

Mitochondrial Calcium Overload Plays a Causal Role in Oxidative Stress in the Failing Heart


 ,
,
Abstract
:1. Introduction
2. Mitochondrial Function in Cardiomyocytes
3. Ca2+ Homeostasis Dysregulation and Mitochondrial Ca2+ Overload in HF
3.1. Cardiac Ca2+ Handling
3.2. Inflammation-Mediated Ca2+ Dyshomeostasis in HF
3.3. Mitochondrial Ca2+ Handling
3.3.1. Role of the Mitochondrial Ca2+ Uniporter
3.3.2. Role of the mNCLX
3.3.3. Factors influencing Mitochondrial Ca2+ Uptake
4. Physical SR–Mitochondria Interaction
4.1. Mitofusin-Mediated Tethering
4.2. IP3R-Grp75-VDAC Complex and Mitochondrial Ca2+ Uptake
5. Mitochondrial Ca2+ Overload and Mitochondrial ROS Production
6. Redox-Regulated Proteins of the Ca2+ Handling Machinery and HF
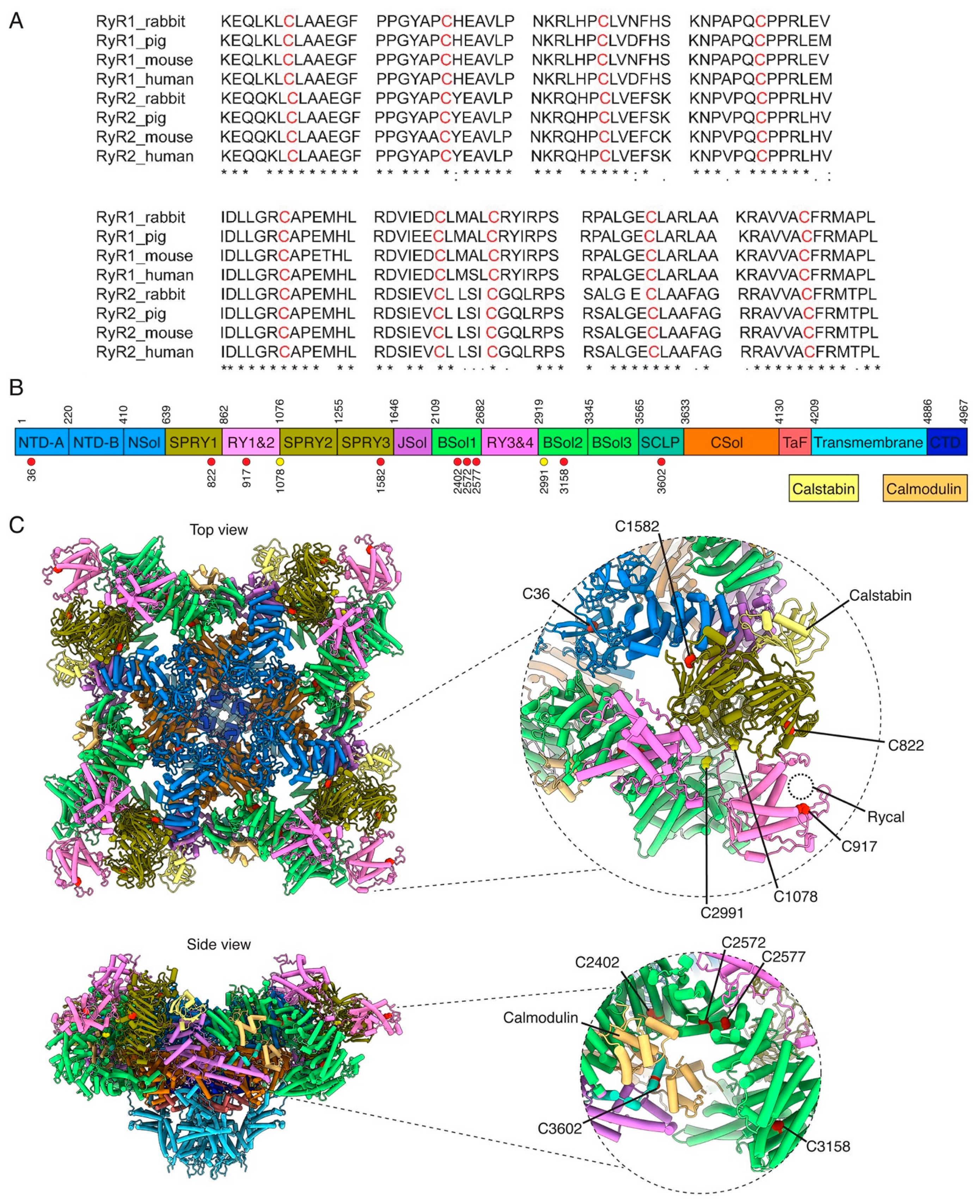
6.1. Ryanodine-Receptor Ca2+ Release Channel
Potentially Oxidized Residues in RyR2

6.2. PKA and CamKII Oxidation and Modulation of SR Ca2+ Efflux
7. Mitochondrial Ca2+ Overload and Cell Death
8. Therapies Targeting Ca2+ Defects/Mitochondrial Oxidative Stress in HF
8.1. Ca2+ Dysregulation-Targeted Therapies: Rycals
8.2. Mitochondria-Targeted Antioxidant SS31

{kind=link}
{kind=link}
| Agent | Target | Action | Diseases | Dose/Species | Development | Outcomes | References |
|---|---|---|---|---|---|---|---|
| AAV1/SERCA2a | SERCA2a | SERCA2a overexpression | Heart failure Ischemic cardiomyopathy | Patients 1 × 1011–1 × 1013 DNase particles | Terminated clinical trials | No effect/ reduced cardiac events 1 year after | [277,302] NCT00534703 NCT01966887 NCT02346422 |
| Alda-1 | Mito ALDH2 | Increased ALDH2 activity | I/R injury HF post-MI | Rat 8.5 mg/kg | Pre-clinical | Reduced infarct size by 60% | [303,304,305,306] |
| Cyclosporine A | MTP | MTP inhibition | I/R injury HF post-MI | Patients 2.5 mg/Kg IV | Phase 3 | Reduced infarct size | [307,308,309] |
| EUK-8 | SOD | SOD/catalase mimetic | DCM Pressure overload-induced HF | Mice 30 mg/kg/day IP | Pre-clinical | Prevented DCM in mice Ameliorated systolic function and survival | [310,311] |
| Idebenone | Coenzyme Q10 | Free radical scavenger | Mitochondrial cardiomyopathy | 225 mg/day In patients | Pre-clinical phase 3 | Increased EF by >50% (a case report) | [312] |
| M40403 | SOD | SOD/catalase mimetic | I/R heart injury | Rats 1 to 10 mg/kg IV | Pre-clinical | Protected tissue damage after I/R in rats | [313,314] |
| mitoTEMPO | Mitochondrial nitroxide | ROS scavenger | Diabetic cardiomyopathy Hypertension | Mice 0.7 mg/kg/day IP | Pre-clinical | Reduced myocardial hypertrophy | [315,316,317] |
| MCI-186 (Edaravone) | Free radicals | Free radical scavenger | HF Acute ischemic stroke | patients 30 mg IV | Pre-clinical phases 2–4 | Reduced enzymatic infarcts/better clinical outcomes | [318,319] |
| MitoQ | ETC | Free radical scavenger | Pressure overload Cardiovascular function | Mice 100 uM/DW Rats Unavailable for patients | Pre-clinical 2 clinical trials | Decreased heart dysfunction | [320,321] NCT03506633 NCT03586414 |
| Metformin | ETC | ETC inhibition | I/R injury HF post-MI HFpEF | Mice and rats 200–250 mg/kg | Phase 2 | Improved cardiac function (rats and mice) | [322,323] NCT03629340 |
| Rycals (S107-ARM210) | Ryanodine receptor (RyR) | Stabilizing RyR | I/R injury HF post-MI | Mice 20–75 mg/kg oral | Preclinical study | Improved cardiac function/reduced arrhythmias | [324] NCT04141670 |
| SS31 | Mitochondria | Cardiolipin protection | HFrEF, HFpEF Congestive HF | Patients 0.25 mg/kg/h | Clinical phases 1 and 2 | Improved cardiac volumes | [298] NCT02814097 NCT02914665 |
| TRO-40303 | MTP | MTP inhibition | I/R injury HF post MI | Patients 2.5 mg/kg | Phase 2 | No effect Reduced infarct size by 38% | [325,326,327] |
| XJB-5-131 | Mitochondrial nitroxide | ROS scavenger | I/R heart injury | Rats 3 mg/Kg IP | Pre-clinical | Improved post-ischemic recovery | [278,279] |
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| HF | Heart failure |
| HFrEF | Heart Failure with reduced ejection fraction |
| SR | Sarcoplasmic reticulum |
| ER | Endoplasmic reticulum |
| ATP | Adenosine triphosphate |
| Ca2+ | Calcium |
| ROS | Reactive oxygen species |
| RyR2 | Ryanodine receptor type 2 |
| IP3Rs | 1,4,5-trisphosphate receptors |
| EC | Excitation–contraction |
| ETC | Electron transport chain |
| NADH | Nicotinamide adenine dinucleotide |
| FADH2 | Flavin adenine dinucleotide |
| ADP | Adenosine diphosphate |
| CK | Creatine kinase |
| CICR | Calcium-induced calcium release |
| SERCA2a | Sarco(endo)plasmic reticulum calcium-ATPase 2a |
| NCX | Sarcolemmal Na+/Ca2+ exchanger |
| TNF-⍺ | Tumor necrosis factor |
| IL-17 | Interleukin-17 |
| IL-6 | Interleukin-6 |
| IL-1ß | Interleukin-1 |
| TGF-ß | Transforming growth factor |
| NOX2 | NADPH oxidase 2 |
| O2·− | Superoxide radical |
| MCU | Mitochondrial Ca2+ uniporter |
| mNCLX | Mitochondrial Na+-Ca2+—Li+ exchanger |
| Letm1 | Mitochondrial proton/calcium exchanger protein |
| TRPC3 | Transient receptor potential canonical 3 |
| MFN2 | Mitofusin2 |
| MFN1 | Mitofusin1 |
| Grp75 | Chaperone glucose-regulated protein 75 |
| VDAC | Voltage-dependent anion channel |
| mPTP | Mitochondrial permeability transition pore |
| MAM | Mitochondria-associated membrane |
| LBD | Ligand-binding domain (LBD) |
| PDZD8 | PDZ-domain-containing 8 |
| Rab7 | Ras-related protein Rab-7a |
| FUNDC1 | FUN14-domain-containing 1 |
| PTPIP51 | Protein tyrosine phosphatase-interacting protein 51 |
| VAPB | Vesicle-associated membrane protein-associated protein B |
| ·OH | Hydroxyl radical |
| H2O2 | Hydrogen peroxide |
| SOD | Superoxide dismutase |
| Prx | Peroxiredoxin |
| Gpx | Glutathione peroxidase |
| Cryo-EM | Cryogenic-electron microscopy |
| NTD | N-terminal domain |
| CPVT | Catecholaminergic polymorphic ventricular tachycardia |
| SPRY1 | SPIa kinase and ryanodine receptor |
| Bsol | Bridging solenoid |
| PKA | Protein kinase A |
| CamKII | Ca2+/calmodulin-dependent protein kinase II |
| cAMP | 3′,5′-cyclic adenosine monophosphate |
| hiPS | Human-induced pluripotent stem |
References
- Black, H.R. The burden of cardiovascular disease: Following the link from hypertension to myocardial infarction and heart failure. Am. J. Hypertens. 2003, 16 Pt 2, 4S–6S. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, E.J.; Muntner, P.; Alonso, A.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Das, S.R.; et al. Heart Disease and Stroke Statistics-2019 Update: A Report from the American Heart Association. Circulation 2019, 139, e56–e528. [Google Scholar] [CrossRef]
- Grossman, W.; Jones, D.; McLaurin, L.P. Wall stress and patterns of hypertrophy in the human left ventricle. J. Clin. Investig. 1975, 56, 56–64. [Google Scholar] [CrossRef]
- Santulli, G. Sympathetic nervous system signaling in heart failure and cardiac aging. In Pathophysiology and Pharmacotherapy of Cardiovascular Disease; Adis: Cham, Switzerland, 2015; pp. 83–105. [Google Scholar] [CrossRef]
- Burkart, F.; Kiowski, W. Circulatory abnormalities and compensatory mechanisms in heart failure. Am. J. Med. 1991, 90, 19S–22S. [Google Scholar] [CrossRef]
- Arai, M.; Alpert, N.R.; MacLennan, D.H.; Barton, P.; Periasamy, M. Alterations in sarcoplasmic reticulum gene expression in human heart failure. A possible mechanism for alterations in systolic and diastolic properties of the failing myocardium. Circ. Res. 1993, 72, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Iemitsu, M.; Miyauchi, T.; Maeda, S.; Sakai, S.; Kobayashi, T.; Fujii, N.; Miyazaki, H.; Matsuda, M.; Yamaguchi, I.; Krzesiak, A.; et al. Physiological and pathological cardiac hypertrophy induce different molecular phenotypes in the rat. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2001, 281, R2029–R2036. [Google Scholar] [CrossRef]
- Bernardo, B.C.; Weeks, K.L.; Pretorius, L.; McMullen, J.R. Molecular distinction between physiological and pathological cardiac hypertrophy: Experimental findings and therapeutic strategies. Pharmacol. Ther. 2010, 128, 191–227. [Google Scholar] [CrossRef]
- Takahashi, T.; Allen, P.D.; Izumo, S. Expression of A-, B-, and C-type natriuretic peptide genes in failing and developing human ventricles. Correlation with expression of the Ca2+-ATPase gene. Circ. Res. 1992, 71, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Mittmann, C.; Eschenhagen, T.; Scholz, H. Cellular and molecular aspects of contractile dysfunction in heart failure. Cardiovasc. Res. 1998, 39, 267–275. [Google Scholar] [CrossRef] [PubMed]
- Sachse, F.B.; Torres, N.S.; Savio-Galimberti, E.; Aiba, T.; Kass, D.A.; Tomaselli, G.F.; Bridge, J.H. Subcellular structures and function of myocytes impaired during heart failure are restored by cardiac resynchronization therapy. Circ. Res. 2012, 110, 588–597. [Google Scholar] [CrossRef]
- Mohammed, S.F.; Hussain, S.; Mirzoyev, S.A.; Edwards, W.D.; Maleszewski, J.J.; Redfield, M.M. Coronary microvascular rarefaction and myocardial fibrosis in heart failure with preserved ejection fraction. Circulation 2015, 131, 550–559. [Google Scholar] [CrossRef] [PubMed]
- Gogiraju, R.; Bochenek, M.L.; Schafer, K. Angiogenic Endothelial Cell Signaling in Cardiac Hypertrophy and Heart Failure. Front. Cardiovasc. Med. 2019, 6, 20. [Google Scholar] [CrossRef]
- Belviso, I.; Angelini, F.; Di Meglio, F.; Picchio, V.; Sacco, A.M.; Nocella, C.; Romano, V.; Nurzynska, D.; Frati, G.; Maiello, C.; et al. The Microenvironment of Decellularized Extracellular Matrix from Heart Failure Myocardium Alters the Balance between Angiogenic and Fibrotic Signals from Stromal Primitive Cells. Int. J. Mol. Sci. 2020, 21, 7903. [Google Scholar] [PubMed]
- Zile, M.R.; O’Meara, E.; Claggett, B.; Prescott, M.F.; Solomon, S.D.; Swedberg, K.; Packer, M.; McMurray, J.J.; Shi, V.; Lefkowitz, M.; et al. Effects of Sacubitril/Valsartan on Biomarkers of Extracellular Matrix Regulation in Patients with HFrEF. J. Am. Coll. Cardiol. 2019, 73, 795–806. [Google Scholar] [CrossRef]
- Richter, M.; Kostin, S. The failing human heart is characterized by decreased numbers of telocytes as result of apoptosis and altered extracellular matrix composition. J. Cell Mol. Med. 2015, 19, 2597–2606. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N.G. The Extracellular Matrix in Ischemic and Nonischemic Heart Failure. Circ. Res. 2019, 125, 117–146. [Google Scholar] [CrossRef]
- Frangogiannis, N.G. Cardiac fibrosis. Cardiovasc. Res. 2021, 117, 1450–1488. [Google Scholar] [CrossRef]
- Kong, P.; Christia, P.; Frangogiannis, N.G. The pathogenesis of cardiac fibrosis. Cell. Mol. Life Sci. 2014, 71, 549–574. [Google Scholar]
- Gargiulo, P.; Marsico, F.; Renga, F.; Dell’Aversana, S.; Esposito, I.; Marciano, C.; Dellegrottaglie, S.; Perrone-Filardi, P.; Paolillo, S. The metabolic syndrome in heart failure: Insights to specific mechanisms. Heart Fail. Rev. 2019, 25, 1–7. [Google Scholar] [CrossRef]
- Harada, T.; Sunaga, H.; Sorimachi, H.; Yoshida, K.; Kato, T.; Kurosawa, K.; Nagasaka, T.; Koitabashi, N.; Iso, T.; Kurabayashi, M.; et al. Pathophysiological role of fatty acid-binding protein 4 in Asian patients with heart failure and preserved ejection fraction. ESC Heart Fail. 2020, 7, 4256–4266. [Google Scholar] [CrossRef]
- Hunter, W.G.; Kelly, J.P.; McGarrah, R.W., 3rd; Khouri, M.G.; Craig, D.; Haynes, C.; Ilkayeva, O.; Stevens, R.D.; Bain, J.R.; Muehlbauer, M.J.; et al. Metabolomic Profiling Identifies Novel Circulating Biomarkers of Mitochondrial Dysfunction Differentially Elevated in Heart Failure with Preserved Versus Reduced Ejection Fraction: Evidence for Shared Metabolic Impairments in Clinical Heart Failure. J. Am. Heart Assoc. 2016, 5, e003190. [Google Scholar] [CrossRef]
- Murray, A.J.; Cole, M.A.; Lygate, C.A.; Carr, C.A.; Stuckey, D.J.; Little, S.E.; Neubauer, S.; Clarke, K. Increased mitochondrial uncoupling proteins, respiratory uncoupling and decreased efficiency in the chronically infarcted rat heart. J. Mol. Cell. Cardiol. 2008, 44, 694–700. [Google Scholar] [CrossRef] [PubMed]
- De Rosa, M.; Gambardella, J.; Shu, J.; Santulli, G. Dietary fat is a key determinant in balancing mitochondrial dynamics in heart failure: A novel mechanism underlying the obesity paradox. Cardiovasc. Res. 2018, 114, 925–927. [Google Scholar] [CrossRef]
- Seki, M.; Powers, J.C.; Maruyama, S.; Zuriaga, M.A.; Wu, C.L.; Kurishima, C.; Kim, L.; Johnson, J.; Poidomani, A.; Wang, T.; et al. Acute and Chronic Increases of Circulating FSTL1 Normalize Energy Substrate Metabolism in Pacing-Induced Heart Failure. Circ. Heart Fail. 2018, 11, e004486. [Google Scholar] [CrossRef]
- Fukushima, A.; Lopaschuk, G.D. Acetylation control of cardiac fatty acid beta-oxidation and energy metabolism in obesity, diabetes, and heart failure. Biochim. Biophys. Acta 2016, 1862, 2211–2220. [Google Scholar] [CrossRef] [PubMed]
- Abdurrachim, D.; Luiken, J.J.; Nicolay, K.; Glatz, J.F.; Prompers, J.J.; Nabben, M. Good and bad consequences of altered fatty acid metabolism in heart failure: Evidence from mouse models. Cardiovasc. Res. 2015, 106, 194–205. [Google Scholar] [CrossRef] [PubMed]
- Neubauer, S. The failing heart—An engine out of fuel. N. Engl. J. Med. 2007, 356, 1140–1151. [Google Scholar] [CrossRef]
- Doenst, T.; Nguyen, T.D.; Abel, E.D. Cardiac metabolism in heart failure: Implications beyond ATP production. Circ. Res. 2013, 113, 709–724. [Google Scholar] [CrossRef]
- Schaper, J.; Meiser, E.; Stammler, G. Ultrastructural morphometric analysis of myocardium from dogs, rats, hamsters, mice, and from human hearts. Circ. Res. 1985, 56, 377–391. [Google Scholar] [CrossRef]
- Booth, D.M.; Enyedi, B.; Geiszt, M.; Varnai, P.; Hajnoczky, G. Redox Nanodomains Are Induced by and Control Calcium Signaling at the ER-Mitochondrial Interface. Mol. Cell 2016, 63, 240–248. [Google Scholar] [CrossRef]
- Ramesh, V.; Sharma, V.K.; Sheu, S.S.; Franzini-Armstrong, C. Structural proximity of mitochondria to calcium release units in rat ventricular myocardium may suggest a role in Ca2+ sequestration. Ann. N. Y. Acad. Sci. 1998, 853, 341–344. [Google Scholar] [CrossRef]
- Szabadkai, G.; Bianchi, K.; Varnai, P.; De Stefani, D.; Wieckowski, M.R.; Cavagna, D.; Nagy, A.I.; Balla, T.; Rizzuto, R. Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J. Cell Biol. 2006, 175, 901–911. [Google Scholar] [CrossRef]
- Boncompagni, S.; Rossi, A.E.; Micaroni, M.; Beznoussenko, G.V.; Polishchuk, R.S.; Dirksen, R.T.; Protasi, F. Mitochondria are linked to calcium stores in striated muscle by developmentally regulated tethering structures. Mol. Biol. Cell 2009, 20, 1058–1067. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Su, D.; O’Rourke, B.; Pogwizd, S.M.; Zhou, L. Mitochondria-derived ROS bursts disturb Ca2+ cycling and induce abnormal automaticity in guinea pig cardiomyocytes: A theoretical study. Am. J. Physiol. Heart Circ. Physiol. 2015, 308, H623–H636. [Google Scholar] [CrossRef]
- Zhou, L.; Aon, M.A.; Liu, T.; O’Rourke, B. Dynamic modulation of Ca2+ sparks by mitochondrial oscillations in isolated guinea pig cardiomyocytes under oxidative stress. J. Mol. Cell. Cardiol. 2011, 51, 632–639. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Liu, J.; Wei, C.; Li, K.; Xie, W.; Wang, Y.; Cheng, H. Bidirectional regulation of Ca2+ sparks by mitochondria-derived reactive oxygen species in cardiac myocytes. Cardiovasc. Res. 2008, 77, 432–441. [Google Scholar] [CrossRef]
- Bragadin, M.; Pozzan, T.; Azzone, G.F. Kinetics of Ca2+ carrier in rat liver mitochondria. Biochemistry 1979, 18, 5972–5978. [Google Scholar] [CrossRef]
- Massari, S.; Pozzan, T. The interaction of organic cations with the mitochondrial membrane. Experientia 1976, 32, 868–869. [Google Scholar] [CrossRef] [PubMed]
- Massari, S.; Pozzan, T. The accumulation ratio of K+, Na+, Ca2+ and tetrapropylammonium in steady-state Mitochondria. Arch. Biochem. Biophys. 1976, 173, 332–340. [Google Scholar] [CrossRef]
- Tromp, J.; Khan, M.A.; Klip, I.T.; Meyer, S.; de Boer, R.A.; Jaarsma, T.; Hillege, H.; van Veldhuisen, D.J.; van der Meer, P.; Voors, A.A. Biomarker Profiles in Heart Failure Patients with Preserved and Reduced Ejection Fraction. J. Am. Heart Assoc. 2017, 6, e003989. [Google Scholar] [CrossRef]
- Bianchi, V.E. Nutrition in chronic heart failure patients: A systematic review. Heart Fail. Rev. 2020, 25, 1017–1026. [Google Scholar] [CrossRef] [PubMed]
- Padmanabhan, A.; Alexanian, M.; Linares-Saldana, R.; Gonzalez-Teran, B.; Andreoletti, G.; Huang, Y.; Connolly, A.J.; Kim, W.; Hsu, A.; Duan, Q.; et al. BRD4 Interacts with GATA4 to Govern Mitochondrial Homeostasis in Adult Cardiomyocytes. Circulation 2020, 142, 2338–2355. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Song, J.; Hu, S. Metabolic remodeling of substrate utilization during heart failure progression. Heart Fail. Rev. 2019, 24, 143–154. [Google Scholar] [CrossRef]
- Santulli, G.; Lewis, D.; des Georges, A.; Marks, A.R.; Frank, J. Ryanodine Receptor Structure and Function in Health and Disease. Subcell Biochem. 2018, 87, 329–352. [Google Scholar]
- Marx, S.O.; Marks, A.R. Dysfunctional ryanodine receptors in the heart: New insights into complex cardiovascular diseases. J. Mol. Cell. Cardiol. 2013, 58, 225–231. [Google Scholar] [CrossRef]
- Marks, A.R. Calcium cycling proteins and heart failure: Mechanisms and therapeutics. J. Clin. Investig. 2013, 123, 46–52. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.; Shan, J.; Reiken, S.; Wehrens, X.H.; Marks, A.R. Analysis of calstabin2 (FKBP12.6)-ryanodine receptor interactions: Rescue of heart failure by calstabin2 in mice. Proc. Natl. Acad. Sci. USA 2006, 103, 3456–3461. [Google Scholar] [CrossRef]
- Gambardella, J.; Trimarco, B.; Iaccarino, G.; Santulli, G. New Insights in Cardiac Calcium Handling and Excitation-Contraction Coupling. Adv. Exp. Med. Biol. 2018, 1067, 373–385. [Google Scholar]
- Shan, J.; Kushnir, A.; Betzenhauser, M.J.; Reiken, S.; Li, J.; Lehnart, S.E.; Lindegger, N.; Mongillo, M.; Mohler, P.J.; Marks, A.R. Phosphorylation of the ryanodine receptor mediates the cardiac fight or flight response in mice. J. Clin. Investig. 2010, 120, 4388–4398. [Google Scholar] [CrossRef]
- Santulli, G.; Xie, W.; Reiken, S.R.; Marks, A.R. Mitochondrial calcium overload is a key determinant in heart failure. Proc. Natl. Acad. Sci. USA 2015, 112, 11389–11394. [Google Scholar] [CrossRef]
- Schwarz, K.; Siddiqi, N.; Singh, S.; Neil, C.J.; Dawson, D.K.; Frenneaux, M.P. The breathing heart—Mitochondrial respiratory chain dysfunction in cardiac disease. Int. J. Cardiol. 2014, 171, 134–143. [Google Scholar] [CrossRef]
- Forini, F.; Nicolini, G.; Kusmic, C.; Iervasi, G. Protective Effects of Euthyroidism Restoration on Mitochondria Function and Quality Control in Cardiac Pathophysiology. Int. J. Mol. Sci. 2019, 20, 3377. [Google Scholar] [CrossRef]
- Li, X.; Lu, W.J.; Li, Y.; Wu, F.; Bai, R.; Ma, S.; Dong, T.; Zhang, H.; Lee, A.S.; Wang, Y.; et al. MLP-deficient human pluripotent stem cell derived cardiomyocytes develop hypertrophic cardiomyopathy and heart failure phenotypes due to abnormal calcium handling. Cell Death Dis. 2019, 10, 610. [Google Scholar] [CrossRef]
- Hamilton, D.J.; Zhang, A.; Li, S.; Cao, T.N.; Smith, J.A.; Vedula, I.; Cordero-Reyes, A.M.; Youker, K.A.; Torre-Amione, G.; Gupte, A.A.; et al. Combination of angiotensin II and l-NG-nitroarginine methyl ester exacerbates mitochondrial dysfunction and oxidative stress to cause heart failure. Am. J. Physiol. Heart Circ. Physiol. 2016, 310, H667–H680. [Google Scholar] [CrossRef] [PubMed]
- Rosca, M.G.; Vazquez, E.J.; Kerner, J.; Parland, W.; Chandler, M.P.; Stanley, W.; Sabbah, H.N.; Hoppel, C.L. Cardiac mitochondria in heart failure: Decrease in respirasomes and oxidative phosphorylation. Cardiovasc. Res. 2008, 80, 30–39. [Google Scholar] [CrossRef]
- Leverve, X.M. Mitochondrial function and substrate availability. Crit. Care Med. 2007, 35 (Suppl. S9), S454–S460. [Google Scholar] [CrossRef]
- Erecinska, M.; Wilson, D.F. Regulation of cellular energy metabolism. J. Membr. Biol. 1982, 70, 1–14. [Google Scholar] [CrossRef]
- Kampjut, D.; Sazanov, L.A. Structure and mechanism of mitochondrial proton-translocating transhydrogenase. Nature 2019, 573, 291–295. [Google Scholar] [CrossRef] [PubMed]
- Kory, N.; Uit de Bos, J.; van der Rijt, S.; Jankovic, N.; Gura, M.; Arp, N.; Pena, I.A.; Prakash, G.; Chan, S.H.; Kunchok, T.; et al. MCART1/SLC25A51 is required for mitochondrial NAD transport. Sci. Adv. 2020, 6, eabe5310. [Google Scholar] [CrossRef] [PubMed]
- Nowinski, S.M.; Solmonson, A.; Rusin, S.F.; Maschek, J.A.; Bensard, C.L.; Fogarty, S.; Jeong, M.Y.; Lettlova, S.; Berg, J.A.; Morgan, J.T.; et al. Mitochondrial fatty acid synthesis coordinates oxidative metabolism in mammalian mitochondria. eLife 2020, 9, e58041. [Google Scholar] [CrossRef]
- Balsa, E.; Perry, E.A.; Bennett, C.F.; Jedrychowski, M.; Gygi, S.P.; Doench, J.G.; Puigserver, P. Defective NADPH production in mitochondrial disease complex I causes inflammation and cell death. Nat. Commun. 2020, 11, 2714. [Google Scholar] [CrossRef] [PubMed]
- Jouaville, L.S.; Pinton, P.; Bastianutto, C.; Rutter, G.A.; Rizzuto, R. Regulation of mitochondrial ATP synthesis by calcium: Evidence for a long-term metabolic priming. Proc. Natl. Acad. Sci. USA 1999, 96, 13807–13812. [Google Scholar] [CrossRef]
- Gong, G.; Liu, X.; Wang, W. Regulation of metabolism in individual mitochondria during excitation-contraction coupling. J. Mol. Cell. Cardiol. 2014, 76, 235–246. [Google Scholar] [CrossRef] [PubMed]
- Piquereau, J.; Veksler, V.; Novotova, M.; Ventura-Clapier, R. Energetic Interactions between Subcellular Organelles in Striated Muscles. Front. Cell Dev. Biol. 2020, 8, 581045. [Google Scholar] [CrossRef]
- Cortassa, S.; Aon, M.A.; Marban, E.; Winslow, R.L.; O’Rourke, B. An integrated model of cardiac mitochondrial energy metabolism and calcium dynamics. Biophys. J. 2003, 84, 2734–2755. [Google Scholar] [CrossRef]
- Glancy, B.; Balaban, R.S. Role of mitochondrial Ca2+ in the regulation of cellular energetics. Biochemistry 2012, 51, 2959–2973. [Google Scholar] [CrossRef]
- Yang, K.C.; Kyle, J.W.; Makielski, J.C.; Dudley, S.C., Jr. Mechanisms of sudden cardiac death: Oxidants and metabolism. Circ. Res. 2015, 116, 1937–1955. [Google Scholar] [CrossRef] [PubMed]
- Guo, A.; Wang, Y.; Chen, B.; Wang, Y.; Yuan, J.; Zhang, L.; Hall, D.; Wu, J.; Shi, Y.; Zhu, Q.; et al. E-C coupling structural protein junctophilin-2 encodes a stress-adaptive transcription regulator. Science 2018, 362, eaan3303. [Google Scholar] [CrossRef]
- Ziman, A.P.; Gomez-Viquez, N.L.; Bloch, R.J.; Lederer, W.J. Excitation-contraction coupling changes during postnatal cardiac development. J. Mol. Cell. Cardiol. 2010, 48, 379–386. [Google Scholar] [CrossRef]
- Firth, J.M.; Yang, H.Y.; Francis, A.J.; Islam, N.; MacLeod, K.T. The Effect of Estrogen on Intracellular Ca2+ and Na+ Regulation in Heart Failure. JACC Basic Transl. Sci. 2020, 5, 901–912. [Google Scholar] [CrossRef]
- Arbel-Ganon, L.; Behar, J.A.; Gomez, A.M.; Yaniv, Y. Distinct mechanisms mediate pacemaker dysfunction associated with catecholaminergic polymorphic ventricular tachycardia mutations: Insights from computational modeling. J. Mol. Cell. Cardiol. 2020, 143, 85–95. [Google Scholar] [CrossRef]
- Abu-Khousa, M.; Fiegle, D.J.; Sommer, S.T.; Minabari, G.; Milting, H.; Heim, C.; Weyand, M.; Tomasi, R.; Dendorfer, A.; Volk, T.; et al. The Degree of t-System Remodeling Predicts Negative Force-Frequency Relationship and Prolonged Relaxation Time in Failing Human Myocardium. Front. Physiol. 2020, 11, 182. [Google Scholar] [CrossRef] [PubMed]
- Scranton, K.; John, S.; Escobar, A.; Goldhaber, J.I.; Ottolia, M. Modulation of the cardiac Na+-Ca2+ exchanger by cytoplasmic protons: Molecular mechanisms and physiological implications. Cell Calcium 2020, 87, 102140. [Google Scholar] [CrossRef]
- Li, S.; Chopra, A.; Keung, W.; Chan, C.W.Y.; Costa, K.D.; Kong, C.W.; Hajjar, R.J.; Chen, C.S.; Li, R.A. Sarco/endoplasmic reticulum Ca2+-ATPase is a more effective calcium remover than sodium-calcium exchanger in human embryonic stem cell-derived cardiomyocytes. Am. J. Physiol. Heart Circ. Physiol. 2019, 317, H1105–H1115. [Google Scholar] [CrossRef] [PubMed]
- Dridi, H.; Kushnir, A.; Zalk, R.; Yuan, Q.; Melville, Z.; Marks, A.R. Intracellular calcium leak in heart failure and atrial fibrillation: A unifying mechanism and therapeutic target. Nat. Rev. Cardiol. 2020, 17, 732–747. [Google Scholar] [PubMed]
- Schroder, F.; Handrock, R.; Beuckelmann, D.J.; Hirt, S.; Hullin, R.; Priebe, L.; Schwinger, R.H.G.; Weil, J.; Herzig, S. Increased availability and open probability of single L-type calcium channels from failing compared with nonfailing human ventricle. Circulation 1998, 98, 969–976. [Google Scholar] [CrossRef] [PubMed]
- Kamp, T.J.; Hell, J.W. Regulation of cardiac L-type calcium channels by protein kinase A and protein kinase C. Circ. Res. 2000, 87, 1095–1102. [Google Scholar] [CrossRef]
- Sipido, K.R.; Volders, P.G.; Vos, M.A.; Verdonck, F. Altered Na/Ca exchange activity in cardiac hypertrophy and heart failure: A new target for therapy? Cardiovasc. Res. 2002, 53, 782–805. [Google Scholar] [CrossRef]
- Hasenfuss, G.; Reinecke, H.; Studer, R.; Meyer, M.; Pieske, B.; Holtz, J.; Holubarsch, C.; Posival, H.; Just, H.; Drexler, H. Relation between myocardial function and expression of sarcoplasmic reticulum Ca2+-ATPase in failing and nonfailing human myocardium. Circ. Res. 1994, 75, 434–442. [Google Scholar] [CrossRef]
- Hobai, I.A.; O’Rourke, B. Decreased sarcoplasmic reticulum calcium content is responsible for defective excitation-contraction coupling in canine heart failure. Circulation 2001, 103, 1577–1584. [Google Scholar] [CrossRef]
- Tu, C.C.; Wan, B.Y.; Zeng, Y. STIM2 knockdown protects against ischemia/reperfusion injury through reducing mitochondrial calcium overload and preserving mitochondrial function. Life Sci. 2020, 247, 116560. [Google Scholar] [CrossRef]
- Santin, Y.; Fazal, L.; Sainte-Marie, Y.; Sicard, P.; Maggiorani, D.; Tortosa, F.; Yücel, Y.Y.; Teyssedre, L.; Rouquette, J.; Marcellin, M.; et al. Mitochondrial 4-HNE derived from MAO-A promotes mitoCa2+ overload in chronic postischemic cardiac remodeling. Cell Death Differ. 2020, 27, 1907–1923. [Google Scholar] [CrossRef]
- Stanciu, A.E. Cytokines in heart failure. Adv. Clin. Chem. 2019, 93, 63–113. [Google Scholar] [PubMed]
- Szabo, T.M.; Frigy, A.; Nagy, E.E. Targeting Mediators of Inflammation in Heart Failure: A Short Synthesis of Experimental and Clinical Results. Int. J. Mol. Sci. 2021, 22, 13053. [Google Scholar] [CrossRef]
- Wu, C.K.; Lee, J.K.; Chiang, F.T.; Yang, C.H.; Huang, S.W.; Hwang, J.J.; Lin, J.-L.; Tseng, C.-D.; Chen, J.-J.; Tsai, C.-T. Plasma levels of tumor necrosis factor-alpha and interleukin-6 are associated with diastolic heart failure through downregulation of sarcoplasmic reticulum Ca2+ ATPase. Crit. Care Med. 2011, 39, 984–992. [Google Scholar] [CrossRef]
- Tsai, C.T.; Wu, C.K.; Lee, J.K.; Chang, S.N.; Kuo, Y.M.; Wang, Y.C.; Lai, L.P.; Chiang, F.T.; Hwang, J.J.; Lin, J.L. TNF-alpha down-regulates sarcoplasmic reticulum Ca2+ ATPase expression and leads to left ventricular diastolic dysfunction through binding of NF-kappaB to promoter response element. Cardiovasc. Res. 2015, 105, 318–329. [Google Scholar] [CrossRef]
- Villegas, S.; Villarreal, F.J.; Dillmann, W.H. Leukemia Inhibitory Factor and Interleukin-6 downregulate sarcoplasmic reticulum Ca2+ ATPase (SERCA2) in cardiac myocytes. Basic Res. Cardiol. 2000, 95, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Xue, G.L.; Li, D.S.; Wang, Z.Y.; Liu, Y.; Yang, J.M.; Li, C.Z.; Li, X.D.; Ma, M.J.; Zhang, M.M.; Liu, Y.J.; et al. Interleukin-17 upregulation participates in the pathogenesis of heart failure in mice via NF-kappaB-dependent suppression of SERCA2a and Cav1.2 expression. Acta Pharmacol. Sin. 2021, 42, 1780–1789. [Google Scholar] [CrossRef] [PubMed]
- Waning, D.L.; Mohammad, K.S.; Reiken, S.; Xie, W.; Andersson, D.C.; John, S.; Chiechi, A.; Wright, L.E.; Umanskaya, A.; Niewolna, M.; et al. Excess TGF-beta mediates muscle weakness associated with bone metastases in mice. Nat. Med. 2015, 21, 1262–1271. [Google Scholar] [CrossRef]
- Campanella, M.; Pinton, P.; Rizzuto, R. Mitochondrial Ca2+ homeostasis in health and disease. Biol. Res. 2004, 37, 653–660. [Google Scholar] [CrossRef]
- Brown, D.A.; O’Rourke, B. Cardiac mitochondria and arrhythmias. Cardiovasc. Res. 2010, 88, 241–249. [Google Scholar] [CrossRef]
- Crompton, M.; Heid, I. The cycling of calcium, sodium, and protons across the inner membrane of cardiac mitochondria. Eur. J. Biochem. 1978, 91, 599–608. [Google Scholar] [CrossRef]
- Palty, R.; Silverman, W.F.; Hershfinkel, M.; Caporale, T.; Sensi, S.L.; Parnis, J.; Nolte, C.; Fishman, D.; Shoshan-Barmatz, V.; Herrmann, S.; et al. NCLX is an essential component of mitochondrial Na+/Ca2+ exchange. Proc. Natl. Acad. Sci. USA 2010, 107, 436–441. [Google Scholar] [CrossRef] [PubMed]
- De Stefani, D.; Raffaello, A.; Teardo, E.; Szabo, I.; Rizzuto, R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 2011, 476, 336–340. [Google Scholar] [CrossRef]
- Fan, M.; Zhang, J.; Tsai, C.W.; Orlando, B.J.; Rodriguez, M.; Xu, Y.; Liao, M.; Tsai, M.-F.; Feng, L. Structure and mechanism of the mitochondrial Ca2+ uniporter holocomplex. Nature 2020, 582, 129–133. [Google Scholar] [CrossRef]
- Raffaello, A.; De Stefani, D.; Sabbadin, D.; Teardo, E.; Merli, G.; Picard, A.; Checchetto, V.; Moro, S.; Szabò, I.; Rizzuto, R. The mitochondrial calcium uniporter is a multimer that can include a dominant-negative pore-forming subunit. EMBO J. 2013, 32, 2362–2376. [Google Scholar] [CrossRef]
- Baughman, J.M.; Perocchi, F.; Girgis, H.S.; Plovanich, M.; Belcher-Timme, C.A.; Sancak, Y.; Bao, X.R.; Strittmatter, L.; Goldberger, O.; Bogorad, R.L.; et al. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 2011, 476, 341–345. [Google Scholar] [CrossRef] [PubMed]
- Selwyn, M.J.; Dawson, A.P.; Dunnett, S.J. Calcium transport in mitochondria. FEBS Lett. 1970, 10, 1–5. [Google Scholar] [CrossRef]
- Vais, H.; Payne, R.; Paudel, U.; Li, C.; Foskett, J.K. Coupled transmembrane mechanisms control MCU-mediated mitochondrial Ca2+ uptake. Proc. Natl. Acad. Sci. USA 2020, 117, 21731–21739. [Google Scholar] [CrossRef] [PubMed]
- Boyman, L.; Lederer, W.J. How the mitochondrial calcium uniporter complex (MCUcx) works. Proc. Natl. Acad. Sci. USA 2020, 117, 22634–22636. [Google Scholar] [CrossRef] [PubMed]
- Fieni, F.; Lee, S.B.; Jan, Y.N.; Kirichok, Y. Activity of the mitochondrial calcium uniporter varies greatly between tissues. Nat. Commun. 2012, 3, 1317. [Google Scholar] [CrossRef]
- Pan, X.; Liu, J.; Nguyen, T.; Liu, C.; Sun, J.; Teng, Y.; Fergusson, M.M.; Rovira, I.I.; Allen, M.; Springer, D.A.; et al. The physiological role of mitochondrial calcium revealed by mice lacking the mitochondrial calcium uniporter. Nat. Cell Biol. 2013, 15, 1464–1472. [Google Scholar] [CrossRef]
- Holmstrom, K.M.; Pan, X.; Liu, J.C.; Menazza, S.; Liu, J.; Nguyen, T.T.; Pan, H.; Parks, R.J.; Anderson, S.; Noguchi, A.; et al. Assessment of cardiac function in mice lacking the mitochondrial calcium uniporter. J. Mol. Cell. Cardiol. 2015, 85, 178–182. [Google Scholar] [CrossRef]
- Dong, Z.; Shanmughapriya, S.; Tomar, D.; Siddiqui, N.; Lynch, S.; Nemani, N.; Breves, S.L.; Zhang, X.; Tripathi, A.; Palaniappan, P.; et al. Mitochondrial Ca2+ Uniporter Is a Mitochondrial Luminal Redox Sensor that Augments MCU Channel Activity. Mol. Cell 2017, 65, 1014–1028.e7. [Google Scholar] [CrossRef]
- Pan, L.; Huang, B.J.; Ma, X.E.; Wang, S.Y.; Feng, J.; Lv, F.; Liu, Y.; Liu, Y.; Li, C.-M.; Liang, D.-D.; et al. MiR-25 protects cardiomyocytes against oxidative damage by targeting the mitochondrial calcium uniporter. Int. J. Mol. Sci. 2015, 16, 5420–5433. [Google Scholar] [CrossRef]
- Kovacs-Bogdan, E.; Sancak, Y.; Kamer, K.J.; Plovanich, M.; Jambhekar, A.; Huber, R.J.; Myre, M.A.; Blower, M.D.; Mootha, V.K. Reconstitution of the mitochondrial calcium uniporter in yeast. Proc. Natl. Acad. Sci. USA 2014, 111, 8985–8990. [Google Scholar] [CrossRef] [PubMed]
- Oxenoid, K.; Dong, Y.; Cao, C.; Cui, T.; Sancak, Y.; Markhard, A.L.; Grabarek, Z.; Kong, L.; Liu, Z.; Ouyang, B.; et al. Architecture of the mitochondrial calcium uniporter. Nature 2016, 533, 269–273. [Google Scholar] [CrossRef]
- Zhuo, W.; Zhou, H.; Guo, R.; Yi, J.; Zhang, L.; Yu, L.; Sui, Y.; Zeng, W.; Wang, P.; Yang, M. Structure of intact human MCU supercomplex with the auxiliary MICU subunits. Protein Cell 2020, 12, 220–229. [Google Scholar] [CrossRef] [PubMed]
- Sancak, Y.; Markhard, A.L.; Kitami, T.; Kovacs-Bogdan, E.; Kamer, K.J.; Udeshi, N.D.; Carr, S.A.; Chaudhuri, D.; Clapham, D.E.; Li, A.A.; et al. EMRE is an essential component of the mitochondrial calcium uniporter complex. Science 2013, 342, 1379–1382. [Google Scholar] [CrossRef]
- Pittis, A.A.; Goh, V.; Cebrian-Serrano, A.; Wettmarshausen, J.; Perocchi, F.; Gabaldon, T. Discovery of EMRE in fungi resolves the true evolutionary history of the mitochondrial calcium uniporter. Nat. Commun. 2020, 11, 4031. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.C.; Syder, N.C.; Ghorashi, N.S.; Willingham, T.B.; Parks, R.J.; Sun, J.; Fergusson, M.M.; Liu, J.; Holmström, K.M.; Menazza, S.; et al. EMRE is essential for mitochondrial calcium uniporter activity in a mouse model. JCI Insight 2020, 5, e134063. [Google Scholar] [CrossRef]
- Wang, Y.; Nguyen, N.X.; She, J.; Zeng, W.; Yang, Y.; Bai, X.C.; Jiang, Y. Structural Mechanism of EMRE-Dependent Gating of the Human Mitochondrial Calcium Uniporter. Cell 2019, 177, 1252–1261.e13. [Google Scholar] [CrossRef] [PubMed]
- Mallilankaraman, K.; Doonan, P.; Cardenas, C.; Chandramoorthy, H.C.; Muller, M.; Miller, R.; Hoffman, N.E.; Gandhirajan, R.K.; Molgo, J.; Birnbaum, M.J.; et al. MICU1 is an essential gatekeeper for MCU-mediated mitochondrial Ca2+ uptake that regulates cell survival. Cell 2012, 151, 630–644. [Google Scholar] [CrossRef] [PubMed]
- Nemani, N.; Dong, Z.; Daw, C.C.; Madaris, T.R.; Ramachandran, K.; Enslow, B.T.; Rubannelsonkumar, C.S.; Shanmughapriya, S.; Mallireddigari, V.; Maity, S.; et al. Mitochondrial pyruvate and fatty acid flux modulate MICU1-dependent control of MCU activity. Sci. Signal 2020, 13, eaaz6206. [Google Scholar] [CrossRef]
- Shah, S.I.; Ullah, G. The Function of Mitochondrial Calcium Uniporter at the Whole-Cell and Single Mitochondrion Levels in WT, MICU1 KO, and MICU2 KO Cells. Cells 2020, 9, 1520. [Google Scholar] [CrossRef]
- Gottschalk, B.; Klec, C.; Leitinger, G.; Bernhart, E.; Rost, R.; Bischof, H.; Madreiter-Sokolowski, C.T.; Radulović, S.; Eroglu, E.; Sattler, W.; et al. MICU1 controls cristae junction and spatially anchors mitochondrial Ca2+ uniporter complex. Nat. Commun. 2019, 10, 3732. [Google Scholar] [CrossRef] [PubMed]
- Phillips, C.B.; Tsai, C.W.; Tsai, M.F. The conserved aspartate ring of MCU mediates MICU1 binding and regulation in the mitochondrial calcium uniporter complex. eLife 2019, 8, e41112. [Google Scholar] [CrossRef] [PubMed]
- Marchi, S.; Corricelli, M.; Branchini, A.; Vitto, V.A.M.; Missiroli, S.; Morciano, G.; Perrone, M.; Ferrarese, M.; Giorgi, C.; Pinotti, M.; et al. Akt-mediated phosphorylation of MICU1 regulates mitochondrial Ca2+ levels and tumor growth. EMBO J. 2019, 38, e99435. [Google Scholar] [CrossRef]
- Paillard, M.; Csordas, G.; Huang, K.T.; Varnai, P.; Joseph, S.K.; Hajnoczky, G. MICU1 Interacts with the D-Ring of the MCU Pore to Control Its Ca2+ Flux and Sensitivity to Ru360. Mol. Cell 2018, 72, 778–785.e3. [Google Scholar] [CrossRef]
- Kamer, K.J.; Sancak, Y.; Fomina, Y.; Meisel, J.D.; Chaudhuri, D.; Grabarek, Z.; Mootha, V.K. MICU1 imparts the mitochondrial uniporter with the ability to discriminate between Ca2+ and Mn2+. Proc. Natl. Acad. Sci. USA 2018, 115, E7960–E7969. [Google Scholar] [CrossRef] [PubMed]
- Vecellio Reane, D.; Vallese, F.; Checchetto, V.; Acquasaliente, L.; Butera, G.; De Filippis, V.; Szabò, I.; Zanotti, G.; Rizzuto, R.; Raffaello, A. A MICU1 Splice Variant Confers High Sensitivity to the Mitochondrial Ca2+ Uptake Machinery of Skeletal Muscle. Mol. Cell 2016, 64, 760–773. [Google Scholar] [CrossRef] [PubMed]
- Logan, C.V.; Szabadkai, G.; Sharpe, J.A.; Parry, D.A.; Torelli, S.; Childs, A.M.; Kriek, M.; Phadke, R.; A Johnson, C.; Roberts, N.Y.; et al. Loss-of-function mutations in MICU1 cause a brain and muscle disorder linked to primary alterations in mitochondrial calcium signaling. Nat. Genet. 2014, 46, 188–193. [Google Scholar] [CrossRef]
- Alam, M.R.; Groschner, L.N.; Parichatikanond, W.; Kuo, L.; Bondarenko, A.I.; Rost, R.; Waldeck-Weiermair, M.; Malli, R.; Graier, G.; Pinotti, M.; et al. Mitochondrial Ca2+ uptake 1 (MICU1) and mitochondrial Ca2+ uniporter (MCU) contribute to metabolism-secretion coupling in clonal pancreatic beta-cells. J. Biol. Chem. 2012, 287, 34445–34454. [Google Scholar] [CrossRef] [PubMed]
- Patron, M.; Checchetto, V.; Raffaello, A.; Teardo, E.; Vecellio Reane, D.; Mantoan, M.; Granatiero, V.; Szabò, I.; De Stefani, D.; Rizzuto, R. MICU1 and MICU2 finely tune the mitochondrial Ca2+ uniporter by exerting opposite effects on MCU activity. Mol. Cell 2014, 53, 726–737. [Google Scholar] [CrossRef] [PubMed]
- Petrungaro, C.; Zimmermann, K.M.; Kuttner, V.; Fischer, M.; Dengjel, J.; Bogeski, I.; Riemer, J. The Ca2+-Dependent Release of the Mia40-Induced MICU1-MICU2 Dimer from MCU Regulates Mitochondrial Ca2+ Uptake. Cell Metab. 2015, 22, 721–733. [Google Scholar] [CrossRef]
- Plovanich, M.; Bogorad, R.L.; Sancak, Y.; Kamer, K.J.; Strittmatter, L.; Li, A.A.; Girgis, H.S.; Kuchimanchi, S.; De Groot, J.; Speciner, L.; et al. MICU2, a paralog of MICU1, resides within the mitochondrial uniporter complex to regulate calcium handling. PLoS ONE 2013, 8, e55785. [Google Scholar] [CrossRef]
- Wu, W.; Shen, Q.; Lei, Z.; Qiu, Z.; Li, D.; Pei, H.; Zheng, J.; Jia, Z. The crystal structure of MICU2 provides insight into Ca2+ binding and MICU1-MICU2 heterodimer formation. EMBO Rep. 2019, 20, e47488. [Google Scholar] [CrossRef] [PubMed]
- Patron, M.; Granatiero, V.; Espino, J.; Rizzuto, R.; De Stefani, D. MICU3 is a tissue-specific enhancer of mitochondrial calcium uptake. Cell Death Differ. 2019, 26, 179–195. [Google Scholar] [CrossRef]
- Wang, B.; Xiong, S.; Lin, S.; Xia, W.; Li, Q.; Zhao, Z.; Wei, X.; Lu, Z.; Wei, X.; Gao, P.; et al. Enhanced Mitochondrial Transient Receptor Potential Channel, Canonical Type 3-Mediated Calcium Handling in the Vasculature from Hypertensive Rats. J. Am. Heart Assoc. 2017, 6, e005812. [Google Scholar] [CrossRef]
- Puente, B.N.; Sun, J.; Parks, R.J.; Fergusson, M.M.; Liu, C.; Springer, D.; Aponte, A.M.; Liu, J.C.; Murphy, E. MICU3 Plays an Important Role in Cardiovascular Function. Circ. Res. 2020, 127, 1571–1573. [Google Scholar] [CrossRef]
- Ashrafi, G.; de Juan-Sanz, J.; Farrell, R.J.; Ryan, T.A. Molecular Tuning of the Axonal Mitochondrial Ca2+ Uniporter Ensures Metabolic Flexibility of Neurotransmission. Neuron 2020, 105, 678–687.e5. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, T.P.; Wu, Y.; Joiner, M.L.; Koval, O.M.; Wilson, N.R.; Luczak, E.D.; Wang, Q.; Chen, B.; Gao, Z.; Zhu, Z.; et al. Inhibition of MCU forces extramitochondrial adaptations governing physiological and pathological stress responses in heart. Proc. Natl. Acad. Sci. USA 2015, 112, 9129–9134. [Google Scholar] [CrossRef] [PubMed]
- Kwong, J.Q.; Lu, X.; Correll, R.N.; Schwanekamp, J.A.; Vagnozzi, R.J.; Sargent, M.A.; York, A.J.; Zhang, J.; Bers, D.M.; Molkentin, J.D. The Mitochondrial Calcium Uniporter Selectively Matches Metabolic Output to Acute Contractile Stress in the Heart. Cell Rep. 2015, 12, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Luongo, T.S.; Lambert, J.P.; Yuan, A.; Zhang, X.; Gross, P.; Song, J.; Shanmughapriya, S.; Gao, E.; Jain, M.; Houser, S.R.; et al. The Mitochondrial Calcium Uniporter Matches Energetic Supply with Cardiac Workload during Stress and Modulates Permeability Transition. Cell Rep. 2015, 12, 23–34. [Google Scholar] [CrossRef]
- Altamimi, T.R.; Karwi, Q.G.; Uddin, G.M.; Fukushima, A.; Kwong, J.Q.; Molkentin, J.D.; Lopaschuk, G.D. Cardiac-specific deficiency of the mitochondrial calcium uniporter augments fatty acid oxidation and functional reserve. J. Mol. Cell. Cardiol. 2019, 127, 223–231. [Google Scholar] [CrossRef]
- Joseph, L.C.; Barca, E.; Subramanyam, P.; Komrowski, M.; Pajvani, U.; Colecraft, H.M.; Hirano, M.; Morrow, J.P. Inhibition of NAPDH Oxidase 2 (NOX2) Prevents Oxidative Stress and Mitochondrial Abnormalities Caused by Saturated Fat in Cardiomyocytes. PLoS ONE 2016, 11, e0145750. [Google Scholar] [CrossRef]
- Yu, Z.; Chen, R.; Li, M.; Yu, Y.; Liang, Y.; Han, F.; Qin, S.; Chen, X.; Su, Y.; Ge, J. Mitochondrial calcium uniporter inhibition provides cardioprotection in pressure overload-induced heart failure through autophagy enhancement. Int. J. Cardiol. 2018, 271, 161–168. [Google Scholar] [CrossRef]
- Zhang, B.; Jia, K.; Tian, J.; Du, H. Cyclophilin D counterbalances mitochondrial calcium uniporter-mediated brain mitochondrial calcium uptake. Biochem. Biophys. Res. Commun. 2020, 529, 314–320. [Google Scholar] [CrossRef]
- Bermont, F.; Hermant, A.; Benninga, R.; Chabert, C.; Jacot, G.; Santo-Domingo, J.; Kraus, M.R.-C.; Feige, J.N.; De Marchi, U. Targeting Mitochondrial Calcium Uptake with the Natural Flavonol Kaempferol, to Promote Metabolism/Secretion Coupling in Pancreatic beta-cells. Nutrients 2020, 12, 538. [Google Scholar] [CrossRef]
- Woods, J.J.; Nemani, N.; Shanmughapriya, S.; Kumar, A.; Zhang, M.; Nathan, S.R.; Thomas, M.; Carvalho, E.; Ramachandran, K.; Srikantan, S.; et al. A Selective and Cell-Permeable Mitochondrial Calcium Uniporter (MCU) Inhibitor Preserves Mitochondrial Bioenergetics after Hypoxia/Reoxygenation Injury. ACS Cent. Sci. 2019, 5, 153–166. [Google Scholar] [CrossRef]
- Hamilton, S.; Terentyeva, R.; Kim, T.Y.; Bronk, P.; Clements, R.T.; O-Uchi, J.; Csordás, G.; Choi, B.-R.; Terentyev, D. Pharmacological Modulation of Mitochondrial Ca2+ Content Regulates Sarcoplasmic Reticulum Ca2+ Release via Oxidation of the Ryanodine Receptor by Mitochondria-Derived Reactive Oxygen Species. Front. Physiol. 2018, 9, 1831. [Google Scholar] [CrossRef]
- Xu, H.; Guan, N.; Ren, Y.L.; Wei, Q.J.; Tao, Y.H.; Yang, G.S.; Liu, X.-Y.; Bu, D.-F.; Zhang, Y.; Zhu, S.-N. IP3R-Grp75-VDAC1-MCU calcium regulation axis antagonists protect podocytes from apoptosis and decrease proteinuria in an Adriamycin nephropathy rat model. BMC Nephrol. 2018, 19, 140. [Google Scholar] [CrossRef]
- Arduino, D.M.; Wettmarshausen, J.; Vais, H.; Navas-Navarro, P.; Cheng, Y.; Leimpek, A.; Ma, Z.; Delrio-Lorenzo, A.; Giordano, A.; Garcia-Perez, C.; et al. Systematic Identification of MCU Modulators by Orthogonal Interspecies Chemical Screening. Mol. Cell 2017, 67, 711–723.e7. [Google Scholar] [CrossRef]
- Kon, N.; Murakoshi, M.; Isobe, A.; Kagechika, K.; Miyoshi, N.; Nagayama, T. DS16570511 is a small-molecule inhibitor of the mitochondrial calcium uniporter. Cell Death Discov. 2017, 3, 17045. [Google Scholar] [CrossRef]
- Ren, T.; Zhang, H.; Wang, J.; Zhu, J.; Jin, M.; Wu, Y.; Guo, X.; Ji, L.; Huang, Q.; Zhang, H.; et al. MCU-dependent mitochondrial Ca2+ inhibits NAD(+)/SIRT3/SOD2 pathway to promote ROS production and metastasis of HCC cells. Oncogene 2017, 36, 5897–5909. [Google Scholar] [CrossRef]
- Pan, S.; Ryu, S.Y.; Sheu, S.S. Distinctive characteristics and functions of multiple mitochondrial Ca2+ influx mechanisms. Sci. China Life Sci. 2011, 54, 763–769. [Google Scholar] [CrossRef] [PubMed]
- Canato, M.; Capitanio, P.; Cancellara, L.; Leanza, L.; Raffaello, A.; Reane, D.V.; Marcucci, L.; Michelucci, A.; Protasi, F.; Reggiani, C. Excessive Accumulation of Ca2+ in Mitochondria of Y522S-RYR1 Knock-in Mice: A Link between Leak from the Sarcoplasmic Reticulum and Altered Redox State. Front. Physiol. 2019, 10, 1142. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, J.; Brustovetsky, T.; Rysted, J.E.; Lin, Z.; Usachev, Y.M.; Brustovetsky, N. Deletion of mitochondrial calcium uniporter incompletely inhibits calcium uptake and induction of the permeability transition pore in brain mitochondria. J. Biol. Chem. 2018, 293, 15652–15663. [Google Scholar] [CrossRef]
- Natarajan, G.K.; Glait, L.; Mishra, J.; Stowe, D.F.; Camara, A.K.S.; Kwok, W.M. Total Matrix Ca2+ Modulates Ca2+ Efflux via the Ca2+/H+ Exchanger in Cardiac Mitochondria. Front. Physiol. 2020, 11, 510600. [Google Scholar] [CrossRef] [PubMed]
- Tsai, M.F.; Jiang, D.; Zhao, L.; Clapham, D.; Miller, C. Functional reconstitution of the mitochondrial Ca2+/H+ antiporter Letm1. J. Gen. Physiol. 2014, 143, 67–73. [Google Scholar] [CrossRef]
- Jiang, D.; Zhao, L.; Clapham, D.E. Genome-wide RNAi screen identifies Letm1 as a mitochondrial Ca2+/H+ antiporter. Science 2009, 326, 144–147. [Google Scholar] [CrossRef]
- Austin, S.; Nowikovsky, K. LETM1: Essential for Mitochondrial Biology and Cation Homeostasis? Trends Biochem Sci. 2019, 44, 648–658. [Google Scholar] [CrossRef] [PubMed]
- Durigon, R.; Mitchell, A.L.; Jones, A.W.; Manole, A.; Mennuni, M.; Hirst, E.M.; Houlden, H.; Maragni, G.; Lattante, S.; Doronzio, P.N.; et al. LETM1 couples mitochondrial DNA metabolism and nutrient preference. EMBO Mol. Med. 2018, 10, e8550. [Google Scholar] [CrossRef] [PubMed]
- Austin, S.; Tavakoli, M.; Pfeiffer, C.; Seifert, J.; Mattarei, A.; De Stefani, D.; Zoratti, M.; Nowikovsky, K. LETM1-Mediated K+ and Na+ Homeostasis Regulates Mitochondrial Ca2+ Efflux. Front. Physiol. 2017, 8, 839. [Google Scholar] [CrossRef] [PubMed]
- Huang, E.; Qu, D.; Huang, T.; Rizzi, N.; Boonying, W.; Krolak, D.; Ciana, P.; Woulfe, J.; Klein, C.; Slack, R.S.; et al. PINK1-mediated phosphorylation of LETM1 regulates mitochondrial calcium transport and protects neurons against mitochondrial stress. Nat. Commun. 2017, 8, 1399. [Google Scholar] [CrossRef]
- Feng, S.; Li, H.; Tai, Y.; Huang, J.; Su, Y.; Abramowitz, J.; Zhu, M.X.; Birnbaumer, L.; Wang, Y. Canonical transient receptor potential 3 channels regulate mitochondrial calcium uptake. Proc. Natl. Acad. Sci. USA 2013, 110, 11011–11016. [Google Scholar] [CrossRef]
- He, X.; Li, S.; Liu, B.; Susperreguy, S.; Formoso, K.; Yao, J.; Kang, J.; Shi, A.; Birnbaumer, L.; Liao, Y. Major contribution of the 3/6/7 class of TRPC channels to myocardial ischemia/reperfusion and cellular hypoxia/reoxygenation injuries. Proc. Natl. Acad. Sci. USA 2017, 114, E4582–E4591. [Google Scholar] [CrossRef] [PubMed]
- Khayyat, N.H.; Tomilin, V.N.; Zaika, O.; Pochynyuk, O. Polymodal roles of TRPC3 channel in the kidney. Channels 2020, 14, 257–267. [Google Scholar] [CrossRef]
- Han, J.W.; Kang, C.; Kim, Y.; Lee, M.G.; Kim, J.Y. Isoproterenol-induced hypertrophy of neonatal cardiac myocytes and H9c2 cell is dependent on TRPC3-regulated CaV1.2 expression. Cell Calcium 2020, 92, 102305. [Google Scholar] [CrossRef]
- Chen, X.; Sooch, G.; Demaree, I.S.; White, F.A.; Obukhov, A.G. Transient Receptor Potential Canonical (TRPC) Channels: Then and Now. Cells 2020, 9, 1983. [Google Scholar] [CrossRef]
- Nishiyama, K.; Tanaka, T.; Nishimura, A.; Nishida, M. TRPC3-based protein signaling complex as a therapeutic target of myocardial atrophy. Curr. Mol. Pharmacol. 2020, 14, 123–131. [Google Scholar] [CrossRef]
- Ma, T.; Lin, S.; Wang, B.; Wang, Q.; Xia, W.; Zhang, H.; Cui, Y.; He, C.; Wu, H.; Sun, F.; et al. TRPC3 deficiency attenuates high salt-induced cardiac hypertrophy by alleviating cardiac mitochondrial dysfunction. Biochem. Biophys. Res. Commun. 2019, 519, 674–681. [Google Scholar] [CrossRef] [PubMed]
- Kirichok, Y.; Krapivinsky, G.; Clapham, D.E. The mitochondrial calcium uniporter is a highly selective ion channel. Nature 2004, 427, 360–364. [Google Scholar] [CrossRef] [PubMed]
- Parks, R.J.; Menazza, S.; Holmstrom, K.M.; Amanakis, G.; Fergusson, M.; Ma, H.; Aponte, A.M.; Bernardi, P.; Finkel, T.; Murphy, E. Cyclophilin D-mediated regulation of the permeability transition pore is altered in mice lacking the mitochondrial calcium uniporter. Cardiovasc. Res. 2019, 115, 385–394. [Google Scholar] [CrossRef]
- Marta, K.; Hasan, P.; Rodriguez-Prados, M.; Paillard, M.; Hajnoczky, G. Pharmacological inhibition of the mitochondrial Ca2+ uniporter: Relevance for pathophysiology and human therapy. J. Mol. Cell. Cardiol. 2020, 151, 135–144. [Google Scholar] [CrossRef]
- Luongo, T.S.; Lambert, J.P.; Gross, P.; Nwokedi, M.; Lombardi, A.A.; Shanmughapriya, S.; Carpenter, A.C.; Kolmetzky, D.; Gao, E.; van Berlo, J.H.; et al. The mitochondrial Na+/Ca2+ exchanger is essential for Ca2+ homeostasis and viability. Nature 2017, 545, 93–97. [Google Scholar] [CrossRef]
- Garbincius, J.F.; Luongo, T.S.; Jadiya, P.; Hildebrand, A.N.; Kolmetzky, D.W.; Mangold, A.S.; Roy, R.; Ibetti, J.; Nwokedi, M.; Koch, W.J.; et al. Enhanced NCLX-dependent mitochondrial Ca2+ efflux attenuates pathological remodeling in heart failure. J. Mol. Cell. Cardiol. 2022, 167, 52–66. [Google Scholar] [CrossRef]
- Sparagna, G.C.; Gunter, K.K.; Sheu, S.S.; Gunter, T.E. Mitochondrial calcium uptake from physiological-type pulses of calcium. A description of the rapid uptake mode. J. Biol. Chem. 1995, 270, 27510–27515. [Google Scholar] [CrossRef] [PubMed]
- Santulli, G.; Marks, A.R. Essential Roles of Intracellular Calcium Release Channels in Muscle, Brain, Metabolism, and Aging. Curr. Mol. Pharmacol. 2015, 8, 206–222. [Google Scholar] [CrossRef]
- Cardenas, C.; Miller, R.A.; Smith, I.; Bui, T.; Molgo, J.; Muller, M.; Vais, H.; Cheung, K.-H.; Yang, J.; Parker, I.; et al. Essential regulation of cell bioenergetics by constitutive InsP3 receptor Ca2+ transfer to mitochondria. Cell 2010, 142, 270–283. [Google Scholar] [CrossRef]
- Rizzuto, R.; Simpson, A.W.; Brini, M.; Pozzan, T. Rapid changes of mitochondrial Ca2+ revealed by specifically targeted recombinant aequorin. Nature 1992, 358, 325–327. [Google Scholar] [CrossRef]
- Jou, M.J.; Peng, T.I.; Sheu, S.S. Histamine induces oscillations of mitochondrial free Ca2+ concentration in single cultured rat brain astrocytes. J. Physiol. 1996, 497 Pt 2, 299–308. [Google Scholar] [CrossRef]
- Llinas, R.; Sugimori, M.; Silver, R.B. Microdomains of high calcium concentration in a presynaptic terminal. Science 1992, 256, 677–679. [Google Scholar] [CrossRef]
- Rizzuto, R.; Brini, M.; Murgia, M.; Pozzan, T. Microdomains with high Ca2+ close to IP3-sensitive channels that are sensed by neighboring mitochondria. Science 1993, 262, 744–747. [Google Scholar] [CrossRef] [PubMed]
- Rizzuto, R.; Pinton, P.; Brini, M.; Chiesa, A.; Filippin, L.; Pozzan, T. Mitochondria as biosensors of calcium microdomains. Cell Calcium 1999, 26, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Szalai, G.; Csordas, G.; Hantash, B.M.; Thomas, A.P.; Hajnoczky, G. Calcium signal transmission between ryanodine receptors and mitochondria. J. Biol. Chem. 2000, 275, 15305–15313. [Google Scholar] [CrossRef] [PubMed]
- Beaumont, V.; Llobet, A.; Lagnado, L. Expansion of calcium microdomains regulates fast exocytosis at a ribbon synapse. Proc. Natl. Acad. Sci. USA 2005, 102, 10700–10705. [Google Scholar] [CrossRef] [PubMed]
- Scriven, D.R.; Klimek, A.; Lee, K.L.; Moore, E.D. The molecular architecture of calcium microdomains in rat cardiomyocytes. Ann. N. Y. Acad. Sci. 2002, 976, 488–499. [Google Scholar] [CrossRef]
- Bautista, D.M.; Lewis, R.S. Modulation of plasma membrane calcium-ATPase activity by local calcium microdomains near CRAC channels in human T cells. J. Physiol. 2004, 556 Pt 3, 805–817. [Google Scholar] [CrossRef]
- Dedkova, E.N.; Blatter, L.A. Mitochondrial Ca2+ and the heart. Cell Calcium 2008, 44, 77–91. [Google Scholar] [CrossRef]
- Dedkova, E.N.; Seidlmayer, L.K.; Blatter, L.A. Mitochondria-mediated cardioprotection by trimetazidine in rabbit heart failure. J. Mol. Cell. Cardiol. 2013, 59, 41–54. [Google Scholar] [CrossRef]
- Moshkforoush, A.; Ashenagar, B.; Tsoukias, N.M.; Alevriadou, B.R. Modeling the role of endoplasmic reticulum-mitochondria microdomains in calcium dynamics. Sci. Rep. 2019, 9, 17072. [Google Scholar] [CrossRef] [PubMed]
- de Brito, O.M.; Scorrano, L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 2008, 456, 605–610. [Google Scholar] [CrossRef]
- Garcia-Perez, C.; Schneider, T.G.; Hajnoczky, G.; Csordas, G. Alignment of sarcoplasmic reticulum-mitochondrial junctions with mitochondrial contact points. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H1907–H1915. [Google Scholar] [CrossRef] [PubMed]
- Zacharioudakis, E.; Biris, N.; Garner, T.; Chen, Y.; Pekson, R.; Dhingra, R.; Santulli, G.; Kirshenbaum, L.A.; Kitsis, R.A.; Gavathiotis, E. Direct Small Molecule Activation of Mitofusins. BioRxiv 2018. [Google Scholar] [CrossRef]
- Paillard, M.; Tubbs, E.; Thiebaut, P.A.; Gomez, L.; Fauconnier, J.; Da Silva, C.C.; Teixeira, G.; Mewton, N.; Belaidi, E.; Durand, A.; et al. Depressing mitochondria-reticulum interactions protects cardiomyocytes from lethal hypoxia-reoxygenation injury. Circulation 2013, 128, 1555–1565. [Google Scholar] [CrossRef]
- Basso, V.; Marchesan, E.; Ziviani, E. A trio has turned into a quartet: DJ-1 interacts with the IP3R-Grp75-VDAC complex to control ER-mitochondria interaction. Cell Calcium 2020, 87, 102186. [Google Scholar] [CrossRef]
- Marchi, S.; Patergnani, S.; Pinton, P. The endoplasmic reticulum-mitochondria connection: One touch, multiple functions. Biochim. Biophys. Acta 2014, 1837, 461–469. [Google Scholar] [CrossRef]
- Giacomello, M.; Drago, I.; Bortolozzi, M.; Scorzeto, M.; Gianelle, A.; Pizzo, P.; Pozzan, T. Ca2+ hot spots on the mitochondrial surface are generated by Ca2+ mobilization from stores, but not by activation of store-operated Ca2+ channels. Mol. Cell 2010, 38, 280–290. [Google Scholar] [CrossRef]
- Rossini, M.; Filadi, R. Sarcoplasmic Reticulum-Mitochondria Kissing in Cardiomyocytes: Ca2+, ATP, and Undisclosed Secrets. Front. Cell Dev. Biol. 2020, 8, 532. [Google Scholar] [CrossRef]
- Chen, Y.; Csordas, G.; Jowdy, C.; Schneider, T.G.; Csordas, N.; Wang, W.; Liu, Y.; Kohlhaas, M.; Meiser, M.; Bergem, S.; et al. Mitofusin 2-containing mitochondrial-reticular microdomains direct rapid cardiomyocyte bioenergetic responses via interorganelle Ca2+ crosstalk. Circ. Res. 2012, 111, 863–875. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhu, X. Endoplasmic reticulum-mitochondria tethering in neurodegenerative diseases. Transl. Neurodegener. 2017, 6, 21. [Google Scholar] [CrossRef]
- Chen, H.; Detmer, S.A.; Ewald, A.J.; Griffin, E.E.; Fraser, S.E.; Chan, D.C. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J. Cell Biol. 2003, 160, 189–200. [Google Scholar] [CrossRef]
- Sood, A.; Jeyaraju, D.V.; Prudent, J.; Caron, A.; Lemieux, P.; McBride, H.M.; Laplante, M.; Tóth, K.; Pellegrini, L. A Mitofusin-2-dependent inactivating cleavage of Opa1 links changes in mitochondria cristae and ER contacts in the postprandial liver. Proc. Natl. Acad. Sci. USA 2014, 111, 16017–16022. [Google Scholar] [CrossRef]
- Munoz, J.P.; Ivanova, S.; Sanchez-Wandelmer, J.; Martinez-Cristobal, P.; Noguera, E.; Sancho, A.; Díaz-Ramos, A.; Hernández-Alvarez, M.I.; Sebastián, D.; Mauvezin, C.; et al. Mfn2 modulates the UPR and mitochondrial function via repression of PERK. EMBO J. 2013, 32, 2348–2361. [Google Scholar] [CrossRef] [PubMed]
- Rizzuto, R.; Bastianutto, C.; Brini, M.; Murgia, M.; Pozzan, T. Mitochondrial Ca2+ homeostasis in intact cells. J. Cell Biol. 1994, 126, 1183–1194. [Google Scholar] [CrossRef]
- Rizzuto, R.; Pinton, P.; Carrington, W.; Fay, F.S.; Fogarty, K.E.; Lifshitz, L.M.; Tuft, R.A.; Pozzan, T. Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science 1998, 280, 1763–1766. [Google Scholar] [CrossRef] [PubMed]
- Cosson, P.; Marchetti, A.; Ravazzola, M.; Orci, L. Mitofusin-2 independent juxtaposition of endoplasmic reticulum and mitochondria: An ultrastructural study. PLoS ONE 2012, 7, e46293. [Google Scholar] [CrossRef]
- Filadi, R.; Greotti, E.; Turacchio, G.; Luini, A.; Pozzan, T.; Pizzo, P. Mitofusin 2 ablation increases endoplasmic reticulum-mitochondria coupling. Proc. Natl. Acad. Sci. USA 2015, 112, E2174–E2181. [Google Scholar] [CrossRef]
- Giorgi, C.; Ito, K.; Lin, H.K.; Santangelo, C.; Wieckowski, M.R.; Lebiedzinska, M.; Bononi, A.; Bonora, M.; Duszynski, J.; Bernardi, R.; et al. PML regulates apoptosis at endoplasmic reticulum by modulating calcium release. Science 2010, 330, 1247–1251. [Google Scholar] [CrossRef]
- Tubbs, E.; Rieusset, J. Study of Endoplasmic Reticulum and Mitochondria Interactions by In Situ Proximity Ligation Assay in Fixed Cells. J. Vis. Exp. 2016, 118, e54899. [Google Scholar]
- Santulli, G.; Nakashima, R.; Yuan, Q.; Marks, A.R. Intracellular calcium release channels: An update. J. Physiol. 2017, 595, 3041–3051. [Google Scholar] [CrossRef]
- Rizzuto, R.; De Stefani, D.; Raffaello, A.; Mammucari, C. Mitochondria as sensors and regulators of calcium signalling. Nat. Rev. Mol. Cell Biol. 2012, 13, 566–578. [Google Scholar] [CrossRef]
- Williams, G.S.; Boyman, L.; Lederer, W.J. Mitochondrial calcium and the regulation of metabolism in the heart. J. Mol. Cell. Cardiol. 2015, 78, 35–45. [Google Scholar] [CrossRef]
- Wust, R.C.; Helmes, M.; Martin, J.L.; van der Wardt, T.J.; Musters, R.J.; van der Velden, J.; Stienen, G.J.M. Rapid frequency-dependent changes in free mitochondrial calcium concentration in rat cardiac myocytes. J. Physiol. 2017, 595, 2001–2019. [Google Scholar] [CrossRef] [PubMed]
- Drago, I.; De Stefani, D.; Rizzuto, R.; Pozzan, T. Mitochondrial Ca2+ uptake contributes to buffering cytoplasmic Ca2+ peaks in cardiomyocytes. Proc. Natl. Acad. Sci. USA 2012, 109, 12986–12991. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, P.; Di Lisa, F. The mitochondrial permeability transition pore: Molecular nature and role as a target in cardioprotection. J. Mol. Cell. Cardiol. 2015, 78, 100–106. [Google Scholar] [CrossRef]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef]
- Hirabayashi, Y.; Kwon, S.K.; Paek, H.; Pernice, W.M.; Paul, M.A.; Lee, J.; Erfani, P.; Raczkowski, A.; Petrey, D.S.; Pon, L.A.; et al. ER-mitochondria tethering by PDZD8 regulates Ca2+ dynamics in mammalian neurons. Science 2017, 358, 623–630. [Google Scholar] [CrossRef] [PubMed]
- Wideman, J.G.; Balacco, D.L.; Fieblinger, T.; Richards, T.A. PDZD8 is not the ‘functional ortholog’ of Mmm1, it is a paralog. F1000Res 2018, 7, 1088. [Google Scholar] [CrossRef]
- Shirane, M.; Wada, M.; Morita, K.; Hayashi, N.; Kunimatsu, R.; Matsumoto, Y.; Matsuzaki, F.; Nakatsumi, H.; Ohta, K.; Tamura, Y.; et al. Protrudin and PDZD8 contribute to neuronal integrity by promoting lipid extraction required for endosome maturation. Nat. Commun. 2020, 11, 4576. [Google Scholar] [CrossRef]
- Elbaz-Alon, Y.; Guo, Y.; Segev, N.; Harel, M.; Quinnell, D.E.; Geiger, T.; Avinoam, O.; Li, D.; Nunnari, J. PDZD8 interacts with Protrudin and Rab7 at ER-late endosome membrane contact sites associated with mitochondria. Nat. Commun. 2020, 11, 3645. [Google Scholar] [CrossRef] [PubMed]
- Qiao, X.; Jia, S.; Ye, J.; Fang, X.; Zhang, C.; Cao, Y.; Xu, C.; Zhao, L.; Zhu, Y.; Wang, L.; et al. PTPIP51 regulates mouse cardiac ischemia/reperfusion through mediating the mitochondria-SR junction. Sci. Rep. 2017, 7, 45379. [Google Scholar] [CrossRef]
- De Vos, K.J.; Morotz, G.M.; Stoica, R.; Tudor, E.L.; Lau, K.F.; Ackerley, S.; Warley, A.; Shaw, C.E.; Miller, C.C.J. VAPB interacts with the mitochondrial protein PTPIP51 to regulate calcium homeostasis. Hum. Mol. Genet. 2012, 21, 1299–1311. [Google Scholar] [CrossRef]
- Stoica, R.; De Vos, K.J.; Paillusson, S.; Mueller, S.; Sancho, R.M.; Lau, K.F.; Vizcay-Barrena, G.; Lin, W.-L.; Xu, Y.-F.; Lewis, J.; et al. ER-mitochondria associations are regulated by the VAPB-PTPIP51 interaction and are disrupted by ALS/FTD-associated TDP-43. Nat. Commun. 2014, 5, 3996. [Google Scholar] [CrossRef] [PubMed]
- Spiteller, G. Are changes of the cell membrane structure causally involved in the aging process? Ann. N. Y. Acad. Sci. 2002, 959, 30–44. [Google Scholar] [CrossRef] [PubMed]
- Cadenas, E.; Davies, K.J. Mitochondrial free radical generation, oxidative stress, and aging. Free Radic. Biol. Med. 2000, 29, 222–230. [Google Scholar] [CrossRef]
- Ozawa, T. Mitochondrial genome mutation in cell death and aging. J. Bioenergy Biomembr. 1999, 31, 377–390. [Google Scholar] [CrossRef]
- Bode, D.; Wen, Y.; Hegemann, N.; Primessnig, U.; Parwani, A.; Boldt, L.H.; Pieske, B.; Heinzel, F.R.; Hohendanner, F. Oxidative Stress and Inflammatory Modulation of Ca2+ Handling in Metabolic HFpEF-Related Left Atrial Cardiomyopathy. Antioxidants 2020, 9, 860. [Google Scholar] [CrossRef]
- Chang, J.C.; Lien, C.F.; Lee, W.S.; Chang, H.R.; Hsu, Y.C.; Luo, Y.P.; Jeng, J.-R.; Hsieh, J.-C.; Yang, K.-T. Intermittent Hypoxia Prevents Myocardial Mitochondrial Ca2+ Overload and Cell Death during Ischemia/Reperfusion: The Role of Reactive Oxygen Species. Cells 2019, 8, 564. [Google Scholar] [CrossRef]
- Mitchell, P. Coupling of phosphorylation to electron and hydrogen transfer by a chemi-osmotic type of mechanism. Nature 1961, 191, 144–148. [Google Scholar] [CrossRef] [PubMed]
- Chance, B.; Sies, H.; Boveris, A. Hydroperoxide metabolism in mammalian organs. Physiol. Rev. 1979, 59, 527–605. [Google Scholar] [CrossRef]
- Aon, M.A.; Cortassa, S.; Marban, E.; O’Rourke, B. Synchronized whole cell oscillations in mitochondrial metabolism triggered by a local release of reactive oxygen species in cardiac myocytes. J. Biol. Chem. 2003, 278, 44735–44744. [Google Scholar] [CrossRef]
- Brand, M.D. The sites and topology of mitochondrial superoxide production. Exp. Gerontol. 2010, 45, 466–472. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, T.; Imai, H. Hydrogen peroxide produced by superoxide dismutase SOD-2 activates sperm in Caenorhabditis elegans. J. Biol. Chem. 2017, 292, 14804–14813. [Google Scholar] [CrossRef] [PubMed]
- Kang, P.T.; Chen, C.L.; Ohanyan, V.; Luther, D.J.; Meszaros, J.G.; Chilian, W.M.; Chen, Y.-R. Overexpressing superoxide dismutase 2 induces a supernormal cardiac function by enhancing redox-dependent mitochondrial function and metabolic dilation. J. Mol. Cell. Cardiol. 2015, 88, 14–28. [Google Scholar] [CrossRef] [PubMed]
- Ribas, V.; Garcia-Ruiz, C.; Fernandez-Checa, J.C. Glutathione and mitochondria. Front. Pharmacol. 2014, 5, 151. [Google Scholar] [CrossRef] [PubMed]
- Lopert, P.; Patel, M. Brain mitochondria from DJ-1 knockout mice show increased respiration-dependent hydrogen peroxide consumption. Redox. Biol. 2014, 2, 667–672. [Google Scholar] [CrossRef]
- Ide, T.; Tsutsui, H.; Kinugawa, S.; Utsumi, H.; Kang, D.; Hattori, N.; Uchida, K.; Arimura, K.-I.; Egashira, K.; Takeshita, A. Mitochondrial electron transport complex I is a potential source of oxygen free radicals in the failing myocardium. Circ. Res. 1999, 85, 357–363. [Google Scholar] [CrossRef]
- Ide, T.; Tsutsui, H.; Kinugawa, S.; Suematsu, N.; Hayashidani, S.; Ichikawa, K.; Utsumi, H.; Machida, Y.; Egashira, K.; Takeshita, A. Direct evidence for increased hydroxyl radicals originating from superoxide in the failing myocardium. Circ. Res. 2000, 86, 152–157. [Google Scholar] [CrossRef]
- Wagner, S.; Rokita, A.G.; Anderson, M.E.; Maier, L.S. Redox regulation of sodium and calcium handling. Antioxid. Redox. Signal 2013, 18, 1063–1077. [Google Scholar] [CrossRef] [PubMed]
- Kiyuna, L.A.; Albuquerque, R.P.E.; Chen, C.H.; Mochly-Rosen, D.; Ferreira, J.C.B. Targeting mitochondrial dysfunction and oxidative stress in heart failure: Challenges and opportunities. Free Radic. Biol. Med. 2018, 129, 155–168. [Google Scholar] [CrossRef] [PubMed]
- Palaniyandi, S.S.; Qi, X.; Yogalingam, G.; Ferreira, J.C.; Mochly-Rosen, D. Regulation of mitochondrial processes: A target for heart failure. Drug Discov. Today Dis. Mech. 2010, 7, e95–e102. [Google Scholar] [CrossRef]
- Mazurek, S.R.; Bovo, E.; Zima, A.V. Regulation of sarcoplasmic reticulum Ca2+ release by cytosolic glutathione in rabbit ventricular myocytes. Free Radic. Biol. Med. 2014, 68, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Shan, J.; Betzenhauser, M.J.; Kushnir, A.; Reiken, S.; Meli, A.C.; Wronska, A.; Dura, M.; Chen, B.; Marks, A.R. Role of chronic ryanodine receptor phosphorylation in heart failure and beta-adrenergic receptor blockade in mice. J. Clin. Investig. 2010, 120, 4375–4387. [Google Scholar] [CrossRef] [PubMed]
- Goonasekera, S.A.; Hammer, K.; Auger-Messier, M.; Bodi, I.; Chen, X.; Zhang, H.; Reiken, S.; Elrod, J.W.; Correll, R.N.; York, A.J.; et al. Decreased cardiac L-type Ca2+ channel activity induces hypertrophy and heart failure in mice. J. Clin. Investig. 2012, 122, 280–290. [Google Scholar] [CrossRef]
- Zalk, R.; Clarke, O.B.; des Georges, A.; Grassucci, R.A.; Reiken, S.; Mancia, F.; Hendrickson, W.A.; Frank, J.; Marks, A.R. Structure of a mammalian ryanodine receptor. Nature 2015, 517, 44–49. [Google Scholar] [CrossRef]
- Efremov, R.G.; Leitner, A.; Aebersold, R.; Raunser, S. Architecture and conformational switch mechanism of the ryanodine receptor. Nature 2015, 517, 39–43. [Google Scholar] [CrossRef]
- Yan, Z.; Bai, X.; Yan, C.; Wu, J.; Li, Z.; Xie, T.; Peng, W.; Yin, C.-C.; Li, X.; Scheres, S.H.W.; et al. Structure of the rabbit ryanodine receptor RyR1 at near-atomic resolution. Nature 2015, 517, 50–55. [Google Scholar] [CrossRef]
- des Georges, A.; Clarke, O.B.; Zalk, R.; Yuan, Q.; Condon, K.J.; Grassucci, R.A.; Hendrickson, W.A.; Marks, A.R.; Frank, J. Structural Basis for Gating and Activation of RyR1. Cell 2016, 167, 145–157.e17. [Google Scholar] [CrossRef]
- Voss, A.A.; Lango, J.; Ernst-Russell, M.; Morin, D.; Pessah, I.N. Identification of hyperreactive cysteines within ryanodine receptor type 1 by mass spectrometry. J. Biol. Chem. 2004, 279, 34514–34520. [Google Scholar] [CrossRef]
- Aracena-Parks, P.; Goonasekera, S.A.; Gilman, C.P.; Dirksen, R.T.; Hidalgo, C.; Hamilton, S.L. Identification of cysteines involved in S-nitrosylation, S-glutathionylation, and oxidation to disulfides in ryanodine receptor type 1. J. Biol. Chem. 2006, 281, 40354–40368. [Google Scholar] [CrossRef]
- Mi, T.; Xiao, Z.; Guo, W.; Tang, Y.; Hiess, F.; Xiao, J.; Wang, Y.; Zhang, J.Z.; Zhang, L.; Wang, R.; et al. Role of Cys3602 in the function and regulation of the cardiac ryanodine receptor. Biochem. J. 2015, 467, 177–190. [Google Scholar] [CrossRef] [PubMed]
- Nikolaienko, R.; Bovo, E.; Rebbeck, R.T.; Kahn, D.; Thomas, D.D.; Cornea, R.L.; Zima, A.V. The functional significance of redox-mediated intersubunit cross-linking in regulation of human type 2 ryanodine receptor. Redox. Biol. 2020, 37, 101729. [Google Scholar] [CrossRef]
- Melville, Z.; Dridi, H.; Yuan, Q.; Reiken, S.; Wronska, A.; Liu, Y.; Clarke, O.; Marks, A. A drug and ATP binding site in type 1 ryanodine receptor. Structure 2022, 30, 1025–1034.e4. [Google Scholar] [CrossRef]
- Miotto, M.C.; Weninger, G.; Dridi, H.; Yuan, Q.; Liu, Y.; Wronska, A.; Melville, Z.; Sittenfeld, L.; Reiken, S.; Marks, A.R. Structural analyses of human ryanodine receptor type 2 channels reveal the mechanisms for sudden cardiac death and treatment. Sci. Adv. 2022, 8, eabo1272. [Google Scholar] [CrossRef]
- Priori, S.G.; Chen, S.R. Inherited dysfunction of sarcoplasmic reticulum Ca2+ handling and arrhythmogenesis. Circ. Res. 2011, 108, 871–883. [Google Scholar] [CrossRef] [PubMed]
- Iyer, K.A.; Hu, Y.; Nayak, A.R.; Kurebayashi, N.; Murayama, T.; Samso, M. Structural mechanism of two gain-of-function cardiac and skeletal RyR mutations at an equivalent site by cryo-EM. Sci. Adv. 2020, 6, eabb2964. [Google Scholar] [CrossRef] [PubMed]
- Zhong, X.; Liu, Y.; Zhu, L.; Meng, X.; Wang, R.; Van Petegem, F.; Wagenknecht, T.; Chen, S.W.; Liu, Z. Conformational dynamics inside amino-terminal disease hotspot of ryanodine receptor. Structure 2013, 21, 2051–2060. [Google Scholar] [CrossRef] [PubMed]
- Lamb, G.D.; Posterino, G.S.; Yamamoto, T.; Ikemoto, N. Effects of a domain peptide of the ryanodine receptor on Ca2+ release in skinned skeletal muscle fibers. Am. J. Physiol. Cell Physiol. 2001, 281, C207–C214. [Google Scholar] [CrossRef]
- Bannister, M.L.; Hamada, T.; Murayama, T.; Harvey, P.J.; Casarotto, M.G.; Dulhunty, A.F.; Ikemoto, N. Malignant hyperthermia mutation sites in the Leu2442-Pro2477 (DP4) region of RyR1 (ryanodine receptor 1) are clustered in a structurally and functionally definable area. Biochem. J. 2007, 401, 333–339. [Google Scholar] [CrossRef]
- Nikolaienko, R.; Bovo, E.; Kahn, D.; Gracia, R.; Jamrozik, T.; Zima, A.V. Cysteines 1078 and 2991 cross-linking plays a critical role in redox regulation of cardiac ryanodine receptor (RyR). Nat. Commun. 2023, 14, 4498. [Google Scholar] [CrossRef] [PubMed]
- Bovo, E.; Mazurek, S.R.; Zima, A.V. The role of RyR2 oxidation in the blunted frequency-dependent facilitation of Ca2+ transient amplitude in rabbit failing myocytes. Pflugers Arch. 2018, 470, 959–968. [Google Scholar] [CrossRef]
- Walker, M.A.; Williams, G.S.B.; Kohl, T.; Lehnart, S.E.; Jafri, M.S.; Greenstein, J.L.; Lederer, W.; Winslow, R.L. Superresolution modeling of calcium release in the heart. Biophys. J. 2014, 107, 3018–3029. [Google Scholar] [CrossRef] [PubMed]
- Cabra, V.; Murayama, T.; Samso, M. Ultrastructural Analysis of Self-Associated RyR2s. Biophys. J. 2016, 110, 2651–2662. [Google Scholar] [CrossRef] [PubMed]
- Marx, S.O.; Ondrias, K.; Marks, A.R. Coupled gating between individual skeletal muscle Ca2+ release channels (ryanodine receptors). Science 1998, 281, 818–821. [Google Scholar] [CrossRef]
- Marx, S.O.; Gaburjakova, J.; Gaburjakova, M.; Henrikson, C.; Ondrias, K.; Marks, A.R. Coupled gating between cardiac calcium release channels (ryanodine receptors). Circ. Res. 2001, 88, 1151–1158. [Google Scholar] [CrossRef]
- Currie, S. Cardiac ryanodine receptor phosphorylation by CaM Kinase II: Keeping the balance right. Front. Biosci. 2009, 14, 5134–5156. [Google Scholar] [CrossRef]
- Campbell, H.M.; Quick, A.P.; Abu-Taha, I.; Chiang, D.Y.; Kramm, C.F.; Word, T.A.; Brandenburg, S.; Hulsurkar, M.; Alsina, K.M.; Liu, H.-B.; et al. Loss of SPEG Inhibitory Phosphorylation of Ryanodine Receptor Type-2 Promotes Atrial Fibrillation. Circulation 2020, 142, 1159–1172. [Google Scholar] [CrossRef]
- Eiringhaus, J.; Herting, J.; Schatter, F.; Nikolaev, V.O.; Sprenger, J.; Wang, Y.; Köhn, M.; Zabel, M.; El-Armouche, A.; Hasenfuss, G.; et al. Protein kinase/phosphatase balance mediates the effects of increased late sodium current on ventricular calcium cycling. Basic Res. Cardiol. 2019, 114, 13. [Google Scholar] [CrossRef]
- Llach, A.; Mazevet, M.; Mateo, P.; Villejouvert, O.; Ridoux, A.; Rucker-Martin, C.; Ribeiro, M.; Fischmeister, R.; Crozatier, B.; Benitah, J.-P.; et al. Progression of excitation-contraction coupling defects in doxorubicin cardiotoxicity. J. Mol. Cell Cardiol. 2019, 126, 129–139. [Google Scholar] [CrossRef]
- Dries, E.; Santiago, D.J.; Johnson, D.M.; Gilbert, G.; Holemans, P.; Korte, S.M.; Roderick, H.L.; Sipido, K.R. Calcium/calmodulin-dependent kinase II and nitric oxide synthase 1-dependent modulation of ryanodine receptors during beta-adrenergic stimulation is restricted to the dyadic cleft. J. Physiol. 2016, 594, 5923–5939. [Google Scholar] [CrossRef] [PubMed]
- Greiser, M.; Kerfant, B.G.; Williams, G.S.; Voigt, N.; Harks, E.; Dibb, K.M.; Giese, A.; Meszaros, J.; Verheule, S.; Ravens, U.; et al. Tachycardia-induced silencing of subcellular Ca2+ signaling in atrial myocytes. J. Clin. Investig. 2014, 124, 4759–4772. [Google Scholar] [CrossRef]
- Di Carlo, M.N.; Said, M.; Ling, H.; Valverde, C.A.; De Giusti, V.C.; Sommese, L.; Palomeque, J.; Aiello, E.A.; Skapura, D.G.; Rinaldi, G.; et al. CaMKII-dependent phosphorylation of cardiac ryanodine receptors regulates cell death in cardiac ischemia/reperfusion injury. J. Mol. Cell. Cardiol. 2014, 74, 274–283. [Google Scholar] [CrossRef]
- Gambardella, J.; Sorriento, D.; Ciccarelli, M.; Del Giudice, C.; Fiordelisi, A.; Napolitano, L.; Trimarco, B.; Iaccarino, G.; Santulli, G. Functional Role of Mitochondria in Arrhythmogenesis. Adv. Exp. Med. Biol. 2017, 982, 191–202. [Google Scholar] [PubMed]
- Kushnir, A.; Santulli, G.; Reiken, S.R.; Coromilas, E.; Godfrey, S.J.; Brunjes, D.L.; Colombo, P.C.; Yuzefpolskaya, M.; Sokol, S.I.; Kitsis, R.N.; et al. Ryanodine Receptor Calcium Leak in Circulating B-Lymphocytes as a Biomarker in Heart Failure. Circulation 2018, 138, 1144–1154. [Google Scholar] [CrossRef]
- Yuan, Q.; Chen, Z.; Santulli, G.; Gu, L.; Yang, Z.G.; Yuan, Z.Q.; Zhao, Y.-T.; Xin, H.-B.; Deng, K.-Y.; Wang, S.-Q.; et al. Functional role of Calstabin2 in age-related cardiac alterations. Sci. Rep. 2014, 4, 7425. [Google Scholar] [CrossRef]
- Xie, W.; Santulli, G.; Reiken, S.R.; Yuan, Q.; Osborne, B.W.; Chen, B.X.; Marks, A.R. Mitochondrial oxidative stress promotes atrial fibrillation. Sci. Rep. 2015, 5, 11427. [Google Scholar] [CrossRef]
- Brennan, J.P.; Bardswell, S.C.; Burgoyne, J.R.; Fuller, W.; Schroder, E.; Wait, R.; Begum, S.; Kentish, J.C.; Eaton, P. Oxidant-induced activation of type I protein kinase A is mediated by RI subunit interprotein disulfide bond formation. J. Biol. Chem. 2006, 281, 21827–21836. [Google Scholar] [CrossRef] [PubMed]
- Erickson, J.R. Mechanisms of CaMKII Activation in the Heart. Front. Pharmacol. 2014, 5, 59. [Google Scholar] [CrossRef]
- Erickson, J.R.; Joiner, M.L.; Guan, X.; Kutschke, W.; Yang, J.; Oddis, C.V.; Bartlett, R.K.; Lowe, J.S.; O’Donnell, S.E.; Aykin-Burns, N.; et al. A dynamic pathway for calcium-independent activation of CaMKII by methionine oxidation. Cell 2008, 133, 462–474. [Google Scholar] [CrossRef]
- Wu, Y.; Wang, Q.; Feng, N.; Granger, J.M.; Anderson, M.E. Myocardial death and dysfunction after ischemia-reperfusion injury require CaMKIIdelta oxidation. Sci. Rep. 2019, 9, 9291. [Google Scholar] [CrossRef]
- Joiner, M.L.; Koval, O.M.; Li, J.; He, B.J.; Allamargot, C.; Gao, Z.; Luczak, E.D.; Hall, D.D.; Fink, B.D.; Chen, B.; et al. CaMKII determines mitochondrial stress responses in heart. Nature 2012, 491, 269–273. [Google Scholar] [CrossRef] [PubMed]
- Fieni, F.; Johnson, D.E.; Hudmon, A.; Kirichok, Y. Mitochondrial Ca2+ uniporter and CaMKII in heart. Nature 2014, 513, E1–E2. [Google Scholar] [CrossRef]
- Nickel, A.G.; Kohlhaas, M.; Bertero, E.; Wilhelm, D.; Wagner, M.; Sequeira, V.; Kreusser, M.M.; Dewenter, M.; Kappl, R.; Hoth, M.; et al. CaMKII does not control mitochondrial Ca2+ uptake in cardiac myocytes. J. Physiol. 2019, 598, 1361–1376. [Google Scholar] [CrossRef]
- Hulot, J.S.; Salem, J.E.; Redheuil, A.; Collet, J.P.; Varnous, S.; Jourdain, P.; Logeart, D.; Gandjbakhch, E.; Bernard, C.; Hatem, S.N.; et al. Effect of intracoronary administration of AAV1/SERCA2a on ventricular remodelling in patients with advanced systolic heart failure: Results from the AGENT-HF randomized phase 2 trial. Eur. J. Heart Fail. 2017, 19, 1534–1541. [Google Scholar] [CrossRef]
- Escobales, N.; Nunez, R.E.; Jang, S.; Parodi-Rullan, R.; Ayala-Pena, S.; Sacher, J.R.; Skoda, E.M.; Wipf, P.; Frontera, W.; Javadov, S. Mitochondria-targeted ROS scavenger improves post-ischemic recovery of cardiac function and attenuates mitochondrial abnormalities in aged rats. J. Mol. Cell. Cardiol. 2014, 77, 136–146. [Google Scholar] [CrossRef]
- Fink, M.P.; Macias, C.A.; Xiao, J.; Tyurina, Y.Y.; Delude, R.L.; Greenberger, J.S.; Kagan, V.E.; Wipf, P. Hemigramicidin-TEMPO conjugates: Novel mitochondria-targeted antioxidants. Crit. Care Med. 2007, 35 (Suppl. S9), S461–S467. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, P.; Vassanelli, S.; Veronese, P.; Colonna, R.; Szabo, I.; Zoratti, M. Modulation of the mitochondrial permeability transition pore. Effect of protons and divalent cations. J. Biol. Chem. 1992, 267, 2934–2939. [Google Scholar] [CrossRef] [PubMed]
- Giorgio, V.; Burchell, V.; Schiavone, M.; Bassot, C.; Minervini, G.; Petronilli, V.; Argenton, F.; Forte, M.; Tosatto, S.; Lippe, G.; et al. Ca2+ binding to F-ATP synthase beta subunit triggers the mitochondrial permeability transition. EMBO Rep. 2017, 18, 1065–1076. [Google Scholar] [CrossRef]
- Halestrap, A.P. The mitochondrial permeability transition: Its molecular mechanism and role in reperfusion injury. Biochem. Soc Symp. 1999, 66, 181–203. [Google Scholar]
- Crompton, M. The mitochondrial permeability transition pore and its role in cell death. Biochem. J. 1999, 341 Pt 2, 233–249. [Google Scholar] [CrossRef] [PubMed]
- Scarabelli, T.M.; Stephanou, A.; Pasini, E.; Gitti, G.; Townsend, P.; Lawrence, K.; Chen-Scarabelli, C.; Saravolatz, L.; Latchman, D.; Knight, R.; et al. Minocycline inhibits caspase activation and reactivation, increases the ratio of XIAP to smac/DIABLO, and reduces the mitochondrial leakage of cytochrome C and smac/DIABLO. J. Am. Coll. Cardiol. 2004, 43, 865–874. [Google Scholar] [CrossRef]
- Li, J.; Li, Y.; Qin, D.; von Harsdorf, R.; Li, P. Mitochondrial fission leads to Smac/DIABLO release quenched by ARC. Apoptosis 2010, 15, 1187–1196. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.A.; Perry, J.B.; Allen, M.E.; Sabbah, H.N.; Stauffer, B.L.; Shaikh, S.R.; Cleland, J.G.; Colucci, W.S.; Butler, J.; Voors, A.A.; et al. Expert consensus document: Mitochondrial function as a therapeutic target in heart failure. Nat. Rev. Cardiol. 2017, 14, 238–250. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.A.; Hartley, R.C.; Cocheme, H.M.; Murphy, M.P. Mitochondrial pharmacology. Trends Pharmacol. Sci. 2012, 33, 341–352. [Google Scholar] [CrossRef] [PubMed]
- Kloner, R.A.; Brown, D.A.; Csete, M.; Dai, W.; Downey, J.M.; Gottlieb, R.A.; Hale, S.L.; Shi, J. New and revisited approaches to preserving the reperfused myocardium. Nat. Rev. Cardiol. 2017, 14, 679–693. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.D.; Li, J.; Tweedie, D.; Yu, H.M.; Chen, L.; Wang, R.; Riordon, D.R.; Brugh, S.A.; Wang, S.; Boheler, K.R.; et al. Crucial role of the sarcoplasmic reticulum in the developmental regulation of Ca2+ transients and contraction in cardiomyocytes derived from embryonic stem cells. FASEB J. 2006, 20, 181–183. [Google Scholar] [CrossRef]
- Sharma, V.K.; Ramesh, V.; Franzini-Armstrong, C.; Sheu, S.S. Transport of Ca2+ from sarcoplasmic reticulum to mitochondria in rat ventricular myocytes. J. Bioenergy Biomembr. 2000, 32, 97–104. [Google Scholar] [CrossRef]
- Rimessi, A.; Pozzato, C.; Carparelli, L.; Rossi, A.; Ranucci, S.; De Fino, I.; Cigana, C.; Talarico, A.; Wieckowski, M.R.; Ribeiro, C.M.P.; et al. Pharmacological modulation of mitochondrial calcium uniporter controls lung inflammation in cystic fibrosis. Sci. Adv. 2020, 6, eaax9093. [Google Scholar] [CrossRef]
- Yano, M.; Kobayashi, S.; Kohno, M.; Doi, M.; Tokuhisa, T.; Okuda, S.; Suetsugu, M.; Hisaoka, T.; Obayashi, M.; Ohkusa, T.; et al. FKBP12.6-mediated stabilization of calcium-release channel (ryanodine receptor) as a novel therapeutic strategy against heart failure. Circulation 2003, 107, 477–484. [Google Scholar] [CrossRef] [PubMed]
- Wehrens, X.H.; Lehnart, S.E.; Reiken, S.; van der Nagel, R.; Morales, R.; Sun, J.; Cheng, Z.; Deng, S.-X.; de Windt, L.J.; Landry, D.W.; et al. Enhancing calstabin binding to ryanodine receptors improves cardiac and skeletal muscle function in heart failure. Proc. Natl. Acad. Sci. USA 2005, 102, 9607–9612. [Google Scholar] [CrossRef] [PubMed]
- Dai, D.F.; Chen, T.; Szeto, H.; Nieves-Cintron, M.; Kutyavin, V.; Santana, L.F.; Rabinovitch, P.S. Mitochondrial targeted antioxidant Peptide ameliorates hypertensive cardiomyopathy. J. Am. Coll. Cardiol. 2011, 58, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Rohani, L.; Machiraju, P.; Sabouny, R.; Meng, G.; Liu, S.; Zhao, T.; Iqbal, F.; Wang, X.; Ravandi, A.; Wu, J.C.; et al. Reversible Mitochondrial Fragmentation in iPSC-Derived Cardiomyocytes from Children with DCMA, a Mitochondrial Cardiomyopathy. Can. J. Cardiol. 2020, 36, 554–563. [Google Scholar] [CrossRef]
- Dai, D.F.; Hsieh, E.J.; Chen, T.; Menendez, L.G.; Basisty, N.B.; Tsai, L.; Beyer, R.P.; Crispin, D.A.; Shulman, N.J.; Szeto, H.H.; et al. Global proteomics and pathway analysis of pressure-overload-induced heart failure and its attenuation by mitochondrial-targeted peptides. Circ. Heart Fail. 2013, 6, 1067–1076. [Google Scholar] [CrossRef]
- Ghionzoli, N.; Gentile, F.; Del Franco, A.M.; Castiglione, V.; Aimo, A.; Giannoni, A.; Burchielli, S.; Cameli, M.; Emdin, M.; Vergaro, G. Current and emerging drug targets in heart failure treatment. Heart Fail. Rev. 2022, 27, 1119–1136. [Google Scholar] [CrossRef]
- Daubert, M.A.; Yow, E.; Dunn, G.; Marchev, S.; Barnhart, H.; Douglas, P.S.; O’Connor, C.; Goldstein, S.; Udelson, J.E.; Sabbah, H.N. Novel Mitochondria-Targeting Peptide in Heart Failure Treatment: A Randomized, Placebo-Controlled Trial of Elamipretide. Circ. Heart Fail. 2017, 10, e004389. [Google Scholar] [CrossRef] [PubMed]
- Gibson, C.M.; Giugliano, R.P.; Kloner, R.A.; Bode, C.; Tendera, M.; Janosi, A.; Merkely, B.; Godlewski, J.; Halaby, R.; Korjian, S.; et al. EMBRACE STEMI study: A Phase 2a trial to evaluate the safety, tolerability, and efficacy of intravenous MTP-131 on reperfusion injury in patients undergoing primary percutaneous coronary intervention. Eur. Heart J. 2016, 37, 1296–1303. [Google Scholar] [CrossRef]
- Cerrato, C.P.; Pirisinu, M.; Vlachos, E.N.; Langel, U. Novel cell-penetrating peptide targeting mitochondria. FASEB J. 2015, 29, 4589–4599. [Google Scholar] [CrossRef]
- Cerrato, C.P.; Langel, U. Effect of a Fusion Peptide by Covalent Conjugation of a Mitochondrial Cell-Penetrating Peptide and a Glutathione Analog Peptide. Mol. Ther. Methods Clin. Dev. 2017, 5, 221–231. [Google Scholar] [CrossRef] [PubMed]
- Zsebo, K.; Yaroshinsky, A.; Rudy, J.J.; Wagner, K.; Greenberg, B.; Jessup, M.; Hajjar, R.J. Long-term effects of AAV1/SERCA2a gene transfer in patients with severe heart failure: Analysis of recurrent cardiovascular events and mortality. Circ. Res. 2014, 114, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.H.; Budas, G.R.; Churchill, E.N.; Disatnik, M.H.; Hurley, T.D.; Mochly-Rosen, D. Activation of aldehyde dehydrogenase-2 reduces ischemic damage to the heart. Science 2008, 321, 1493–1495. [Google Scholar] [CrossRef] [PubMed]
- Gomes, K.M.; Campos, J.C.; Bechara, L.R.; Queliconi, B.; Lima, V.M.; Disatnik, M.H.; Magno, P.; Chen, C.-H.; Brum, P.C.; Kowaltowski, A.J.; et al. Aldehyde dehydrogenase 2 activation in heart failure restores mitochondrial function and improves ventricular function and remodelling. Cardiovasc. Res. 2014, 103, 498–508. [Google Scholar] [CrossRef]
- Gomes, K.M.; Bechara, L.R.; Lima, V.M.; Ribeiro, M.A.; Campos, J.C.; Dourado, P.M.; Kowaltowski, A.J.; Mochly-Rosen, D.; Ferreira, J.C. Aldehydic load and aldehyde dehydrogenase 2 profile during the progression of post-myocardial infarction cardiomyopathy: Benefits of Alda-1. Int. J. Cardiol. 2015, 179, 129–138. [Google Scholar] [CrossRef]
- Luo, X.J.; Liu, B.; Ma, Q.L.; Peng, J. Mitochondrial aldehyde dehydrogenase, a potential drug target for protection of heart and brain from ischemia/reperfusion injury. Curr. Drug Targets 2014, 15, 948–955. [Google Scholar] [CrossRef]
- Piot, C.; Croisille, P.; Staat, P.; Thibault, H.; Rioufol, G.; Mewton, N.; Elbelghiti, R.; Cung, T.T.; Bonnefoy, E.; Angoulvant, D.; et al. Effect of cyclosporine on reperfusion injury in acute myocardial infarction. N. Engl. J. Med. 2008, 359, 473–481. [Google Scholar] [CrossRef] [PubMed]
- Ottani, F.; Latini, R.; Staszewsky, L.; La Vecchia, L.; Locuratolo, N.; Sicuro, M.; Masson, S.; Barlera, S.; Milani, V.; Lombardi, M.; et al. Cyclosporine A in Reperfused Myocardial Infarction: The Multicenter, Controlled, Open-Label CYCLE Trial. J. Am. Coll. Cardiol. 2016, 67, 365–374. [Google Scholar] [CrossRef]
- Mewton, N.; Croisille, P.; Gahide, G.; Rioufol, G.; Bonnefoy, E.; Sanchez, I.; Cung, T.T.; Sportouch, C.; Angoulvant, D.; Finet, G.; et al. Effect of cyclosporine on left ventricular remodeling after reperfused myocardial infarction. J. Am. Coll. Cardiol. 2010, 55, 1200–1205. [Google Scholar] [CrossRef]
- Kawakami, S.; Matsuda, A.; Sunagawa, T.; Noda, Y.; Kaneko, T.; Tahara, S.; Hiraumi, Y.; Adachi, S.; Matsui, H.; Ando, K.; et al. Antioxidant, EUK-8, prevents murine dilated cardiomyopathy. Circ. J. 2009, 73, 2125–2134. [Google Scholar] [CrossRef]
- van Empel, V.P.; Bertrand, A.T.; van Oort, R.J.; van der Nagel, R.; Engelen, M.; van Rijen, H.V.; Doevendans, P.A.; Crijns, H.J.; Ackerman, S.L.; Sluiter, W.; et al. EUK-8, a superoxide dismutase and catalase mimetic, reduces cardiac oxidative stress and ameliorates pressure overload-induced heart failure in the harlequin mouse mutant. J. Am. Coll. Cardiol. 2006, 48, 824–832. [Google Scholar] [CrossRef]
- Lerman-Sagie, T.; Rustin, P.; Lev, D.; Yanoov, M.; Leshinsky-Silver, E.; Sagie, A.; Ben-Gal, T.; Munnich, A. Dramatic improvement in mitochondrial cardiomyopathy following treatment with idebenone. J. Inherit. Metab. Dis. 2001, 24, 28–34. [Google Scholar] [CrossRef]
- Salvemini, D.; Wang, Z.Q.; Zweier, J.L.; Samouilov, A.; Macarthur, H.; Misko, T.P.; Currie, M.G.; Cuzzocrea, S.; Sikorski, J.A.; Riley, D.P.; et al. A nonpeptidyl mimic of superoxide dismutase with therapeutic activity in rats. Science 1999, 286, 304–306. [Google Scholar] [CrossRef]
- Masini, E.; Cuzzocrea, S.; Mazzon, E.; Marzocca, C.; Mannaioni, P.F.; Salvemini, D. Protective effects of M40403, a selective superoxide dismutase mimetic, in myocardial ischaemia and reperfusion injury in vivo. Br. J. Pharmacol. 2002, 136, 905–917. [Google Scholar] [CrossRef]
- Ni, R.; Cao, T.; Xiong, S.; Ma, J.; Fan, G.C.; Lacefield, J.C.; Lu, Y.; Le Tissier, S.; Peng, T. Therapeutic inhibition of mitochondrial reactive oxygen species with mito-TEMPO reduces diabetic cardiomyopathy. Free Radic. Biol. Med. 2016, 90, 12–23. [Google Scholar] [CrossRef] [PubMed]
- Dikalova, A.E.; Bikineyeva, A.T.; Budzyn, K.; Nazarewicz, R.R.; McCann, L.; Lewis, W.; Harrison, D.G.; Dikalova, A. Therapeutic targeting of mitochondrial superoxide in hypertension. Circ. Res. 2010, 107, 106–116. [Google Scholar] [CrossRef] [PubMed]
- Dikalova, A.E.; Kirilyuk, I.A.; Dikalov, S.I. Antihypertensive effect of mitochondria-targeted proxyl nitroxides. Redox. Biol. 2015, 4, 355–362. [Google Scholar] [CrossRef] [PubMed]
- Tsujita, K.; Shimomura, H.; Kawano, H.; Hokamaki, J.; Fukuda, M.; Yamashita, T.; Hida, S.; Nakamura, Y.; Nagayoshi, Y.; Sakamoto, T.; et al. Effects of edaravone on reperfusion injury in patients with acute myocardial infarction. Am. J. Cardiol. 2004, 94, 481–484. [Google Scholar] [CrossRef]
- Edaravone Acute Infarction Study Group. Effect of a novel free radical scavenger, edaravone (MCI-186), on acute brain infarction. Randomized, placebo-controlled, double-blind study at multicenters. Cerebrovasc. Dis. 2003, 15, 222–229. [Google Scholar] [CrossRef]
- Junior, R.F.R.; Dabkowski, E.R.; Shekar, K.C.; O’Connell, K.A.; Hecker, P.A.; Murphy, M.P. MitoQ improves mitochondrial dysfunction in heart failure induced by pressure overload. Free Radic. Biol. Med. 2018, 117, 18–29. [Google Scholar] [CrossRef]
- Adlam, V.J.; Harrison, J.C.; Porteous, C.M.; James, A.M.; Smith, R.A.; Murphy, M.P.; Sammut, I.A. Targeting an antioxidant to mitochondria decreases cardiac ischemia-reperfusion injury. FASEB J. 2005, 19, 1088–1095. [Google Scholar] [CrossRef]
- Yin, M.; van der Horst, I.C.; van Melle, J.P.; Qian, C.; van Gilst, W.H.; Sillje, H.H.; de Boer, R.A. Metformin improves cardiac function in a nondiabetic rat model of post-MI heart failure. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H459–H468. [Google Scholar] [CrossRef]
- Sun, D.; Yang, F. Metformin improves cardiac function in mice with heart failure after myocardial infarction by regulating mitochondrial energy metabolism. Biochem. Biophys. Res. Commun. 2017, 486, 329–335. [Google Scholar] [CrossRef] [PubMed]
- Dridi, H.; Wu, W.; Reiken, S.R.; Ofer, R.M.; Liu, Y.; Yuan, Q.; Sittenfeld, L.; Kushner, J.; Muchir, A.; Worman, H.J.; et al. Ryanodine receptor remodeling in cardiomyopathy and muscular dystrophy caused by lamin A/C gene mutation. Hum. Mol. Genet. 2021, 29, 3919–3934. [Google Scholar] [CrossRef] [PubMed]
- Atar, D.; Arheden, H.; Berdeaux, A.; Bonnet, J.L.; Carlsson, M.; Clemmensen, P.; Cuvier, V.; Danchin, N.; Dubois-Randé, J.-L.; Engblom, H.; et al. Effect of intravenous TRO40303 as an adjunct to primary percutaneous coronary intervention for acute ST-elevation myocardial infarction: MITOCARE study results. Eur. Heart J. 2015, 36, 112–119. [Google Scholar] [CrossRef]
- Schaller, S.; Paradis, S.; Ngoh, G.A.; Assaly, R.; Buisson, B.; Drouot, C.; Ostuni, M.A.; Lacapere, J.J.; Bassissi, F.; Bordet, T.; et al. TRO40303, a new cardioprotective compound, inhibits mitochondrial permeability transition. J. Pharmacol. Exp. Ther. 2010, 333, 696–706. [Google Scholar] [CrossRef] [PubMed]
- Butt, N.; Bache-Mathiesen, L.K.; Nordrehaug, J.E.; Tuseth, V.; Munk, P.S.; Bonarjee, V.; Hall, T.S.; Jensen, S.E.; Halvorsen, S.; Firat, H.; et al. Administration of the Mitochondrial Permeability Transition Pore Inhibitor, TRO40303, prior to Primary Percutaneous Coronary Intervention, Does Not Affect the Levels of Pro-Inflammatory Cytokines or Acute-Phase Proteins. Cardiology 2017, 138, 122–132. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dridi, H.; Santulli, G.; Bahlouli, L.; Miotto, M.C.; Weninger, G.; Marks, A.R. Mitochondrial Calcium Overload Plays a Causal Role in Oxidative Stress in the Failing Heart. Biomolecules 2023, 13, 1409. https://doi.org/10.3390/biom13091409
Dridi H, Santulli G, Bahlouli L, Miotto MC, Weninger G, Marks AR. Mitochondrial Calcium Overload Plays a Causal Role in Oxidative Stress in the Failing Heart. Biomolecules. 2023; 13(9):1409. https://doi.org/10.3390/biom13091409
Chicago/Turabian StyleDridi, Haikel, Gaetano Santulli, Laith Bahlouli, Marco C. Miotto, Gunnar Weninger, and Andrew R. Marks. 2023. "Mitochondrial Calcium Overload Plays a Causal Role in Oxidative Stress in the Failing Heart" Biomolecules 13, no. 9: 1409. https://doi.org/10.3390/biom13091409
APA StyleDridi, H., Santulli, G., Bahlouli, L., Miotto, M. C., Weninger, G., & Marks, A. R. (2023). Mitochondrial Calcium Overload Plays a Causal Role in Oxidative Stress in the Failing Heart. Biomolecules, 13(9), 1409. https://doi.org/10.3390/biom13091409