

Recent Advances in Metalloproteomics

 , ,
, ,  and
and
Abstract
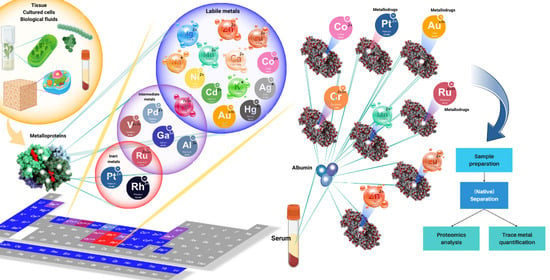
:
1. Introduction
2. Labile Complexes
2.1. General Considerations
2.2. Separation Techniques to Analyse Complex Metalloprotein Mixtures
2.3. Matching Metals with Proteins
2.4. Alternative Chemoproteomic Approaches
2.5. Selected Examples for Metalloproteomic Speciation Studies on Labile Metals in Complex Mixtures
2.5.1. Prokaryotic Microbes
2.5.2. Plants
2.5.3. Blood Plasma/Serum
2.5.4. Other Biofluids
2.5.5. Animal Cells and Tissues
2.5.6. MRI Contrast Agents and Related Lanthanide Compounds
3. Inert Complexes
3.1. Platinum Complexes
3.2. Ruthenium Complexes
4. Gold Complexes
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 2D-GE | 2-Dimensional Gel Electrophoresis |
| Ac | Acetate |
| AEX | Anion Exchange Chromatography |
| BSA | Bovine Serum Albumin |
| CE | Capillary Electrophoresis |
| CFS | Cerebrospinal fluid |
| DDM | n-Dodecyl-β-D-maltoside |
| ESI-MS | Electrospray Ionisation Mass Spectrometry |
| EDTA | 2,2′,2′′,2′′′-(Ethane-1,2-diyldinitrilo)tetraacetic acid |
| FBS/FCS | Fetal Bovine Serum/Fetal Calf Serum |
| FPLC | Fast Protein Liquid Chromatography |
| GE | Gel Electrophoresis |
| HEPES | 2-[4-(2-Hydroxyethyl)piperazin-1-yl]ethane-1-sulfonic acid |
| HMW | High-molecular-weight |
| HPLC | High-Performance Liquid Chromatography |
| HSA | Human Serum Albumin |
| ICP-OES | Inductively-Coupled Plasma Optical Emission Spectroscopy |
| ICP-MS | Inductively-Coupled Plasma Mass Spectrometry |
| IDA | Isotope Dilution Analysis |
| IDMS | Isotope Dilution Mass Spectrometry |
| IEF | Isolectric Focusing |
| LA | Laser Ablation |
| LMW | Low-molecular-weight |
| MALDI | Matrix-Assisted Laser Desorption Ionisation |
| MOPS | 3-(Morpholin-4-yl)propane-1-sulfonic acid |
| PAGE | Polyacrylamide Gel Electrophoresis |
| PBS | Phosphate-Buffered Saline |
| PEEK | Polyether ether ketone |
| SAX | Strong Anion Exchange |
| SDS | Sodium dodecyl sulfate |
| SEC | Size-Exclusion Chromatography |
| TCEP | 3,3′,3′′-Phosphanetriyltripropanoic acid |
| Tris | 2-Amino-2-(hydroxymethyl)propane-1,3-diol |
| TPEN | N1,N1,N2,N2-Tetrakis[(pyridin-2-yl)methyl]ethane-1,2-diamine |
References
- Valasatava, Y.; Rosato, A.; Furnham, N.; Thornton, J.M.; Andreini, C. To What Extent Do Structural Changes in Catalytic Metal Sites Affect Enzyme Function? J. Inorg. Biochem. 2018, 179, 40–53. [Google Scholar] [CrossRef] [PubMed]
- Freisinger, E.; Sigel, R.K.O. From Nucleotides to Ribozymes—A Comparison of Their Metal Ion Binding Properties. Coord. Chem. Rev. 2007, 251, 1834–1851. [Google Scholar] [CrossRef]
- Krzywoszyńska, K.; Witkowska, D.; Swiatek-Kozlowska, J.; Szebesczyk, A.; Kozłowski, H. General Aspects of Metal Ions as Signaling Agents in Health and Disease. Biomolecules 2020, 10, 1417. [Google Scholar] [CrossRef] [PubMed]
- Anthony, E.J.; Bolitho, E.M.; Bridgewater, H.E.; Carter, O.W.L.; Donnelly, J.M.; Imberti, C.; Lant, E.C.; Lermyte, F.; Needham, R.J.; Palau, M.; et al. Metallodrugs Are Unique: Opportunities and Challenges of Discovery and Development. Chem. Sci. 2020, 11, 12888–12917. [Google Scholar] [CrossRef] [PubMed]
- Peana, M.; Pelucelli, A.; Medici, S.; Cappai, R.; Nurchi, V.M.; Zoroddu, M.A. Metal Toxicity and Speciation: A Review. Curr. Med. Chem. 2021, 28, 7190–7208. [Google Scholar] [CrossRef] [PubMed]
- Barnett, J.P.; Scanlan, D.J.; Blindauer, C.A. Protein Fractionation and Detection for Metalloproteomics: Challenges and Approaches. Anal. Bioanal. Chem. 2012, 402, 3311–3322. [Google Scholar] [CrossRef] [PubMed]
- Fu, D.; Finney, L. Metalloproteomics: Challenges and Prospective for Clinical Research Applications. Expert Rev. Proteom. 2014, 11, 13–19. [Google Scholar] [CrossRef]
- Zeng, X.; Cheng, Y.; Wang, C. Global Mapping of Metalloproteomes. Biochemistry 2021, 60, 3507–3514. [Google Scholar] [CrossRef]
- Roberts, E.A.; Sarkar, B. Metalloproteomics: Focus on Metabolic Issues Relating to Metals. Curr. Opin. Clin. Nutr. Metab. Care 2014, 17, 425–430. [Google Scholar] [CrossRef]
- Blindauer, C.A. Chapter 10—Metalloproteomics. In Clinical and Translational Perspectives on Wilson Disease; Kerkar, N., Roberts, E.A., Eds.; Academic Press: Cambridge, MA, USA, 2019; pp. 85–100. [Google Scholar]
- Xu, X.H.; Wang, H.B.; Li, H.Y.; Sun, H.Z. Metalloproteomic Approaches for Matching Metals to Proteins: The Power of Inductively Coupled Plasma Mass Spectrometry (ICP-MS). Chem. Lett. 2020, 49, 697–704. [Google Scholar] [CrossRef]
- Lothian, A.; Hare, D.J.; Grimm, R.; Ryan, T.M.; Masters, C.L.; Roberts, B.R. Metalloproteomics: Principles, Challenges and Applications to Neurodegeneration. Front. Aging Neurosci. 2013, 5, 35. [Google Scholar] [CrossRef] [PubMed]
- Hauser-Davis, R.A.; Lopes, R.M.; Mota, F.B.; Moreira, J.C. The Evolution of Environmental Metalloproteomics over the Last 15 Years through Bibliometric Techniques. Ecotoxicol. Environ. Saf. 2017, 140, 279–287. [Google Scholar] [CrossRef]
- da Silva, M.A.; Sussulini, A.; Arruda, M.A. Metalloproteomics as an Interdisciplinary Area Involving Proteins and Metals. Expert Rev. Proteom. 2010, 7, 387–400. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Li, H.; Sun, H. Metalloproteomics for Biomedical Research: Methodology and Applications. Annu. Rev. Biochem. 2022, 91, 449–473. [Google Scholar] [CrossRef] [PubMed]
- Yannone, S.M.; Hartung, S.; Menon, A.L.; Adams, M.W.W.; Tainer, J.A. Metals in Biology: Defining Metalloproteomes. Curr. Opin. Biotechnol. 2012, 23, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Maret, W. Metallomics: The Science of Biometals and Biometalloids. Adv. Exp. Med. Biol. 2018, 1055, 1–20. [Google Scholar] [CrossRef]
- Maret, W.; Caruso, J.A.; Contag, C.H.; Giedroc, D.P.; Hagedoorn, P.L.; Matusch, A.; Skaar, E.P.; Thompson, R.B. Methods and Technologies for Studying Metals in Biological Systems. In Trace Metals and Infectious Diseases; Nriagu, J.O., Skaar, E.P., Eds.; MIT Press © 2015 Massachusetts Institute of Technology and the Frankfurt Institute for Advanced Studies: Cambridge, MA, USA, 2015. [Google Scholar]
- Arruda, M.A.Z. (Ed.) Metallomics: The Science of Biometals, 1st ed.; Springer International Publishing AG: Cham, Switzerland, 2018; p. 279. [Google Scholar]
- Montes-Bayon, M.; Sharar, M.; Corte-Rodriguez, M. Trends on (Elemental and Molecular) Mass Spectrometry Based Strategies for Speciation and Metallomics. Trends Anal. Chem. 2018, 104, 4–10. [Google Scholar] [CrossRef]
- Lobinski, R.; Moulin, C.; Ortega, R. Imaging and Speciation of Trace Elements in Biological Environment. Biochimie 2006, 88, 1591–1604. [Google Scholar] [CrossRef]
- de Jesus, J.R.; da Costa, L.F.; Lehmann, E.L.; Galazzi, R.M.; Madrid, K.C.; Arruda, M.A.Z. Chemical Speciation and Metallomics. Adv. Exp. Med. Biol. 2018, 1055, 183–211. [Google Scholar] [CrossRef]
- Arruda, M.A.Z.; de Jesus, J.R.; Blindauer, C.A.; Stewart, A.J. Speciomics as a Concept Involving Chemical Speciation and Omics. J. Proteom. 2022, 263, 104615. [Google Scholar] [CrossRef]
- Shi, W.; Chance, M.R. Metalloproteomics: Forward and Reverse Approaches in Metalloprotein Structural and Functional Characterization. Curr. Opin. Chem. Biol. 2011, 15, 144–148. [Google Scholar] [CrossRef] [PubMed]
- Michalke, B. Review About Powerful Combinations of Advanced and Hyphenated Sample Introduction Techniques with Inductively Coupled Plasma-Mass Spectrometry (ICP-MS) for Elucidating Trace Element Species in Pathologic Conditions on a Molecular Level. Int. J. Mol. Sci. 2022, 23, 6109. [Google Scholar] [CrossRef] [PubMed]
- Bishop, D.P.; Hare, D.J.; Clases, D.; Doble, P.A. Applications of Liquid Chromatography-Inductively Coupled Plasma-Mass Spectrometry in the Biosciences: A Tutorial Review and Recent Developments. Trends Anal. Chem. 2018, 104, 11–21. [Google Scholar] [CrossRef]
- Clases, D.; de Vega, R.G. Facets of ICP-MS and Their Potential in the Medical Sciences-Part 1: Fundamentals, Stand-Alone and Hyphenated Techniques. Anal. Bioanal. Chem. 2022, 414, 7337–7361. [Google Scholar] [CrossRef] [PubMed]
- Hagege, A.; Huynh, T.N.S.; Hebrant, M. Separative Techniques for Metalloproteomics Require Balance between Separation and Perturbation. TrAC Trends Anal. Chem. 2015, 64, 64–74. [Google Scholar] [CrossRef]
- Smith, S.D.; She, Y.M.; Roberts, E.A.; Sarkar, B. Using Immobilized Metal Affinity Chromatography, Two-Dimensional Electrophoresis and Mass Spectrometry to Identify Hepatocellular Proteins with Copper-Binding Ability. J. Proteome Res. 2004, 3, 834–840. [Google Scholar] [CrossRef] [PubMed]
- Bettmer, J. Metalloproteomics: A Challenge for Analytical Chemists. Anal. Bioanal. Chem. 2005, 383, 370–371. [Google Scholar] [CrossRef] [PubMed]
- Prange, A.; Pröfrock, D. Application of CE-ICP-MS and CE-ESI-MS in Metalloproteomics: Challenges, Developments, and Limitations. Anal. Bioanal. Chem. 2005, 383, 372–389. [Google Scholar] [CrossRef]
- Szpunar, J. Advances in Analytical Methodology for Bioinorganic Speciation Analysis: Metallomics, Metalloproteomics and Heteroatom-Tagged Proteomics and Metabolomics. Analyst 2005, 130, 442–465. [Google Scholar] [CrossRef]
- Munoz, A.H.S.; Kubachka, K.; Wrobel, K.; Corona, F.G.; Yathavakilla, S.K.V.; Caruso, J.A.; Wrobel, K. Metallomics Approach to Trace Element Analysis in Ustilago maydis Using Cellular Fractionation, Atomic Absorption Spectrometry, and Size Exclusion Chromatography with ICP-MS Detection. J. Agric. Food Chem. 2005, 53, 5138–5143. [Google Scholar] [CrossRef]
- Lincoln, S.F. Mechanistic Studies of Metal Aqua Ions: A Semi-Historical Perspective. Helv. Chim. Acta 2005, 88, 523–545. [Google Scholar] [CrossRef]
- Andreini, C.; Bertini, I.; Rosato, A. Metalloproteomes: A Bioinformatic Approach. Acc. Chem. Res. 2009, 42, 1471–1479. [Google Scholar] [CrossRef]
- Wehrspan, Z.J.; McDonnell, R.T.; Elcock, A.H. Identification of Iron-Sulfur (Fe-S) Cluster and Zinc (Zn) Binding Sites within Proteomes Predicted by Deepmind’s Alphafold2 Program Dramatically Expands the Metalloproteome. J. Mol. Biol. 2022, 434, 167377. [Google Scholar] [CrossRef] [PubMed]
- Foster, A.W.; Young, T.R.; Chivers, P.T.; Robinson, N.J. Protein Metalation in Biology. Curr. Opin. Chem. Biol. 2022, 66, 102095. [Google Scholar] [CrossRef] [PubMed]
- Cvetkovic, A.; Menon, A.L.; Thorgersen, M.P.; Scott, J.W.; Poole, F.L., 2nd; Jenney, F.E., Jr.; Lancaster, W.A.; Praissman, J.L.; Shanmukh, S.; Vaccaro, B.J.; et al. Microbial Metalloproteomes Are Largely Uncharacterized. Nature 2010, 466, 779–782. [Google Scholar] [CrossRef] [PubMed]
- Mazzotta, M.G.; McIlvin, M.R.; Saito, M.A. Characterization of the Fe Metalloproteome of a Ubiquitous Marine Heterotroph, Pseudoalteromonas (Bb2-At2): Multiple Bacterioferritin Copies Enable Significant Fe Storage. Metallomics 2020, 12, 654–667. [Google Scholar] [CrossRef]
- Tottey, S.; Waldron, K.J.; Firbank, S.J.; Reale, B.; Bessant, C.; Sato, K.; Cheek, T.R.; Gray, J.; Banfield, M.J.; Dennison, C.; et al. Protein-Folding Location Can Regulate Manganese-Binding Versus Copper- or Zinc-Binding. Nature 2008, 455, 1138–1142. [Google Scholar] [CrossRef]
- Nyborg, J.K.; Peersen, O.B. That Zincing Feeling: The Effects of EDTA on the Behaviour of Zinc-Binding Transcriptional Regulators. Biochem. J. 2004, 381, e3. [Google Scholar] [CrossRef]
- Hare, D.J.; Grubman, A.; Ryan, T.M.; Lothian, A.; Liddell, J.R.; Grimm, R.; Matsuda, T.; Doble, P.A.; Cherny, R.A.; Bush, A.I.; et al. Profiling the Iron, Copper and Zinc Content in Primary Neuron and Astrocyte Cultures by Rapid Online Quantitative Size Exclusion Chromatography-Inductively Coupled Plasma-Mass Spectrometry. Metallomics 2013, 5, 1656–1662. [Google Scholar] [CrossRef]
- Dupree, E.J.; Jayathirtha, M.; Yorkey, H.; Mihasan, M.; Petre, B.A.; Darie, C.C. A Critical Review of Bottom-up Proteomics: The Good, the Bad, and the Future of This Field. Proteomes 2020, 8, 14. [Google Scholar] [CrossRef]
- Chery, C.C.; Moens, L.; Cornelis, R.; Vanhaecke, F. Capabilities and Limitations of Gel Electrophoresis for Elemental Speciation: A Laboratory’s Experience. Pure Appl. Chem. 2006, 78, 91–103. [Google Scholar] [CrossRef]
- Meyer, S.; Clases, D.; de Vega, R.G.; Padula, M.P.; Doble, P.A. Separation of Intact Proteins by Capillary Electrophoresis. Analyst 2022, 147, 2988–2996. [Google Scholar] [CrossRef] [PubMed]
- Jahromi, E.Z.; White, W.; Wu, Q.; Yamdagni, R.; Gailer, J. Remarkable Effect of Mobile Phase Buffer on the SEC-ICP-AES Derived Cu, Fe and Zn-Metalloproteome Pattern of Rabbit Blood Plasma. Metallomics 2010, 2, 460–468. [Google Scholar] [CrossRef] [PubMed]
- Lago, L.; Thomas, O.R.B.; Roberts, B.R. Choice of Mobile Phase: Implications for Size Exclusion Chromatography-Inductively Coupled Plasma-Mass Spectrometry Analyses of Copper, Zinc and Iron Metalloproteins. J. Chromatogr. A 2020, 1616, 460806. [Google Scholar] [CrossRef] [PubMed]
- Marcus, K.; Lelong, C.; Rabilloud, T. What Room for Two-Dimensional Gel-Based Proteomics in a Shotgun Proteomics World? Proteomes 2020, 8, 17. [Google Scholar] [CrossRef] [PubMed]
- Sussulini, A.; Kratzin, H.; Jahn, O.; Banzato, C.E.M.; Arruda, M.A.Z.; Becker, J.S. Metallomics Studies of Human Blood Serum from Treated Bipolar Disorder Patients. Anal. Chem. 2010, 82, 5859–5864. [Google Scholar] [CrossRef] [PubMed]
- Nowakowski, A.B.; Wobig, W.J.; Petering, D.H. Native SDS-PAGE: High Resolution Electrophoretic Separation of Proteins with Retention of Native Properties Including Bound Metal Ions. Metallomics 2014, 6, 1068–1078. [Google Scholar] [CrossRef]
- Wang, Y.; Hu, L.; Xu, F.; Quan, Q.; Lai, Y.T.; Xia, W.; Yang, Y.; Chang, Y.Y.; Yang, X.; Chai, Z.; et al. Integrative Approach for the Analysis of the Proteome-Wide Response to Bismuth Drugs in Helicobacter pylori. Chem. Sci. 2017, 8, 4626–4633. [Google Scholar] [CrossRef]
- Sun, X.; Xiao, C.L.; Ge, R.; Yin, X.; Li, H.; Li, N.; Yang, X.; Zhu, Y.; He, X.; He, Q.Y. Putative Copper- and Zinc-Binding Motifs in Streptococcus pneumoniae Identified by Immobilized Metal Affinity Chromatography and Mass Spectrometry. Proteomics 2011, 11, 3288–3298. [Google Scholar] [CrossRef]
- Barnett, J.P.; Scanlan, D.J.; Blindauer, C.A. Fractionation and Identification of Metalloproteins from a Marine Cyanobacterium. Anal. Bioanal. Chem. 2012, 402, 3371–3377. [Google Scholar] [CrossRef]
- Ludwig, C.; Gillet, L.; Rosenberger, G.; Amon, S.; Collins, B.C.; Aebersold, R. Data-Independent Acquisition-Based SWATH-MS for Quantitative Proteomics: A Tutorial. Mol. Syst. Biol. 2018, 14, e8126. [Google Scholar] [CrossRef]
- Jayathirtha, M.; Dupree, E.J.; Manzoor, Z.; Larose, B.; Sechrist, Z.; Neagu, A.N.; Petre, B.A.; Darie, C.C. Mass Spectrometric (MS) Analysis of Proteins and Peptides. Curr. Protein Pept. Sci. 2021, 22, 92–120. [Google Scholar] [CrossRef] [PubMed]
- Rotello, R.J.; Veenstra, T.D. Mass Spectrometry Techniques: Principles and Practices for Quantitative Proteomics. Curr. Protein Pept. Sci. 2021, 22, 121–133. [Google Scholar] [CrossRef]
- Sivanich, M.K.; Gu, T.J.; Tabang, D.N.; Li, L. Recent Advances in Isobaric Labeling and Applications in Quantitative Proteomics. Proteomics 2022, 22, e2100256. [Google Scholar] [CrossRef] [PubMed]
- Sarpong-Kumankomah, S.; Knox, K.B.; Kelly, M.E.; Hunter, G.; Popescu, B.; Nichol, H.; Kopciuk, K.; Ntanda, H.; Gailer, J. Quantification of Human Plasma Metalloproteins in Multiple Sclerosis, Ischemic Stroke and Healthy Controls Reveals an Association of Haptoglobin-Hemoglobin Complexes with Age. PLoS ONE 2022, 17, e0262160. [Google Scholar] [CrossRef] [PubMed]
- Miller, K.; Sarpong-Kumankomah, S.; Egorov, A.; Gailer, J. Sample Preparation of Blood Plasma Enables Baseline Separation of Iron Metalloproteins by SEC-GFAAS. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2020, 1147, 122147. [Google Scholar] [CrossRef]
- Sussulini, A.; Becker, J.S.; Becker, J.S. Laser Ablation ICP-MS: Application in Biomedical Research. Mass Spectrom. Rev. 2017, 36, 47–57. [Google Scholar] [CrossRef]
- Raimunda, D.; Khare, T.; Giometti, C.; Vogt, S.; Arguello, J.M.; Finney, L. Identifying Metalloproteins through X-Ray Fluorescence Mapping and Mass Spectrometry. Metallomics 2012, 4, 921–927. [Google Scholar] [CrossRef]
- Verbi, F.M.; Arruda, S.C.C.; Rodriguez, A.P.M.; Perez, C.A.; Arruda, M.A.Z. Metal-Binding Proteins Scanning and Determination by Combining Gel Electrophoresis, Synchrotron Radiation X-Ray Fluorescence and Atomic Spectrometry. J. Biochem. Biophys. Methods 2005, 62, 97–109. [Google Scholar] [CrossRef]
- HommaTakeda, S.; Shinyashiki, M.; Nakai, I.; Tohyama, C.; Kumagai, Y.; Shimojo, N. Direct Detection of Mercury-Bound Metalloproteins (Metallothionein and Cu,Zn-Superoxide Dismutase) Using a Combination of Gel Electrophoresis and One Dimensional Synchrotron Radiation X-Ray Fluorescence Analysis. Anal. Lett. 1996, 29, 601–611. [Google Scholar] [CrossRef]
- Ferrer, M.; Golyshina, O.V.; Beloqui, A.; Golyshin, P.N.; Timmis, K.N. The Cellular Machinery of Ferroplasma acidiphilum Is Iron-Protein-Dominated. Nature 2007, 445, 91–94. [Google Scholar] [CrossRef]
- Sevcenco, A.M.; Hagen, W.R.; Hagedoorn, P.L. Microbial Metalloproteomes Explored Using Mirage. Chem. Biodivers. 2012, 9, 1967–1980. [Google Scholar] [CrossRef]
- Hagedoorn, P.L. Microbial Metalloproteomics. Proteomes 2015, 3, 424–439. [Google Scholar] [CrossRef]
- Sevcenco, A.M.; Pinkse, M.W.; Wolterbeek, H.T.; Verhaert, P.D.; Hagen, W.R.; Hagedoorn, P.L. Exploring the Microbial Metalloproteome Using Mirage. Metallomics 2011, 3, 1324–1330. [Google Scholar] [CrossRef]
- Coverdale, J.P.C.; Harrington, C.F.; Solovyev, N. Review: Advances in the Accuracy and Traceability of Metalloprotein Measurements Using Isotope Dilution Inductively Coupled Plasma Mass Spectrometry. Crit. Rev. Anal. Chem. 2023, 1–18. [Google Scholar] [CrossRef]
- Robinson, N.J.; Waldron, K.; Tottey, S.; Bessant, C. Metalloprotein Metal Pools: Identification and Quantification by Coupling Native and Non-Native Separations through Principal Component Analysis. Protoc. Exch. 2008. [Google Scholar] [CrossRef]
- Lancaster, W.A.; Praissman, J.L.; Poole, F.L., 2nd; Cvetkovic, A.; Menon, A.L.; Scott, J.W.; Jenney, F.E., Jr.; Thorgersen, M.P.; Kalisiak, E.; Apon, J.V.; et al. A Computational Framework for Proteome-Wide Pursuit and Prediction of Metalloproteins Using ICP-MS-Ms and MS/MS data. BMC Bioinf. 2011, 12, 64. [Google Scholar] [CrossRef]
- Mazzotta, M.G.; McIlvin, M.R.; Moran, D.M.; Wang, D.T.; Bidle, K.D.; Lamborg, C.H.; Saito, M.A. Characterization of the Metalloproteome of Pseudoalteromonas (BB2-AT2): Biogeochemical Underpinnings for Zinc, Manganese, Cobalt, and Nickel Cycling in a Ubiquitous Marine Heterotroph. Metallomics 2021, 13, mfab060. [Google Scholar] [CrossRef]
- Meissner, F.; Geddes-McAlister, J.; Mann, M.; Bantscheff, M. The Emerging Role of Mass Spectrometry-Based Proteomics in Drug Discovery. Nat. Rev. Drug Discov. 2022, 21, 637–654. [Google Scholar] [CrossRef] [PubMed]
- Babak, M.V.; Meier, S.M.; Huber, K.V.M.; Reynisson, J.; Legin, A.A.; Jakupec, M.A.; Roller, A.; Stukalov, A.; Gridling, M.; Bennett, K.L.; et al. Target Profiling of an Antimetastatic RAPTA Agent by Chemical Proteomics: Relevance to the Mode of Action. Chem. Sci. 2015, 6, 2449–2456. [Google Scholar] [CrossRef] [PubMed]
- Iacobucci, I.; La Manna, S.; Cipollone, I.; Monaco, V.; Canè, L.; Cozzolino, F. From the Discovery of Targets to Delivery Systems: How to Decipher and Improve the Metallodrugs’ Actions at a Molecular Level. Pharmaceutics 2023, 15, 1997. [Google Scholar] [CrossRef]
- Skos, L.; Borutzki, Y.; Gerner, C.; Meier-Menches, S.M. Methods to Identify Protein Targets of Metal-Based Drugs. Curr. Opin. Chem. Biol. 2023, 73, 102257. [Google Scholar] [CrossRef]
- Fung, S.K.; Zou, T.T.; Cao, B.; Lee, P.Y.; Fung, Y.M.E.; Hu, D.; Lok, C.N.; Che, C.M. Cyclometalated Gold(III) Complexes Containing N-Heterocyclic Carbene Ligands Engage Multiple Anti-Cancer Molecular Targets. Angew. Chem. Int. Ed. 2017, 56, 3892–3896. [Google Scholar] [CrossRef]
- Meier, S.M.; Kreutz, D.; Winter, L.; Klose, M.H.M.; Cseh, K.; Weiss, T.; Bileck, A.; Alte, B.; Mader, J.C.; Jana, S.; et al. An Organoruthenium Anticancer Agent Shows Unexpected Target Selectivity for Plectin. Angew. Chem. Int. Ed. 2017, 56, 8267–8271. [Google Scholar] [CrossRef]
- Hu, D.; Liu, Y.G.; Lai, Y.T.; Tong, K.C.; Fung, Y.M.; Lok, C.N.; Che, C.M. Anticancer Gold(III) Porphyrins Target Mitochondrial Chaperone Hsp60. Angew. Chem. Int. Ed. 2016, 55, 1387–1391. [Google Scholar] [CrossRef]
- Neuditschko, B.; King, A.P.; Huang, Z.Y.; Janker, L.; Bileck, A.; Borutzki, Y.; Marker, S.C.; Gerner, C.; Wilson, J.J.; Meier-Menches, S.M. An Anticancer Rhenium Tricarbonyl Targets Fe-S Cluster Biogenesis in Ovarian Cancer Cells. Angew. Chem. Int. Ed. 2022, 61, e202209136. [Google Scholar] [CrossRef]
- Lai, Y.T.; Chang, Y.Y.; Hu, L.G.; Yang, Y.; Chao, A.L.; Du, Z.Y.; Tanner, J.A.; Chye, M.L.; Qian, C.M.; Ng, K.M.; et al. Rapid Labeling of Intracellular His-Tagged Proteins in Living Cells. Proc. Natl. Acad. Sci. USA 2015, 112, 2948–2953. [Google Scholar] [CrossRef]
- Lai, Y.T.; Yang, Y.; Hu, L.; Cheng, T.; Chang, Y.Y.; Koohi-Moghadam, M.; Wang, Y.; Xia, J.; Wang, J.; Li, H.; et al. Integration of Fluorescence Imaging with Proteomics Enables Visualization and Identification of Metallo-Proteomes in Living Cells. Metallomics 2017, 9, 38–47. [Google Scholar] [CrossRef]
- Hu, X.; Li, H.; Ip, T.K.; Cheung, Y.F.; Koohi-Moghadam, M.; Wang, H.; Yang, X.; Tritton, D.N.; Wang, Y.; Wang, Y.; et al. Arsenic Trioxide Targets Hsp60, Triggering Degradation of p53 and Survivin. Chem. Sci. 2021, 12, 10893–10900. [Google Scholar] [CrossRef]
- Jiang, N.; Cheng, T.; Wang, M.; Chan, G.C.; Jin, L.; Li, H.; Sun, H. Tracking Iron-Associated Proteomes in Pathogens by a Fluorescence Approach. Metallomics 2018, 10, 77–82. [Google Scholar] [CrossRef]
- Wang, H.B.; Hu, L.G.; Li, H.Y.; Lai, Y.T.; Wei, X.Y.; Xu, X.H.; Cao, Z.K.; Cao, H.M.; Wan, Q.Y.; Chang, Y.Y.; et al. Mitochondrial ATP Synthase as a Direct Molecular Target of Chromium(III) to Ameliorate Hyperglycaemia Stress. Nat. Commun. 2023, 14, 1738. [Google Scholar] [CrossRef]
- Jaime-Perez, N.; Kaftan, D.; Bina, D.; Bokhari, S.N.H.; Shreedhar, S.; Küpper, H. Mechanisms of Sublethal Copper Toxicity Damage to the Photosynthetic Apparatus of Rhodospirillum rubrum. Biochim. Biophys. Acta Bioenerg. 2019, 1860, 640–650. [Google Scholar] [CrossRef]
- Küpper, H.; Bokhari, S.N.H.; Jaime-Perez, N.; Lyubenova, L.; Ashraf, N.; Andresen, E. Ultratrace Metal Speciation Analysis by Coupling of Sector-Field ICP-MS to High-Resolution Size Exclusion and Reversed-Phase Liquid Chromatography. Anal. Chem. 2019, 91, 10961–10969. [Google Scholar] [CrossRef]
- Yan, X.T.; He, B.; Wang, D.Y.; Hu, L.G.; Liu, L.H.; Liao, C.Y.; Jiang, G.B. Two-Dimensional (Weak Anion Exchange Chromatography-Gel Electrophoresis) Separations Coupling to Inductively Coupled Plasma Mass Spectrometry Strategy for Analysis of Metalloproteins. Talanta 2018, 184, 404–410. [Google Scholar] [CrossRef]
- Neville, S.L.; Eijkelkamp, B.A.; Lothian, A.; Paton, J.C.; Roberts, B.R.; Rosch, J.W.; McDevitt, C.A. Cadmium Stress Dictates Central Carbon Flux and Alters Membrane Composition in Streptococcus pneumoniae. Commun. Biol. 2020, 3, 694. [Google Scholar] [CrossRef]
- Budhraja, R.; Karande, S.; Ding, C.; Ullrich, M.K.; Wagner, S.; Reemtsma, T.; Adrian, L. Characterization of Membrane-Bound Metalloproteins in the Anaerobic Ammonium-Oxidizing Bacterium “Candidatus Kuenenia Stuttgartiensis” Strain CSTR1. Talanta 2021, 223, 121711. [Google Scholar] [CrossRef]
- Li, Y.L.; Zhang, M.N.; Zheng, C.B.; Hu, L.G.; Wang, C.; Jiang, J.; He, B.; Jiang, G.B. Analysis of Silver-Associated Proteins in Pathogen Via Combination of Native Sds-Page, Fluorescent Staining, and Inductively Coupled Plasma Mass Spectrometry. J. Chromatogr. A 2019, 1607, 460393. [Google Scholar] [CrossRef]
- Betts, H.D.; Neville, S.L.; McDevitt, C.A.; Sumby, C.J.; Harris, H.H. The Biochemical Fate of Ag+ Ions in Staphylococcus aureus, Escherichia coli, and Biological Media. J. Inorg. Biochem. 2021, 225, 111598. [Google Scholar] [CrossRef]
- Wang, H.; Yan, A.; Liu, Z.; Yang, X.; Xu, Z.; Wang, Y.; Wang, R.; Koohi-Moghadam, M.; Hu, L.; Xia, W.; et al. Deciphering Molecular Mechanism of Silver by Integrated Omic Approaches Enables Enhancing Its Antimicrobial Efficacy in E. coli. PLOS Biol. 2019, 17, e3000292. [Google Scholar] [CrossRef]
- Kroukamp, E.M.; Wondimu, T.; Forbes, P.B.C. Metal and Metalloid Speciation in Plants: Overview, Instrumentation, Approaches and Commonly Assessed Elements. Trends Anal. Chem. 2016, 77, 87–99. [Google Scholar] [CrossRef]
- Galazzi, R.M.; de Jesus, J.R.; Arruda, M.A.Z. Sample Preparation Focusing on Plant Omics. Adv. Exp. Med. Biol. 2019, 1073, 161–185. [Google Scholar] [CrossRef] [PubMed]
- Vacchina, V.; Polec, K.; Szpunar, J. Speciation of Cadmium in Plant Tissues by Size-Exclusion Chromatography with ICP-MS Detection. J. Anal. At. Spectrom. 1999, 14, 1557–1566. [Google Scholar] [CrossRef]
- Thomas, G.; Andresen, E.; Mattusch, J.; Hubáček, T.; Küpper, H. Deficiency and Toxicity of Nanomolar Copper in Low Irradiance—A Physiological and Metalloproteomic Study in the Aquatic Plant Ceratophyllum demersum. Aquat. Toxicol. 2016, 177, 226–236. [Google Scholar] [CrossRef] [PubMed]
- Andresen, E.; Kappel, S.; Stark, H.J.; Riegger, U.; Borovec, J.; Mattusch, J.; Heinz, A.; Schmelzer, C.E.H.; Matouskova, S.; Dickinson, B.; et al. Cadmium Toxicity Investigated at the Physiological and Biophysical Levels under Environmentally Relevant Conditions Using the Aquatic Model Plant Ceratophyllum demersum. New Phytol. 2016, 210, 1244–1258. [Google Scholar] [CrossRef] [PubMed]
- Andresen, E.; Flores-Sanchez, I.J.; Brückner, D.; Bokhari, S.N.H.; Falkenberg, G.; Küpper, H. Sublethal and Lethal Cd Toxicity in Soybean Roots Specifically Affects the Metabolome, Cd Binding to Proteins and Cellular Distribution of Cd. J. Hazard. Mater. 2023, 442, 130062. [Google Scholar] [CrossRef] [PubMed]
- Andresen, E.; Lyubenova, L.; Hubáček, T.; Bokhari, S.N.H.; Matoušková, Š.; Mijovilovich, A.; Rohovec, J.; Küpper, H. Chronic Exposure of Soybean Plants to Nanomolar Cadmium Reveals Specific Additional High-Affinity Targets of Cadmium Toxicity. J. Exp. Bot. 2020, 71, 1628–1644. [Google Scholar] [CrossRef] [PubMed]
- Manley, S.A.; Byrns, S.; Lyon, A.W.; Brown, P.; Gailer, J. Simultaneous Cu-, Fe-, and Zn-Specific Detection of Metalloproteins Contained in Rabbit Plasma by Size-Exclusion Chromatography-Inductively Coupled Plasma Atomic Emission Spectroscopy. J. Biol. Inorg. Chem. 2009, 14, 61–74. [Google Scholar] [CrossRef]
- Sarpong-Kumankomah, S.; Gailer, J. Identification of a Haptoglobin-Hemoglobin Complex in Human Blood Plasma. J. Inorg. Biochem. 2019, 201, 110802. [Google Scholar] [CrossRef]
- Coverdale, J.P.C.; Barnett, J.P.; Adamu, A.H.; Griffiths, E.J.; Stewart, A.J.; Blindauer, C.A. A Metalloproteomic Analysis of Interactions between Plasma Proteins and Zinc: Elevated Fatty Acid Levels Affect Zinc Distribution. Metallomics 2019, 11, 1805–1819. [Google Scholar] [CrossRef]
- Sarkar, B. Metal-Protein Interactions in Transport, Accumulation, and Excretion of Metals. Biol. Trace Elem. Res. 1989, 21, 137–144. [Google Scholar] [CrossRef]
- Bhattacharya, A.A.; Grüne, T.; Curry, S. Crystallographic Analysis Reveals Common Modes of Binding of Medium and Long-Chain Fatty Acids to Human Serum Albumin. J. Mol. Biol. 2000, 303, 721–732. [Google Scholar] [CrossRef] [PubMed]
- Arya, S.; Gourley, A.J.; Penedo, J.C.; Blindauer, C.A.; Stewart, A.J. Fatty Acids May Influence Insulin Dynamics through Modulation of Albumin-Zn2+ Interactions. Bioessays 2021, 43, 2100172. [Google Scholar] [CrossRef] [PubMed]
- Sobczak, A.I.S.; Katundu, K.G.H.; Phoenix, F.A.; Khazaipoul, S.; Yu, R.; Lampiao, F.; Stefanowicz, F.; Blindauer, C.A.; Pitt, S.J.; Smith, T.K.; et al. Albumin-Mediated Alteration of Plasma Zinc Speciation by Fatty Acids Modulates Blood Clotting in Type-2 Diabetes. Chem. Sci. 2021, 12, 4079–4093. [Google Scholar] [CrossRef] [PubMed]
- Mbiydzenyuy, N.E.; Ninsiima, H.I.; Valladares, M.B.; Pieme, C.A. Zinc and Linoleic Acid Pre-Treatment Attenuates Biochemical and Histological Changes in the Midbrain of Rats with Rotenone-Induced Parkinsonism. BMC Neurosci. 2018, 19, 29. [Google Scholar] [CrossRef] [PubMed]
- Kirsipuu, T.; Zadoroznaja, A.; Smirnova, J.; Friedemann, M.; Plitz, T.; Tougu, V.; Palumaa, P. Copper(II)-Binding Equilibria in Human Blood. Sci. Rep. 2020, 10, 5686. [Google Scholar] [CrossRef] [PubMed]
- Roberts, E.A.; Schilsky, M.L. Current and Emerging Issues in Wilson’s Disease. N. Engl. J. Med. 2023, 389, 922–938. [Google Scholar] [CrossRef] [PubMed]
- Quarles, C.D.; Macke, M.; Michalke, B.; Zischka, H.; Karst, U.; Sullivan, P.; Field, M.P. LC-ICP-MS Method for the Determination of “Extractable Copper” in Serum. Metallomics 2020, 12, 1348–1355. [Google Scholar] [CrossRef]
- Solovyev, N.; Ala, A.; Schilsky, M.; Mills, C.; Willis, K.; Harrington, C.F. Biomedical Copper Speciation in Relation to Wilson’s Disease Using Strong Anion Exchange Chromatography Coupled to Triple Quadrupole Inductively Coupled Plasma Mass Spectrometry. Anal. Chim. Acta 2020, 1098, 27–36. [Google Scholar] [CrossRef]
- Busto, M.E.D.; Cuello-Nunez, S.; Ward-Deitrich, C.; Morley, T.; Goenaga-Infante, H. A Fit-for-Purpose Copper Speciation Method for the Determination of Exchangeable Copper Relevant to Wilson’s Disease. Anal. Bioanal. Chem. 2022, 414, 561–573. [Google Scholar] [CrossRef]
- Michalke, B. Review About the Manganese Speciation Project Related to Neurodegeneration: An Analytical Chemistry Approach to Increase the Knowledge About Manganese Related Parkinsonian Symptoms. J. Trace Elem. Med. Biol. 2016, 37, 50–61. [Google Scholar] [CrossRef]
- Michalke, B.; Willkommen, D.; Drobyshev, E.; Solovyev, N. The Importance of Speciation Analysis in Neurodegeneration Research. Trends Anal. Chem. 2018, 104, 160–170. [Google Scholar] [CrossRef]
- Michalke, B.; Lucio, M.; Berthele, A.; Kanawati, B. Manganese Speciation in Paired Serum and Csf Samples Using SEC-DRC-ICP-MS and CE-ICP-DRC-MS. Anal. Bioanal. Chem. 2013, 405, 2301–2309. [Google Scholar] [CrossRef] [PubMed]
- Michalke, B.; Berthele, A.; Venkataramani, V. Simultaneous Quantification and Speciation of Trace Metals in Paired Serum and CSF Samples by Size Exclusion Chromatography-Inductively Coupled Plasma-Dynamic Reaction Cell-Mass Spectrometry (SEC-DRC-ICP-MS). Int. J. Mol. Sci. 2021, 22, 8892. [Google Scholar] [CrossRef]
- Neth, K.; Lucio, M.; Walker, A.; Kanawati, B.; Zorn, J.; Schmitt-Kopplin, P.; Michalke, B. Diverse Serum Manganese Species Affect Brain Metabolites Depending on Exposure Conditions. Chem. Res. Toxicol. 2015, 28, 1434–1442. [Google Scholar] [CrossRef] [PubMed]
- Ajsuvakova, O.P.; Tinkov, A.A.; Willkommen, D.; Skalnaya, A.A.; Danilov, A.B.; Pilipovich, A.A.; Aschner, M.; Skalny, A.V.; Michalke, B.; Skalnaya, M.G. Assessment of Copper, Iron, Zinc and Manganese Status and Speciation in Patients with Parkinson’s Disease: A Pilot Study. J. Trace Elem. Med. Biol. 2020, 59, 126423. [Google Scholar] [CrossRef] [PubMed]
- Barnett, J.P.; Blindauer, C.A.; Kassaar, O.; Khazaipoul, S.; Martin, E.M.; Sadler, P.J.; Stewart, A.J. Allosteric Modulation of Zinc Speciation by Fatty Acids. Biochim. Biophys. Acta Gen. Subj. 2013, 1830, 5456–5464. [Google Scholar] [CrossRef] [PubMed]
- Ajsuvakova, O.P.; Skalnaya, M.G.; Michalke, B.; Tinkov, A.A.; Serebryansky, E.P.; Karganov, M.Y.; Medvedeva, Y.S.; Skalny, A.V. Alteration of Iron (Fe), Copper (Cu), Zinc (Zn), and Manganese (Mn) Tissue Levels and Speciation in Rats with Desferioxamine-Induced Iron Deficiency. Biometals 2021, 34, 923–936. [Google Scholar] [CrossRef]
- Miroshnikov, S.A.; Notova, S.V.; Skalnaya, M.G.; Sizova, E.A.; Marshinskaia, O.V.; Kazakova, T.V.; Skalny, A.V.; Michalke, B.; Ajsuvakova, O.P.; Tinkov, A.A. Speciation of Serum Copper and Zinc-Binding High- and Low-Molecular Mass Ligands in Dairy Cows Using HPLC-ICP-MS Technique. Biol. Trace Elem. Res. 2022, 200, 591–599. [Google Scholar] [CrossRef]
- Notova, S.V.; Lebedev, S.V.; Marshinskaia, O.V.; Kazakova, T.V.; Ajsuvakova, O.P. Speciation Analysis of Manganese against the Background of Its Different Content in the Blood Serum of Dairy Cows. Biometals 2023, 36, 35–48. [Google Scholar] [CrossRef]
- Garcia-Fernandez, J.; Bettmer, J.; Jakubowski, N.; Panne, U.; Anon, E.; Montes-Bayon, M.; Sanz-Medel, A. The Fate of Iron Nanoparticles Used for Treatment of Iron Deficiency in Blood Using Mass-Spectrometry Based Strategies. Microchim. Acta 2017, 184, 3673–3680. [Google Scholar] [CrossRef]
- Kerger, B.D.; Gerads, R.; Gurleyuk, H.; Thuett, K.A.; Finley, B.L.; Paustenbach, D.J. Cobalt Speciation Assay for Human Serum, Part I. Method for Measuring Large and Small Molecular Cobalt and Protein-Binding Capacity Using Size Exclusion Chromatography with Inductively Coupled Plasma-Mass Spectroscopy Detection. Toxicol. Environ. Chem. 2013, 95, 687–708. [Google Scholar] [CrossRef]
- Loeschner, K.; Harrington, C.F.; Kearney, J.L.; Langton, D.J.; Larsen, E.H. Feasibility of Asymmetric Flow Field-Flow Fractionation Coupled to ICP-MS for the Characterization of Wear Metal Particles and Metalloproteins in Biofluids from Hip Replacement Patients. Anal. Bioanal. Chem. 2015, 407, 4541–4554. [Google Scholar] [CrossRef] [PubMed]
- Ordonez, Y.N.; Montes-Bayon, M.; Blanco-Gonzalez, E.; Paz-Jimenez, J.; Tejerina-Lobo, J.M.; Pena-Lopez, J.M.; Sanz-Medel, A. Metal Release in Patients with Total Hip Arthroplasty by DF-ICP-MS and Their Association to Serum Proteins. J. Anal. Atom. Spect. 2009, 24, 1037–1043. [Google Scholar] [CrossRef]
- Huynh, T.N.S.; Bourgeois, D.; Basset, C.; Vidaud, C.; Hagege, A. Assessment of CE-ICP/MS Hyphenation for the Study of Uranyl/Protein Interactions. Electrophoresis 2015, 36, 1374–1382. [Google Scholar] [CrossRef] [PubMed]
- Michalke, B.; Nischwitz, V. Review on Metal Speciation Analysis in Cerebrospinal Fluid-Current Methods and Results a Review. Anal. Chim. Acta 2010, 682, 23–36. [Google Scholar] [CrossRef] [PubMed]
- Roos, P.M. Ultraclean Paired Sampling for Metal Analysis in Neurodegenerative Disorders. J. Trace Elem. Med. Biol. 2019, 52, 48–52. [Google Scholar] [CrossRef] [PubMed]
- Willkommen, D.; Lucio, M.; Schmitt-Kopplin, P.; Gazzaz, M.; Schroeter, M.; Sigaroudi, A.; Michalke, B. Species Fractionation in a Case-Control Study Concerning Parkinson’s Disease: Cu-Amino Acids Discriminate CSF of PD from Controls. J. Trace Elem. Med. Biol. 2018, 49, 164–170. [Google Scholar] [CrossRef]
- Fernandez-Menendez, S.; Fernandez-Sanchez, M.L.; Fernandez-Colomer, B.; St Remy, R.R.D.; Cotallo, G.D.C.; Freire, A.S.; Braz, B.F.; Santelli, R.E.; Sanz-Medel, A. Total Zinc Quantification by Inductively Coupled Plasma-Mass Spectrometry and Its Speciation by Size Exclusion Chromatography-Inductively Coupled Plasma-Mass Spectrometry in Human Milk and Commercial Formulas: Importance in Infant Nutrition. J. Chromatogr. A 2016, 1428, 246–254. [Google Scholar] [CrossRef]
- Trinta, V.D.; Padilha, P.D.; Petronilho, S.; Santelli, R.E.; Braz, B.F.; Freire, A.S.; Saunders, C.; da Rocha, H.F.; Sanz-Medel, A.; Fernandez-Sanchez, M.L. Total Metal Content and Chemical Speciation Analysis of Iron, Copper, Zinc and Iodine in Human Breast Milk Using High-Performance Liquid Chromatography Separation and Inductively Coupled Plasma Mass Spectrometry Detection. Food Chem. 2020, 326, 126978. [Google Scholar] [CrossRef]
- Acosta, M.; Torres, S.; Mariño-Repizo, L.; Martinez, L.D.; Gil, R.A. Novel Method for Metalloproteins Determination in Human Breast Milk by Size Exclusion Chromatography Coupled to Inductively Coupled Plasma Mass Spectrometry. J. Pharm. Biomed. Anal. 2018, 158, 209–213. [Google Scholar] [CrossRef]
- Thomas, O.R.B.; Ganio, K.; Roberts, B.R.; Swearer, S.E. Trace Element-Protein Interactions in Endolymph from the Inner Ear of Fish: Implications for Environmental Reconstructions Using Fish Otolith Chemistry. Metallomics 2017, 9, 239–249. [Google Scholar] [CrossRef] [PubMed]
- Hare, D.J.; Rembach, A.; Roberts, B.R. The Emerging Role of Metalloproteomics in Alzheimer’s Disease Research. Methods Mol. Biol. 2016, 1303, 379–389. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.S.; Lago, L.; de Vega, R.G.; Bray, L.; Hare, D.J.; Clases, D.; Doble, P.A.; Adlard, P.A. Characterising the Spatial and Temporal Brain Metal Profile in a Mouse Model of Tauopathy. Metallomics 2020, 12, 301–313. [Google Scholar] [CrossRef] [PubMed]
- Lothian, A.; Roberts, B.R. Standards for Quantitative Metalloproteomic Analysis Using Size Exclusion ICP-MS. J. Vis. Exp. 2016, 53737. [Google Scholar] [CrossRef]
- Hancock, S.M.; Portbury, S.D.; Gunn, A.P.; Roberts, B.R.; Bush, A.I.; Adlard, P.A. Zinc Transporter-3 Knockout Mice Demonstrate Age-Dependent Alterations in the Metalloproteome. Int. J. Mol. Sci. 2020, 21, 839. [Google Scholar] [CrossRef]
- Larner, F.; McLean, C.A.; Halliday, A.N.; Roberts, B.R. Copper Isotope Compositions of Superoxide Dismutase and Metallothionein from Post-Mortem Human Frontal Cortex. Inorganics 2019, 7, 86. [Google Scholar] [CrossRef]
- Garcia, J.A.; Fernandez, D.T.; Alvarez, E.A.; Gonzalez, E.B.; Montes-Bayon, M.; Sanz-Medel, A. Iron Speciation, Ferritin Concentrations and Fe: Ferritin Ratios in Different Malignant Breast Cancer Cell Lines: On the Search for Cancer Biomarkers. Metallomics 2016, 8, 1090–1096. [Google Scholar] [CrossRef]
- Hare, D.J.; Roberts, B.R.; McColl, G. Profiling Changes to Natively-Bound Metals During Caenorhabditis elegans Development. RSC Adv. 2016, 6, 113689–113693. [Google Scholar] [CrossRef]
- Nong, Q.; Dong, H.; Liu, Y.; Liu, L.; He, B.; Huang, Y.; Jiang, J.; Luan, T.; Chen, B.; Hu, L. Characterization of the Mercury-Binding Proteins in Tuna and Salmon Sashimi: Implications for Health Risk of Mercury in Food. Chemosphere 2021, 263, 128110. [Google Scholar] [CrossRef]
- de Queiroz, J.V.; Cavecci-Mendonça, B.; Vieira, J.C.S.; Martins, R.A.; de Almeida Assunção, A.S.; Cavallini, N.G.; Dos Santos, F.A.; de Magalhães Padilha, P. Metalloproteomic Strategies for Identifying Proteins as Biomarkers of Mercury Exposure in Serrasalmus rhombeus from the Amazon Region. Biol. Trace Elem. Res. 2021, 199, 712–720. [Google Scholar] [CrossRef]
- Bucher, G.; Frelon, S.; Simon, O.; Lobinski, R.; Mounicou, S. Development of Non-Denaturing Off-Gel Isoelectric Focusing for the Separation of Uranium-Protein Complexes in Fish. Anal. Bioanal. Chem. 2014, 406, 3517–3520. [Google Scholar] [CrossRef] [PubMed]
- Bucher, G.; Mounicou, S.; Simon, O.; Floriani, M.; Lobinski, R.; Frelon, S. Insights into the Nature of Uranium Target Proteins within Zebrafish Gills after Chronic and Acute Waterborne Exposures. Environ. Toxicol. Chem. 2016, 35, 736–741. [Google Scholar] [CrossRef] [PubMed]
- Aryal, B.P.; Paunesku, T.; Woloschak, G.E.; He, C.; Jensen, M.P. A Proteomic Approach to Identification of Plutonium-Binding Proteins in Mammalian Cells. J. Proteom. 2012, 75, 1505–1514. [Google Scholar] [CrossRef] [PubMed]
- Loreti, V.; Bettmer, J. Determination of the MRI Contrast Agent Gd-DTPA by SEC-ICP-MS. Anal. Bioanal. Chem. 2004, 379, 1050–1054. [Google Scholar] [CrossRef] [PubMed]
- Telgmann, L.; Lindner, U.; Lingott, J.; Jakubowski, N. Analysis and Speciation of Lanthanoides by ICP-MS. Phys. Sci. Rev. 2016, 1, 20160058. [Google Scholar] [CrossRef]
- Gianolio, E.; Bardini, P.; Arena, F.; Stefania, R.; Di Gregorio, E.; Iani, R.; Aime, S. Gadolinium Retention in the Rat Brain: Assessment of the Amounts of Insoluble Gadolinium-Containing Species and Intact Gadolinium Complexes after Repeated Administration of Gadolinium-Based Contrast Agents. Radiology 2017, 285, 839–849. [Google Scholar] [CrossRef] [PubMed]
- Strzeminska, I.; Factor, C.; Jimenez-Lamana, J.; Lacomme, S.; Subirana, M.A.; Le Coustumer, P.; Schaumlöffel, D.; Robert, P.; Szpunar, J.; Corot, C.; et al. Comprehensive Speciation Analysis of Residual Gadolinium in Deep Cerebellar Nuclei in Rats Repeatedly Administered with Gadoterate Meglumine or Gadodiamide. Investig. Radiol. 2022, 57, 283–292. [Google Scholar] [CrossRef]
- Bucker, P.; Funke, S.K.I.; Factor, C.; Rasschaert, M.; Robert, P.; Sperling, M.; Karst, U. Combined Speciation Analysis and Elemental Bioimaging Provide New Insight into Gadolinium Retention in Kidney. Metallomics 2022, 14, mfac004. [Google Scholar] [CrossRef]
- Rasschaert, M.; Weller, R.O.; Schroeder, J.A.; Brochhausen, C.; Idee, J.M. Retention of Gadolinium in Brain Parenchyma: Pathways for Speciation, Access, and Distribution. A Critical Review. J. Magn. Reson. Imaging 2020, 52, 1293–1305. [Google Scholar] [CrossRef]
- Frenzel, T.; Apte, C.; Jost, G.; Schockel, L.; Lohrke, J.; Pietsch, H. Quantification and Assessment of the Chemical Form of Residual Gadolinium in the Brain after Repeated Administration of Gadolinium-Based Contrast Agents Comparative Study in Rats. Investig. Radiol. 2017, 52, 396–404. [Google Scholar] [CrossRef]
- Robert, P.; Fingerhut, S.; Factor, C.; Vives, V.; Letien, J.; Sperling, M.; Rasschaert, M.; Santus, R.; Ballet, S.; Idee, J.M.; et al. One-Year Retention of Gadolinium in the Brain: Comparison of Gadodiamide and Gadoterate Meglumine in a Rodent Model. Radiology 2018, 288, 424–433. [Google Scholar] [CrossRef] [PubMed]
- Strzeminska, I.; Factor, C.; Robert, P.; Grindel, A.L.; Comby, P.O.; Szpunar, J.; Corot, C.; Lobinski, R. Long-Term Evaluation of Gadolinium Retention in Rat Brain after Single Injection of a Clinically Relevant Dose of Gadolinium-Based Contrast Agents. Investig. Radiol. 2020, 55, 138–143. [Google Scholar] [CrossRef] [PubMed]
- Strzeminska, I.; Factor, C.; Robert, P.; Szpunar, J.; Corot, C.; Lobinski, R. Speciation Analysis of Gadolinium in the Water-Insoluble Rat Brain Fraction after Administration of Gadolinium-Based Contrast Agents. Investig. Radiol. 2021, 56, 535–544. [Google Scholar] [CrossRef] [PubMed]
- Nong, Q.Y.; Chen, X.; Hu, L.G.; Huang, Y.S.; Luan, T.G.; Liu, H.T.; Chen, B.W. Identification and Characterization of Gd-Binding Proteins in Nih-3t3 Cells. Talanta 2020, 219, 121281. [Google Scholar] [CrossRef] [PubMed]
- Clases, D.; de Vega, R.G.; Bishop, D.; Doble, P. SEC-ICP-MS and on-Line Isotope Dilution Analysis for Characterisation and Quantification of Immunochemical Assays. Anal. Bioanal. Chem. 2019, 411, 3553–3560. [Google Scholar] [CrossRef]
- Wang, H.; Zhou, Y.; Xu, X.; Li, H.; Sun, H. Metalloproteomics in Conjunction with Other Omics for Uncovering the Mechanism of Action of Metallodrugs: Mechanism-Driven New Therapy Development. Curr. Opin. Chem. Biol. 2020, 55, 171–179. [Google Scholar] [CrossRef]
- Wang, Y.; Li, H.; Sun, H. Metalloproteomics for Unveiling the Mechanism of Action of Metallodrugs. Inorg. Chem. 2019, 58, 13673–13685. [Google Scholar] [CrossRef]
- Steel, T.R.; Hartinger, C.G. Metalloproteomics for Molecular Target Identification of Protein-Binding Anticancer Metallodrugs. Metallomics 2020, 12, 1627–1636. [Google Scholar] [CrossRef]
- Wootton, C.A.; Lam, Y.P.Y.; Willetts, M.; van Agthoven, M.A.; Barrow, M.P.; Sadler, P.J.; O’Connor, P.B. Automatic Assignment of Metal-Containing Peptides in Proteomic LC-MS and MS/MS Data Sets. Analyst 2017, 142, 2029–2037. [Google Scholar] [CrossRef]
- Wang, J.; Tao, J.; Jia, S.; Wang, M.; Jiang, H.; Du, Z. The Protein-Binding Behavior of Platinum Anticancer Drugs in Blood Revealed by Mass Spectrometry. Pharmaceuticals 2021, 14, 104. [Google Scholar] [CrossRef]
- Will, J.; Wolters, D.A.; Sheldrick, W.S. Characterisation of Cisplatin Binding Sites in Human Serum Proteins Using Hyphenated Multidimensional Liquid Chromatography and ESI Tandem Mass Spectrometry. ChemMedChem 2008, 3, 1696–1707. [Google Scholar] [CrossRef]
- Moreno-Gordaliza, E.; Canas, B.; Palacios, M.A.; Gomez-Gomez, M.M. Characterization of Pt-Protein Complexes by nHPLC-ESI-LTQ MS/MS Using a Gel-Based Bottom-up Approach. Talanta 2012, 88, 599–608. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Gordaliza, E.; Esteban-Fernandez, D.; Giesen, C.; Lehmann, K.; Lazaro, A.; Tejedor, A.; Scheler, C.; Canas, B.; Jakubowski, N.; Linscheid, M.W.; et al. LA-ICP-MS and nHPLC-ESI-LTQ-FT-MS/MS for the Analysis of Cisplatin-Protein Complexes Separated by Two Dimensional Gel Electrophoresis in Biological Samples. J. Anal. At. Spectrom. 2012, 27, 1474–1483. [Google Scholar] [CrossRef]
- Moraleja, I.; Moreno-Gordaliza, E.; Esteban-Fernández, D.; Mena, M.L.; Linscheid, M.W.; Gómez-Gómez, M.M. A Shotgun Approach for the Identification of Platinum–Protein Complexes. Anal. Bioanal. Chem. 2015, 407, 2393–2403. [Google Scholar] [CrossRef] [PubMed]
- Harper, B.W.J.; Morris, T.T.; Gailer, J.; Aldrich-Wright, J.R. Probing the Interaction of Bisintercalating (2,2′:6′,2″-Terpyridine)Platinum(II) Complexes with Glutathione and Rabbit Plasma. J. Inorg. Biochem. 2016, 163, 95–102. [Google Scholar] [CrossRef]
- Galvez, L.; Theiner, S.; Grabarics, M.; Kowol, C.R.; Keppler, B.K.; Hann, S.; Koellensperger, G. Critical Assessment of Different Methods for Quantitative Measurement of Metallodrug-Protein Associations. Anal. Bioanal. Chem. 2018, 410, 7211–7220. [Google Scholar] [CrossRef]
- Timerbaev, A.R. Role of Metallomic Strategies in Developing Ruthenium Anticancer Drugs. Trends Anal. Chem. 2016, 80, 547–554. [Google Scholar] [CrossRef]
- Klose, M.H.M.; Schoberl, A.; Heffeter, P.; Berger, W.; Hartinger, C.G.; Koellensperger, G.; Meier-Menches, S.M.; Keppler, B.K. Serum-Binding Properties of Isosteric Ruthenium and Osmium Anticancer Agents Elucidated by SEC-ICP-MS. Monatsh. Chem. 2018, 149, 1719–1726. [Google Scholar] [CrossRef]
- Sulyok, M.; Hann, S.; Hartinger, C.G.; Keppler, B.K.; Stingeder, G.; Koellensperger, G. Two Dimensional Separation Schemes for Investigation of the Interaction of an Anticancer Ruthenium(III) Compound with Plasma Proteins. J. Anal. At. Spectrom. 2005, 20, 856–863. [Google Scholar] [CrossRef]
- Groessl, M.; Hartinger, C.G.; Polec-Pawlak, K.; Jarosz, M.; Keppler, B.K. Capillary Electrophoresis Hyphenated to Inductively Coupled Plasma-Mass Spectrometry: A Novel Approach for the Analysis of Anticancer Metallodrugs in Human Serum and Plasma. Electrophoresis 2008, 29, 2224–2232. [Google Scholar] [CrossRef]
- Levina, A.; Chetcuti, A.R.M.; Lay, P.A. Controversial Role of Transferrin in the Transport of Ruthenium Anticancer Drugs. Biomolecules 2022, 12, 1319. [Google Scholar] [CrossRef] [PubMed]
- Aleksenko, S.S.; Matczuk, M.; Lu, X.; Foteeva, L.S.; Pawlak, K.; Timerbaev, A.R.; Jarosz, M. Metallomics for Drug Development: An Integrated CE-ICP-MS and ICP-MS Approach Reveals the Speciation Changes for an Investigational Ruthenium(III) Drug Bound to Holo-Transferrin in Simulated Cancer Cytosol. Metallomics 2013, 5, 955–963. [Google Scholar] [CrossRef]
- Matczuk, M.; Kupiec, M.; Legat, J.; Pawlak, K.; Timerbaev, A.R.; Jarosz, M. A Shotgun Metalloproteomic Approach Enables Identification of Proteins Involved in the Speciation of a Ruthenium Anticancer Drug in the Cytosol of Cancer Cells. Analyst 2015, 140, 3492–3499. [Google Scholar] [CrossRef] [PubMed]
- Holtkamp, H.U.; Movassaghi, S.; Morrow, S.J.; Kubanik, M.; Hartinger, C.G. Metallomic Study on the Metabolism of Rapta-C and Cisplatin in Cell Culture Medium and Its Impact on Cell Accumulation. Metallomics 2018, 10, 455–462. [Google Scholar] [CrossRef] [PubMed]
- Groessl, M.; Terenghi, M.; Casini, A.; Elviri, L.; Lobinski, R.; Dyson, P.J. Reactivity of Anticancer Metallodrugs with Serum Proteins: New Insights from Size Exclusion Chromatography-ICP-MS and ESI-MS. J. Anal. At. Spectrom. 2010, 25, 305–313. [Google Scholar] [CrossRef] [PubMed]
- Michelucci, E.; Pieraccini, G.; Moneti, G.; Gabbiani, C.; Pratesi, A.; Messori, L. Mass Spectrometry and Metallomics: A General Protocol to Assess Stability of Metallodrug-Protein Adducts in Bottom-up MS Experiments. Talanta 2017, 167, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Artner, C.; Holtkamp, H.U.; Hartinger, C.G.; Meier-Menches, S.M. Characterizing Activation Mechanisms and Binding Preferences of Ruthenium Metallo-Prodrugs by a Competitive Binding Assay. J. Inorg. Biochem. 2017, 177, 322–327. [Google Scholar] [CrossRef]
- Moreno-Alcantar, G.; Picchetti, P.; Casini, A. Gold Complexes in Anticancer Therapy: From New Design Principles to Particle-Based Delivery Systems. Angew. Chem. Int. Ed. 2023, 135, e202218000. [Google Scholar] [CrossRef]
- Brescia, F.; Ronconi, L. Gold: Not Just Jewelry. Future Med. Chem. 2023, 15, 647–650. [Google Scholar] [CrossRef]
- Geri, A.; Massai, L.; Messori, L. Protein Metalation by Medicinal Gold Compounds: Identification of the Main Features of the Metalation Process through ESI MS Experiments. Molecules 2023, 28, 5196. [Google Scholar] [CrossRef]
- Matz, S.G.; Elder, R.C.; Tepperman, K. Liquid Chromatography with an Inductively Coupled Plasma Mass Spectrometric Detector for Simultaneous Determination of Gold Drug Metabolites and Related Metals in Human Blood. J. Anal. At. Spectrom. 1989, 4, 767–771. [Google Scholar] [CrossRef]
- Matczuk, M.; Anecka, K.; Scaletti, F.; Messori, L.; Keppler, B.K.; Timerbaev, A.R.; Jarosz, M. Speciation of Metal-Based Nanomaterials in Human Serum Characterized by Capillary Electrophoresis Coupled to ICP-MS: A Case Study of Gold Nanoparticles. Metallomics 2015, 7, 1364–1370. [Google Scholar] [CrossRef] [PubMed]
- Massai, L.; Zoppi, C.; Cirri, D.; Pratesi, A.; Messori, L. Reactions of Medicinal Gold(III) Compounds with Proteins and Peptides Explored by Electrospray Ionization Mass Spectrometry and Complementary Biophysical Methods. Front. Chem. 2020, 8, 581648. [Google Scholar] [CrossRef]
- Zoppi, C.; Messori, L.; Pratesi, A. ESI MS Studies Highlight the Selective Interaction of Auranofin with Protein Free Thiols. Dalton Trans. 2020, 49, 5906–5913. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Challenge | Applies to | Mandates to |
|---|---|---|
| Metal sites require folded proteins | Intra- and extracellular metalloproteomes | Work under near-physiological conditions |
| Protons compete for metal sites | Intra- and extracellular metalloproteomes | Avoid low pH |
| The inside of cells is a reducing environment, and exposure to oxygen can lead to oxidation of metals and/or certain ligands (e.g., thiolates) | Intracellular Metalloproteomes | Work under inert atmosphere or add reducing agent |
| Removal of intracellular barriers can lead to metal redistribution | Intracellular metalloproteomes | Carry out sub-cellular fractionation |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Coverdale, J.P.C.; Polepalli, S.; Arruda, M.A.Z.; da Silva, A.B.S.; Stewart, A.J.; Blindauer, C.A. Recent Advances in Metalloproteomics. Biomolecules 2024, 14, 104. https://doi.org/10.3390/biom14010104
Coverdale JPC, Polepalli S, Arruda MAZ, da Silva ABS, Stewart AJ, Blindauer CA. Recent Advances in Metalloproteomics. Biomolecules. 2024; 14(1):104. https://doi.org/10.3390/biom14010104
Chicago/Turabian StyleCoverdale, James P. C., Sirilata Polepalli, Marco A. Z. Arruda, Ana B. Santos da Silva, Alan J. Stewart, and Claudia A. Blindauer. 2024. "Recent Advances in Metalloproteomics" Biomolecules 14, no. 1: 104. https://doi.org/10.3390/biom14010104
APA StyleCoverdale, J. P. C., Polepalli, S., Arruda, M. A. Z., da Silva, A. B. S., Stewart, A. J., & Blindauer, C. A. (2024). Recent Advances in Metalloproteomics. Biomolecules, 14(1), 104. https://doi.org/10.3390/biom14010104