, 652–660. [Google Scholar] [CrossRef] [PubMed]
- Kearney, M.; Franks, L.; Lee, S.; Tiersten, A.; Makower, D.F.; Cigler, T.; Mundi, P.; Chi, D.C.; Goel, A.; Klein, P.; et al. Phase I/II trial of ruxolitinib in combination with trastuzumab in metastatic HER2 positive breast cancer. Breast Cancer Res. Treat. 2021, 189, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Lynce, F.; Williams, J.T.; Regan, M.M.; Bunnell, C.A.; Freedman, R.A.; Tolaney, S.M.; Chen, W.Y.; Mayer, E.L.; Partridge, A.H.; Winer, E.P.; et al. Phase I study of JAK1/2 inhibitor ruxolitinib with weekly paclitaxel for the treatment of HER2-negative metastatic breast cancer. Cancer Chemother. Pharmacol. 2021, 87, 673–679. [Google Scholar] [CrossRef] [PubMed]
- Stover, D.G.; Gil Del Alcazar, C.R.; Brock, J.; Guo, H.; Overmoyer, B.; Balko, J.; Xu, Q.; Bardia, A.; Tolaney, S.M.; Gelman, R.; et al. Phase II study of ruxolitinib, a selective JAK1/2 inhibitor, in patients with metastatic triple-negative breast cancer. NPJ Breast Cancer 2018, 4, 10. [Google Scholar] [CrossRef]
- Tyagi, S.; Gupta, P.; Saini, A.S.; Kaushal, C.; Sharma, S. The peroxisome proliferator-activated receptor: A family of nuclear receptors role in various diseases. J. Adv. Pharm. Technol. Res. 2011, 2, 236–240. [Google Scholar] [CrossRef]
- Qian, Z.; Chen, L.; Liu, J.; Jiang, Y.; Zhang, Y. The emerging role of PPAR-alpha in breast cancer. Biomed. Pharmacother. 2023, 161, 114420. [Google Scholar] [CrossRef]
- Park, P.W. Isolation and functional analysis of syndecans. Methods Cell Biol. 2018, 143, 317–333. [Google Scholar] [CrossRef]
- Tkachenko, E.; Rhodes, J.M.; Simons, M. Syndecans: New kids on the signaling block. Circ. Res. 2005, 96, 488–500. [Google Scholar] [CrossRef]
- Motta, J.M.; Hassan, H.; Ibrahim, S.A. Revisiting the Syndecans: Master Signaling Regulators with Prognostic and Targetable Therapeutic Values in Breast Carcinoma. Cancers 2023, 15, 1794. [Google Scholar] [CrossRef]
- Gomes, A.M.; Stelling, M.P.; Pavao, M.S. Heparan sulfate and heparanase as modulators of breast cancer progression. Biomed. Res. Int. 2013, 2013, 852093. [Google Scholar] [CrossRef] [PubMed]
- Hammond, E.; Brandt, R.; Dredge, K. PG545, a heparan sulfate mimetic, reduces heparanase expression in vivo, blocks spontaneous metastases and enhances overall survival in the 4T1 breast carcinoma model. PLoS ONE 2012, 7, e52175. [Google Scholar] [CrossRef] [PubMed]
- Miao, H.Q.; Elkin, M.; Aingorn, E.; Ishai-Michaeli, R.; Stein, C.A.; Vlodavsky, I. Inhibition of heparanase activity and tumor metastasis by laminarin sulfate and synthetic phosphorothioate oligodeoxynucleotides. Int. J. Cancer 1999, 83, 424–431. [Google Scholar] [CrossRef]
- Schonfeld, K.; Herbener, P.; Zuber, C.; Hader, T.; Bernoster, K.; Uherek, C.; Schuttrumpf, J. Activity of Indatuximab Ravtansine against Triple-Negative Breast Cancer in Preclinical Tumor Models. Pharm. Res. 2018, 35, 118. [Google Scholar] [CrossRef]
- Zhao, W.; Yang, H.; Chai, J.; Xing, L. RUNX2 as a promising therapeutic target for malignant tumors. Cancer Manag. Res. 2021, 13, 2539–2548. [Google Scholar] [CrossRef]
- Lin, T.C. RUNX2 and Cancer. Int. J. Mol. Sci. 2023, 24, 7001. [Google Scholar] [CrossRef]
- Vishal, M.; Swetha, R.; Thejaswini, G.; Arumugam, B.; Selvamurugan, N. Role of Runx2 in breast cancer-mediated bone metastasis. Int. J. Biol. Macromol. 2017, 99, 608–614. [Google Scholar] [CrossRef]
- Vansteenkiste, J.; Van Cutsem, E.; Dumez, H.; Chen, C.; Ricker, J.L.; Randolph, S.S.; Schoffski, P. Early phase II trial of oral vorinostat in relapsed or refractory breast, colorectal, or non-small cell lung cancer. Invest. New Drugs 2008, 26, 483–488. [Google Scholar] [CrossRef]
- Luu, T.H.; Morgan, R.J.; Leong, L.; Lim, D.; McNamara, M.; Portnow, J.; Frankel, P.; Smith, D.D.; Doroshow, J.H.; Wong, C.; et al. A phase II trial of vorinostat (suberoylanilide hydroxamic acid) in metastatic breast cancer: A California Cancer Consortium study. Clin. Cancer Res. 2008, 14, 7138–7142. [Google Scholar] [CrossRef]
- Munster, P.N.; Thurn, K.T.; Thomas, S.; Raha, P.; Lacevic, M.; Miller, A.; Melisko, M.; Ismail-Khan, R.; Rugo, H.; Moasser, M.; et al. A phase II study of the histone deacetylase inhibitor vorinostat combined with tamoxifen for the treatment of patients with hormone therapy-resistant breast cancer. Br. J. Cancer 2011, 104, 1828–1835. [Google Scholar] [CrossRef]
- Luu, T.; Kim, K.P.; Blanchard, S.; Anyang, B.; Hurria, A.; Yang, L.; Beumer, J.H.; Somlo, G.; Yen, Y. Phase IB trial of ixabepilone and vorinostat in metastatic breast cancer. Breast Cancer Res. Treat. 2018, 167, 469–478. [Google Scholar] [CrossRef] [PubMed]
- Peterson, L.M.; Kurland, B.F.; Yan, F.; Jiresova, A.N.; Gadi, V.K.; Specht, J.M.; Gralow, J.R.; Schubert, E.K.; Link, J.M.; Krohn, K.A.; et al. (18)F-Fluoroestradiol PET Imaging in a Phase II Trial of Vorinostat to Restore Endocrine Sensitivity in ER+/HER2- Metastatic Breast Cancer. J. Nucl. Med. 2021, 62, 184–190. [Google Scholar] [CrossRef]
- Yardley, D.A.; Ismail-Khan, R.R.; Melichar, B.; Lichinitser, M.; Munster, P.N.; Klein, P.M.; Cruickshank, S.; Miller, K.D.; Lee, M.J.; Trepel, J.B. Randomized phase II, double-blind, placebo-controlled study of exemestane with or without entinostat in postmenopausal women with locally recurrent or metastatic estrogen receptor-positive breast cancer progressing on treatment with a nonsteroidal aromatase inhibitor. J. Clin. Oncol. 2013, 31, 2128–2135. [Google Scholar] [CrossRef] [PubMed]
- Connolly, R.M.; Zhao, F.; Miller, K.D.; Lee, M.J.; Piekarz, R.L.; Smith, K.L.; Brown-Glaberman, U.A.; Winn, J.S.; Faller, B.A.; Onitilo, A.A.; et al. E2112: Randomized Phase III Trial of Endocrine Therapy Plus Entinostat or Placebo in Hormone Receptor-Positive Advanced Breast Cancer. A Trial of the ECOG-ACRIN Cancer Research Group. J. Clin. Oncol. 2021, 39, 3171–3181. [Google Scholar] [CrossRef]
- Xu, B.; Zhang, Q.; Hu, X.; Li, Q.; Sun, T.; Li, W.; Ouyang, Q.; Wang, J.; Tong, Z.; Yan, M.; et al. Entinostat, a class I selective histone deacetylase inhibitor, plus exemestane for Chinese patients with hormone receptor-positive advanced breast cancer: A multicenter, randomized, double-blind, placebo-controlled, phase 3 trial. Acta Pharm. Sin. B 2023, 13, 2250–2258. [Google Scholar] [CrossRef] [PubMed]
- Connolly, R.M.; Li, H.; Jankowitz, R.C.; Zhang, Z.; Rudek, M.A.; Jeter, S.C.; Slater, S.A.; Powers, P.; Wolff, A.C.; Fetting, J.H.; et al. Combination Epigenetic Therapy in Advanced Breast Cancer with 5-Azacitidine and Entinostat: A Phase II National Cancer Institute/Stand Up to Cancer Study. Clin. Cancer Res. 2017, 23, 2691–2701. [Google Scholar] [CrossRef] [PubMed]
- Tan, W.W.; Allred, J.B.; Moreno-Aspitia, A.; Northfelt, D.W.; Ingle, J.N.; Goetz, M.P.; Perez, E.A. Phase I Study of Panobinostat (LBH589) and Letrozole in Postmenopausal Metastatic Breast Cancer Patients. Clin. Breast Cancer 2016, 16, 82–86. [Google Scholar] [CrossRef]
- Marinho, A.; Nunes, C.; Reis, S. Hyaluronic Acid: A Key Ingredient in the Therapy of Inflammation. Biomolecules 2021, 11, 1518. [Google Scholar] [CrossRef]
- Markowska, A.; Antoszczak, M.; Markowska, J.; Huczynski, A. Role of Hyaluronic Acid in Selected Malignant Neoplasms in Women. Biomedicines 2023, 11, 304. [Google Scholar] [CrossRef]
- Wu, W.; Chen, L.; Wang, Y.; Jin, J.; Xie, X.; Zhang, J. Hyaluronic acid predicts poor prognosis in breast cancer patients: A protocol for systematic review and meta analysis. Medicine (Baltimore) 2020, 99, e20438. [Google Scholar] [CrossRef]
- Sacks, J.D.; Barbolina, M.V. Expression and Function of CD44 in Epithelial Ovarian Carcinoma. Biomolecules 2015, 5, 3051–3066. [Google Scholar] [CrossRef] [PubMed]
- Orian-Rousseau, V.; Sleeman, J. CD44 is a multidomain signaling platform that integrates extracellular matrix cues with growth factor and cytokine signals. Adv. Cancer Res. 2014, 123, 231–254. [Google Scholar] [CrossRef]
- Della Sala, F.; Fabozzi, A.; di Gennaro, M.; Nuzzo, S.; Makvandi, P.; Solimando, N.; Pagliuca, M.; Borzacchiello, A. Advances in Hyaluronic-Acid-Based (Nano)Devices for Cancer Therapy. Macromol. Biosci. 2022, 22, e2100304. [Google Scholar] [CrossRef] [PubMed]
- Rupp, U.; Schoendorf-Holland, E.; Eichbaum, M.; Schuetz, F.; Lauschner, I.; Schmidt, P.; Staab, A.; Hanft, G.; Huober, J.; Sinn, H.P.; et al. Safety and pharmacokinetics of bivatuzumab mertansine in patients with CD44v6-positive metastatic breast cancer: Final results of a phase I study. Anticancer. Drugs 2007, 18, 477–485. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Richardson, D.R. The role of the NDRG1 in the pathogenesis and treatment of breast cancer. Biochim. Biophys. Acta Rev. Cancer 2023, 1878, 188871. [Google Scholar] [CrossRef]
- Villodre, E.S.; Hu, X.; Eckhardt, B.L.; Larson, R.; Huo, L.; Yoon, E.C.; Gong, Y.; Song, J.; Liu, S.; Ueno, N.T.; et al. NDRG1 in Aggressive Breast Cancer Progression and Brain Metastasis. J. Natl. Cancer Inst. 2022, 114, 579–591. [Google Scholar] [CrossRef]
- Feins, S.; Kong, W.; Williams, E.F.; Milone, M.C.; Fraietta, J.A. An introduction to chimeric antigen receptor (CAR) T-cell immunotherapy for human cancer. Am. J. Hematol. 2019, 94, S3–S9. [Google Scholar] [CrossRef]
- Alnefaie, A.; Albogami, S.; Asiri, Y.; Ahmad, T.; Alotaibi, S.S.; Al-Sanea, M.M.; Althobaiti, H. Chimeric Antigen Receptor T-Cells: An Overview of Concepts, Applications, Limitations, and Proposed Solutions. Front. Bioeng. Biotechnol. 2022, 10, 797440. [Google Scholar] [CrossRef]
- Nasiri, F.; Kazemi, M.; Mirarefin, S.M.J.; Mahboubi Kancha, M.; Ahmadi Najafabadi, M.; Salem, F.; Dashti Shokoohi, S.; Evazi Bakhshi, S.; Safarzadeh Kozani, P.; Safarzadeh Kozani, P. CAR-T cell therapy in triple-negative breast cancer: Hunting the invisible devil. Front. Immunol. 2022, 13, 1018786. [Google Scholar] [CrossRef]
- Yang, Y.H.; Liu, J.W.; Lu, C.; Wei, J.F. CAR-T Cell Therapy for Breast Cancer: From Basic Research to Clinical Application. Int. J. Biol. Sci. 2022, 18, 2609–2626. [Google Scholar] [CrossRef]
- Corti, C.; Venetis, K.; Sajjadi, E.; Zattoni, L.; Curigliano, G.; Fusco, N. CAR-T cell therapy for triple-negative breast cancer and other solid tumors: Preclinical and clinical progress. Expert. Opin. Investig. Drugs 2022, 31, 593–605. [Google Scholar] [CrossRef] [PubMed]
- Dees, S.; Ganesan, R.; Singh, S.; Grewal, I.S. Emerging CAR-T Cell Therapy for the Treatment of Triple-Negative Breast Cancer. Mol. Cancer Ther. 2020, 19, 2409–2421. [Google Scholar] [CrossRef]
- Bozorgi, A.; Bozorgi, M.; Khazaei, M. Immunotherapy and immunoengineering for breast cancer; a comprehensive insight into CAR-T cell therapy advancements, challenges and prospects. Cell Oncol. (Dordr) 2022, 45, 755–777. [Google Scholar] [CrossRef] [PubMed]
- Schepisi, G.; Gianni, C.; Palleschi, M.; Bleve, S.; Casadei, C.; Lolli, C.; Ridolfi, L.; Martinelli, G.; De Giorgi, U. The New Frontier of Immunotherapy: Chimeric Antigen Receptor T (CAR-T) Cell and Macrophage (CAR-M) Therapy against Breast Cancer. Cancers 2023, 15, 1597. [Google Scholar] [CrossRef] [PubMed]





| Drug | Target | Indication | Combination (Target) | Clinical Trials | Citation |
|---|---|---|---|---|---|
| Avelumab | PD-L1 | HER2-Positive or TNBC | - Palbociclib (CDK4/6) Trastuzumab (HER2) and Utomilumab (4-1BB/CD137) Fulvestrant (ER) Talazoparib (PARP) Binimetinib (MEK1/2) TRX518 (GITR) | NCT01772004 (Phase Ib) | [71] |
| NCT04841148 (Phase II) | - | ||||
| NCT03414658 (Phase II) | - | ||||
| NCT03147287 (Phase II) | - | ||||
| NCT03330405 (Phase II) | - | ||||
| NCT03971409 (Phase II) | - | ||||
| NCT03861403 (Phase I/II) | - | ||||
| Durvalumab | PD-L1 | HER2-Negative, HER-2-Positive, or TNBC | Tremelimumab (CTLA-4) | Multiple | [72,73] |
| Olaparib (PARP) | Multiple | [73,74,75] | |||
| Bevacizumab (VEGF) | NCT02802098 (Phase I) | [76] | |||
| Cediranib (VEGFR) | NCT02484404 (Phase I/II) | [77,78] | |||
| Trastuzumab (HER2) | Multiple | [79] | |||
| Ibrutinib (BTK) | NCT02401048 (Phase I/II) | [80] | |||
| Camrelizumab | PD-1 | TNBC | Apatinib (VEGFR) | NCT03394287 (Phase II) | [81] |
| NCT04303741 (Phase II) | [82] | ||||
| Fuzuloparib (PARP) | NCT03945604 (Phase Ib) | [83] | |||
| Famitinib (VEGFR) | NCT04129996 (Phase II) | [84] | |||
| Nivolumab | PD-1 | HR-Positive, HER2-Negative, or TNBC | Ipilimumab (CTLA-4) | NCT02834013 (Phase II) | [85,86] |
| NCT03409198 (Phase IIb) | [87] | ||||
| Bevacizumab (VEGF) | WJOG9917B | [88] | |||
| Palbociclib (CDK4/6)/Anastrozole (aromatase) | NCT04075604 (Phase II) | - | |||
| Spartalizumab | PD-1 | TNBC | LAG525 (LAG3) | NCT02460224 (Phase I/II) | [69] |
| NCT03499899 (Phase II) | - | ||||
| Cemiplimab | PD-1 | Advanced breast cancer | SNS-101 (VISTA) | NCT05864144 (Phase I/II) | - |
| Sintilimab | PD-1 | HER2-Positive or TNBC | Apatinib (VEGFR) | NCT04722718 (Phase II) | - |
| Bevacizumab (VEGF) | NCT05386524 (Phase II) | - | |||
| Anlotinib (Multiple RTKS, including VEGFR) | NCT04877821 (Phase II) | - | |||
| Trastuzumab/Pertuzumab (HER2) | NCT05429684 (Phase III) | - | |||
| Ipilimumab | CTLA-4 | HER2-Negative breast cancer | Nivolumab (PD-1) | NCT02834013 (Phase II) NCT03409198 (Phase IIb) | [85,86] [87] |
| Tremelimumab | CTLA-4 | Metastatic breast cancer | Durvalumab (PD-L1) | Multiple | [72,73] |
| LAG525 (Ieramilimab) | LAG3 | TNBC | Spartalizumab (PD-1) | NCT02460224 (Phase I/II) | [69] |
| NCT03499899 (Phase II) | - | ||||
| Sabatolimab | TIM-3 | Advanced breast cancer | Spartalizumab (PD-1) | NCT02608268 (Phase I/Ib) | [70] |
| Drug | Targeted Protein | Indication | Used in Combination | Clinical Trials |
|---|---|---|---|---|
| Inavolisib | PI3Kα | HER2-Postivie | Pertuzumab/trastuzumab and Endocrine Therapy | NCT05306041 (Phase II) |
| Copanlisib | Pan-PI3K * | HER2-Positive | Pertuzumab and trastuzumab | NCT04108858 (Phase II) |
| Buparlisib (BKM120) | Pan-class I PI3K | HER2-Positive | Trastuzumab and paclitaxel | NCT01816594 (Phase II) |
| Eganelisib (IPI-549) | PI3Kγ | TNBC | Atezolizumab (PD-L1 mAb) and nab-Paclitaxel | NCT03961698 (Phase II) |
| Bimiralisib (PQR309) | PIEK/mTORC1/2 [119] | TNBC and HER2-negative | Eribulin (Microtubule Targeting Agent) | NCT02723877 (Phase II) |
| Panitumumab | EGFR | TNBC | Carboplatin and gemcitabine | NCT00894504 (Phase II) |
| Ipatasertib | AKT | TNBC or Hormone Receptor-Positive, HER2-Negative | Paclitaxel | NCT03337724 (Phase III) |
| Ipatasertib | AKT | TNBC | Atezolizumab and paclitaxel | NCT04177108 (Phase III) |
| Uprosertib (GSK2141795) | AKT | TNBC | Trametinib (MEK1/2 inhibitor) | NCT01964924 (Phase II) |
| Capivasertib (AZD5363) | AKT | TNBC | Olaparib | NCT02208375 (Phase II) |
| Rapamycin | mTORC1 | HER2-Positive | Trastuzumab | NCT00411788 (Phase II) |
| Ridaforolimus | mTOR | HER2-Positive | Trastuzumab | NCT00736970 (Phase II) |
| Temsirolimus | mTOR | TNBC | Neratinib | |
| Vistusertib (AZD2014) | mTORC1/2 | TNBC | Olaparib | NCT02208375 (Phase II) |
| Ribavirin | eIF4 | Metastatic breast cancer | - | NCT01056757 (Phase II) |
| Drug | Indication | Used in Combination | Outcome | Clinical Trials |
|---|---|---|---|---|
| Dasatinib * | TNBC | - | Did not increase baseline membrane EGFR | NCT02720185 (Phase II) |
| Dasatinib * | Advanced † breast cancer | - | No progression-free patient after 16 weeks | NCT00546104 (Phase II) |
| Dasatinib * | HER2-Positive | Trastuzumab and paclitaxel | ~80% of patient showed partial or complete response | NCT01306942 (Phase I/II) |
| Dasatinib * | Metastatic breast cancer | Selumetinib (AZD6244; MEK inhibitor) | Closed early for futility (one out of 30 patients had clinical benefit) [131] | NCT00780676 (Phase II) |
| Tirbanibulin (KX2-391) ** | Breast cancer with prior taxanes therapy | Paclitaxel | - | NCT01764087 (Phase II) |
| Saracatinib | HR-Positive | Anastrozole | No significant positive impact for patients receiving saracatinib in addition to anastrozole | NCT01216176 (Phase I/II) |
| Saracatinib | Metastatic or advanced breast cancer | - | Out of 9 patients, 3 stable and 6 with progression of cancer | NCT00559507 (Phase II) |
| Saracatinib | Advanced solid tumors | AZD2171 (VEGFR inhibitor) | - | NCT00475956 (Phase I) |
| Bosutinib * | Advanced HER2-negative | Exemestane | Terminated (unfavorable risk/benefit ratio) | NCT00793546 (Phase II) |
| Bosutinib * | Advanced HER2-negative | Letrozole | Terminated (unfavorable risk/benefit ratio) | NCT00880009 (Phase II) |
| Bosutinib * | Advanced breast cancer | - | ~40% progression-free survival and 26.4% overall survival | NCT00319254 (Phase II) |
| Bosutinib * | Metastatic/advanced breast cancer | Capecitabine | Terminated (unfavorable risk/benefit ratio) | NCT00959946 (Phase I/II) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Montazeri Aliabadi, H.; Manda, A.; Sidgal, R.; Chung, C. Targeting Breast Cancer: The Familiar, the Emerging, and the Uncharted Territories. Biomolecules 2023, 13, 1306. https://doi.org/10.3390/biom13091306
Montazeri Aliabadi H, Manda A, Sidgal R, Chung C. Targeting Breast Cancer: The Familiar, the Emerging, and the Uncharted Territories. Biomolecules. 2023; 13(9):1306. https://doi.org/10.3390/biom13091306
Chicago/Turabian StyleMontazeri Aliabadi, Hamidreza, Arthur Manda, Riya Sidgal, and Co Chung. 2023. "Targeting Breast Cancer: The Familiar, the Emerging, and the Uncharted Territories" Biomolecules 13, no. 9: 1306. https://doi.org/10.3390/biom13091306
APA StyleMontazeri Aliabadi, H., Manda, A., Sidgal, R., & Chung, C. (2023). Targeting Breast Cancer: The Familiar, the Emerging, and the Uncharted Territories. Biomolecules, 13(9), 1306. https://doi.org/10.3390/biom13091306