
N-Glycosylation as a Modulator of Protein Conformation and Assembly in Disease
 ,
,  , ,
, , 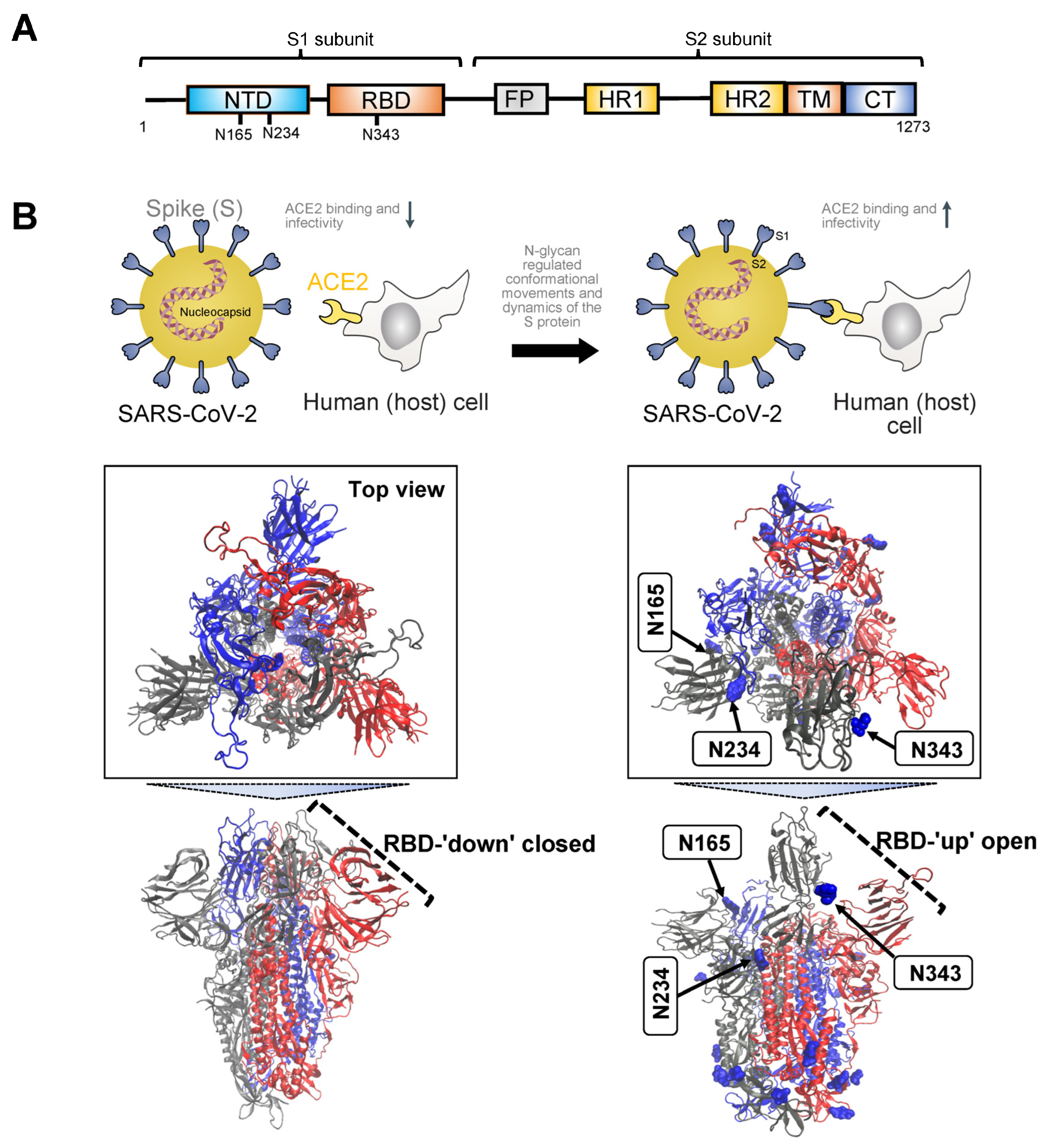{kind=link}
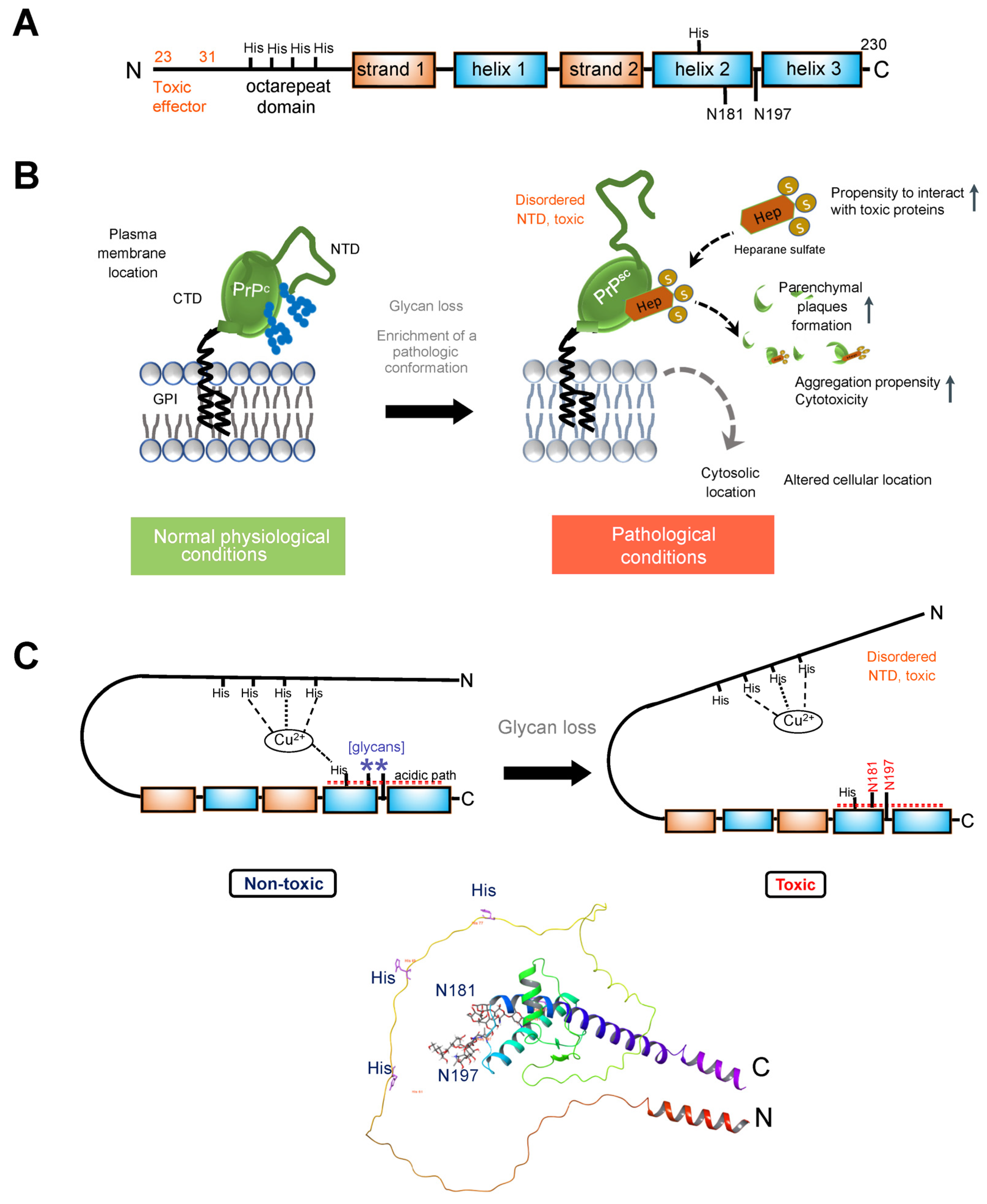{kind=link}
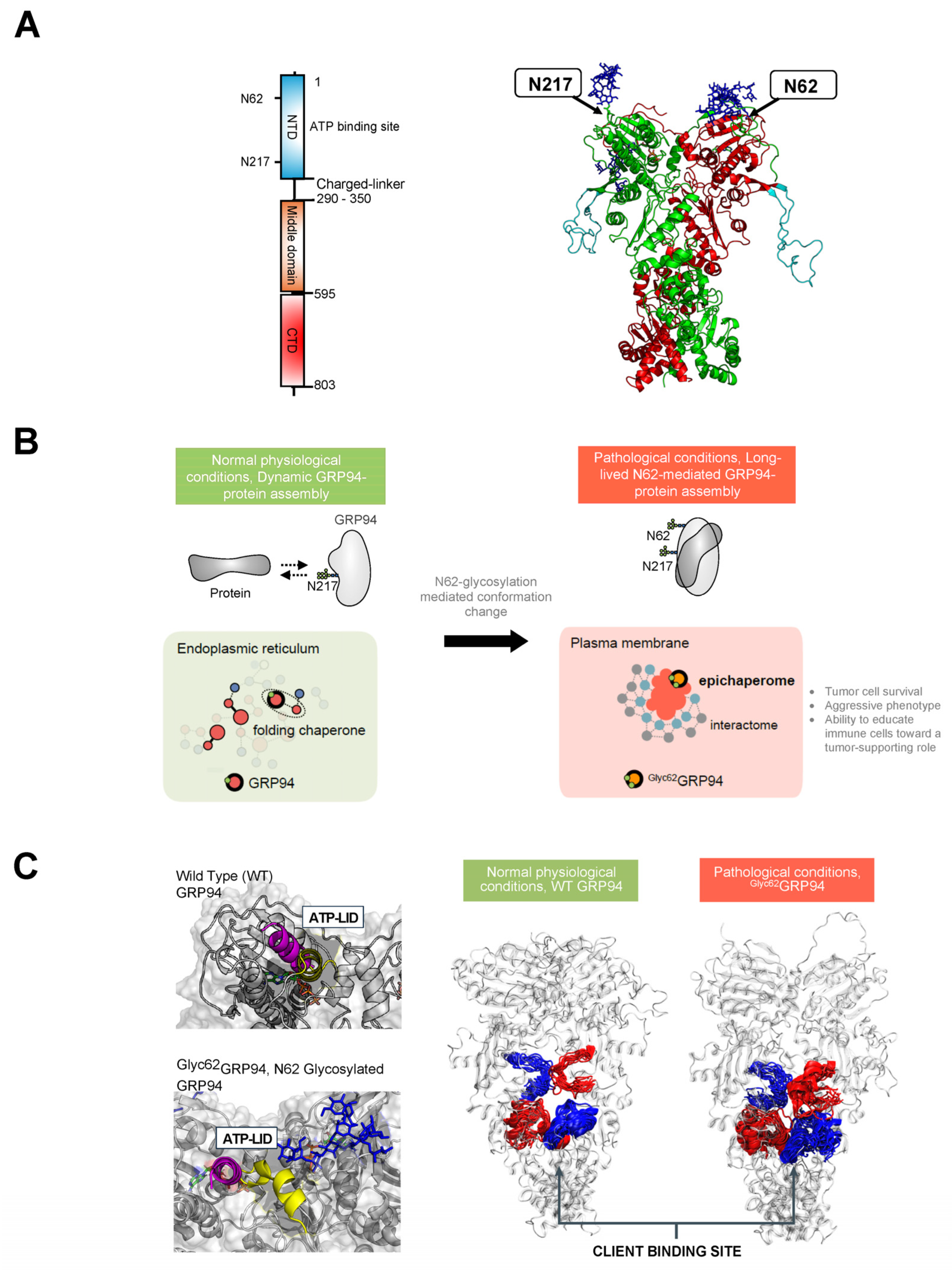{kind=link}
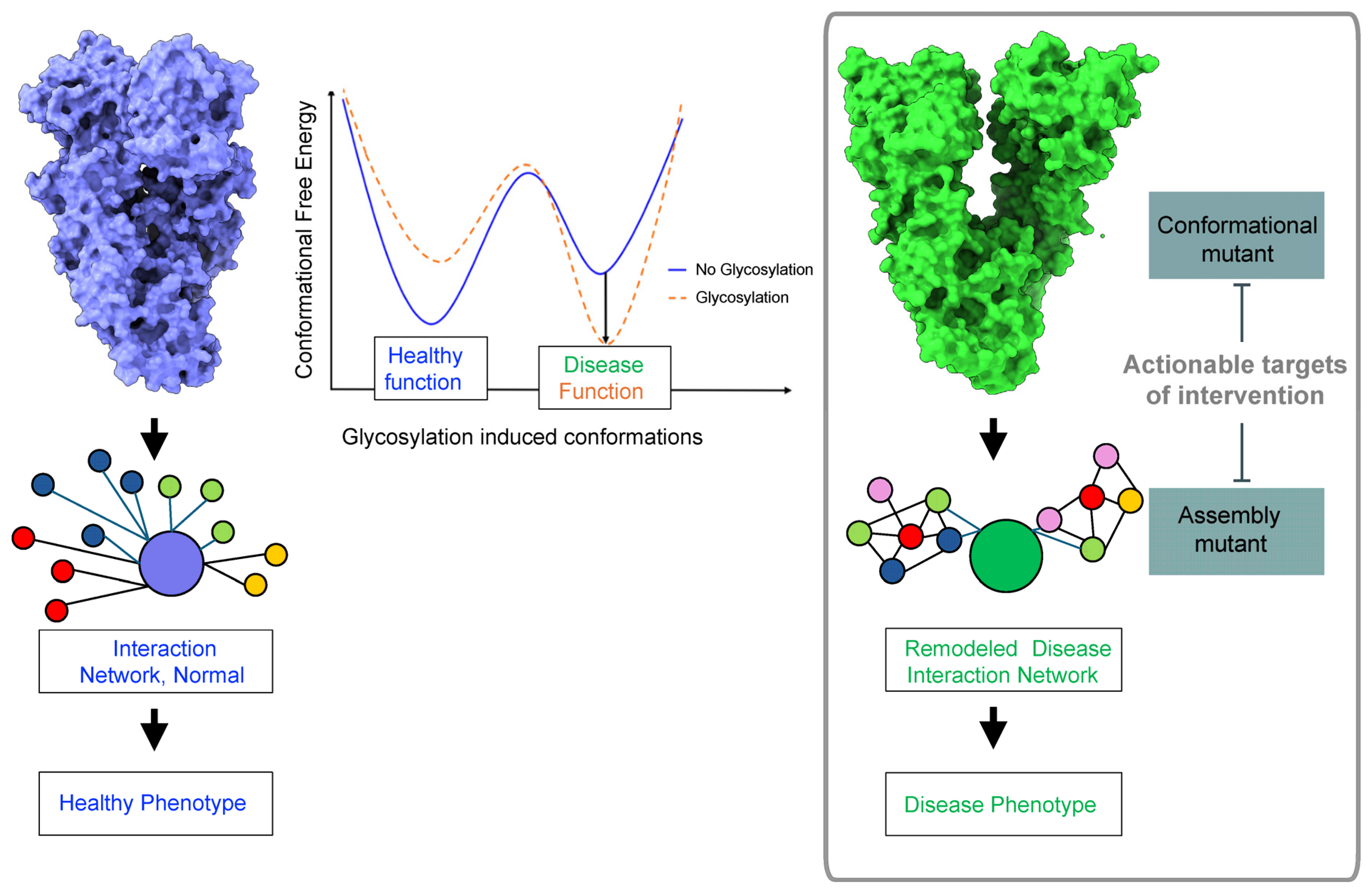{kind=link}
Abstract
:1. Introduction
2. Glycosylation and Protein Conformation
3. N-Glycans’ Effect on Pathologic Protein Conformations in Disease
3.1. Severe Acute Respiratory Syndrome SARS Proteins
3.2. Prion Protein
3.3. Glucose-Regulated Protein 94 (GRP94)
4. Therapeutic Implications—Targeting Conformational and Assembly Mutants in Disease
4.1. Targeting the Viral Protein Conformations
4.2. Correcting the Prion Protein Conformation
4.3. Targeting Pathologic GRP94 Conformers and Assemblies
5. Conclusions and Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Moremen, K.W.; Tiemeyer, M.; Nairn, A.V. Vertebrate protein glycosylation: Diversity, synthesis and function. Nat. Rev. Mol. Cell Biol. 2012, 13, 448–462. [Google Scholar] [CrossRef] [PubMed]
- Reily, C.; Stewart, T.J.; Renfrow, M.B.; Novak, J. Glycosylation in health and disease. Nat. Rev. Nephrol. 2019, 15, 346–366. [Google Scholar] [CrossRef] [PubMed]
- Schjoldager, K.T.; Narimatsu, Y.; Joshi, H.J.; Clausen, H. Global view of human protein glycosylation pathways and functions. Nat. Rev. Mol. Cell Biol. 2020, 21, 729–749. [Google Scholar] [CrossRef]
- Varki, A. Biological roles of glycans. Glycobiology 2017, 27, 3–49. [Google Scholar] [CrossRef] [PubMed]
- Rini, J.M.; Moremen, K.W.; Davis, B.G.; Esko, J.D. Glycosyltransferases and Glycan-Processing Enzymes. In Essentials of Glycobiology, 4th ed.; Varki, A., Cummings, R.D., Esko, J.D., Stanley, P., Hart, G.W., Aebi, M., Mohnen, D., Kinoshita, T., Packer, N.H., Prestegard, J.H., et al., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2022; pp. 67–78. [Google Scholar]
- Corfield, A.P.; Berry, M. Glycan variation and evolution in the eukaryotes. Trends Biochem. Sci. 2015, 40, 351–359. [Google Scholar] [CrossRef]
- Lebrilla, C.B.; Liu, J.; Widmalm, G.; Prestegard, J.H. Oligosaccharides and Polysaccharides. In Essentials of Glycobiology, 4th ed.; Varki, A., Cummings, R.D., Esko, J.D., Stanley, P., Hart, G.W., Aebi, M., Mohnen, D., Kinoshita, T., Packer, N.H., Prestegard, J.H., et al., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2022; pp. 33–42. [Google Scholar]
- Seeberger, P.H. Monosaccharide Diversity. In Essentials of Glycobiology, 4th ed.; Varki, A., Cummings, R.D., Esko, J.D., Stanley, P., Hart, G.W., Aebi, M., Mohnen, D., Kinoshita, T., Packer, N.H., Prestegard, J.H., et al., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2022; pp. 21–32. [Google Scholar]
- Rudd, P.M.; Karlsson, N.G.; Khoo, K.H.; Thaysen-Andersen, M.; Wells, L.; Packer, N.H. Glycomics and Glycoproteomics. In Essentials of Glycobiology, 4th ed.; Varki, A., Cummings, R.D., Esko, J.D., Stanley, P., Hart, G.W., Aebi, M., Mohnen, D., Kinoshita, T., Packer, N.H., Prestegard, J.H., et al., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2022; pp. 689–704. [Google Scholar]
- Chao, Q.; Ding, Y.; Chen, Z.H.; Xiang, M.H.; Wang, N.; Gao, X.D. Recent Progress in Chemo-Enzymatic Methods for the Synthesis of N-Glycans. Front. Chem. 2020, 8, 513. [Google Scholar] [CrossRef] [PubMed]
- de Haas, P.; Hendriks, W.; Lefeber, D.J.; Cambi, A. Biological and Technical Challenges in Unraveling the Role of N-Glycans in Immune Receptor Regulation. Front. Chem. 2020, 8, 55. [Google Scholar] [CrossRef]
- Fang, P.; Ji, Y.; Oellerich, T.; Urlaub, H.; Pan, K.T. Strategies for Proteome-Wide Quantification of Glycosylation Macro- and Micro-Heterogeneity. Int. J. Mol. Sci. 2022, 23, 1609. [Google Scholar] [CrossRef]
- Schmaltz, R.M.; Hanson, S.R.; Wong, C.H. Enzymes in the synthesis of glycoconjugates. Chem. Rev. 2011, 111, 4259–4307. [Google Scholar] [CrossRef]
- Zacchi, L.F.; Schulz, B.L. N-glycoprotein macroheterogeneity: Biological implications and proteomic characterization. Glycoconj. J. 2016, 33, 359–376. [Google Scholar] [CrossRef]
- Springer, S.A.; Gagneux, P. Glycan evolution in response to collaboration, conflict, and constraint. J. Biol. Chem. 2013, 288, 6904–6911. [Google Scholar] [CrossRef]
- Gong, Y.; Qin, S.; Dai, L.; Tian, Z. The glycosylation in SARS-CoV-2 and its receptor ACE2. Signal Transduct. Target. Ther. 2021, 6, 396. [Google Scholar] [CrossRef]
- Otaki, M.; Hirane, N.; Natsume-Kitatani, Y.; Nogami Itoh, M.; Shindo, M.; Kurebayashi, Y.; Nishimura, S.I. Mouse tissue glycome atlas 2022 highlights inter-organ variation in major N-glycan profiles. Sci. Rep. 2022, 12, 17804. [Google Scholar] [CrossRef]
- Lumibao, J.C.; Tremblay, J.R.; Hsu, J.; Engle, D.D. Altered glycosylation in pancreatic cancer and beyond. J. Exp. Med. 2022, 219, e20211505. [Google Scholar] [CrossRef]
- Saleh, F.M.; Chandra, P.K.; Lin, D.; Robinson, J.E.; Izadpanah, R.; Mondal, D.; Bollensdorff, C.; Alt, E.U.; Zhu, Q.; Marasco, W.A.; et al. A New Humanized Mouse Model Mimics Humans in Lacking alpha-Gal Epitopes and Secreting Anti-Gal Antibodies. J. Immunol. 2020, 204, 1998–2005. [Google Scholar] [CrossRef]
- Riley, N.M.; Hebert, A.S.; Westphall, M.S.; Coon, J.J. Capturing site-specific heterogeneity with large-scale N-glycoproteome analysis. Nat. Commun. 2019, 10, 1311. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Wang, Z.; Guo, L.; Zhu, Z.; Zhang, Y. Proteome-Wide Analysis of N-Glycosylation Stoichiometry Using SWATH Technology. J. Proteome Res. 2017, 16, 3830–3840. [Google Scholar] [CrossRef] [PubMed]
- Bagdonaite, I.; Malaker, S.A.; Polasky, D.A.; Riley, N.M.; Schjoldager, K.; Vakhrushev, S.Y.; Halim, A.; Aoki-Kinoshita, K.F.; Nesvizhskii, A.I.; Bertozzi, C.R.; et al. Glycoproteomics. Nat. Rev. Methods Prim. 2022, 2, 48. [Google Scholar] [CrossRef]
- Hu, Y.; Pan, J.; Shah, P.; Ao, M.; Thomas, S.N.; Liu, Y.; Chen, L.; Schnaubelt, M.; Clark, D.J.; Rodriguez, H.; et al. Integrated Proteomic and Glycoproteomic Characterization of Human High-Grade Serous Ovarian Carcinoma. Cell Rep. 2020, 33, 108276. [Google Scholar] [CrossRef]
- Campbell, M.P.; Peterson, R.; Mariethoz, J.; Gasteiger, E.; Akune, Y.; Aoki-Kinoshita, K.F.; Lisacek, F.; Packer, N.H. UniCarbKB: Building a knowledge platform for glycoproteomics. Nucleic Acids Res. 2014, 42, D215–D221. [Google Scholar] [CrossRef]
- Li, X.; Xu, Z.; Hong, X.; Zhang, Y.; Zou, X. Databases and Bioinformatic Tools for Glycobiology and Glycoproteomics. Int. J. Mol. Sci. 2020, 21, 6727. [Google Scholar] [CrossRef] [PubMed]
- Toukach, P.V.; Egorova, K.S. Carbohydrate structure database merged from bacterial, archaeal, plant and fungal parts. Nucleic Acids Res. 2016, 44, D1229–D1236. [Google Scholar] [CrossRef] [PubMed]
- Scherbinina, S.I.; Toukach, P.V. Three-Dimensional Structures of Carbohydrates and Where to Find Them. Int. J. Mol. Sci. 2020, 21, 7702. [Google Scholar] [CrossRef] [PubMed]
- Bohm, M.; Bohne-Lang, A.; Frank, M.; Loss, A.; Rojas-Macias, M.A.; Lutteke, T. Glycosciences.DB: An annotated data collection linking glycomics and proteomics data (2018 update). Nucleic Acids Res. 2019, 47, D1195–D1201. [Google Scholar] [CrossRef] [PubMed]
- Aoki-Kinoshita, K.F.; Kanehisa, M. Glycomic Analysis Using KEGG GLYCAN. In Glycoinformatics; Lütteke, T., Frank, M., Eds.; Springer: New York, NY, USA, 2015; pp. 97–107. [Google Scholar]
- Maeda, M.; Fujita, N.; Suzuki, Y.; Sawaki, H.; Shikanai, T.; Narimatsu, H. JCGGDB: Japan Consortium for Glycobiology and Glycotechnology Database. In Glycoinformatics; Lütteke, T., Frank, M., Eds.; Springer: New York, NY, USA, 2015; pp. 161–179. [Google Scholar]
- Toukach, P.V.; Egorova, K.S. Source files of the Carbohydrate Structure Database: The way to sophisticated analysis of natural glycans. Sci. Data 2022, 9, 131. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Hu, Y.; Ao, M.; Shah, P.; Chen, J.; Yang, W.; Jia, X.; Tian, Y.; Thomas, S.; Zhang, H. N-GlycositeAtlas: A database resource for mass spectrometry-based human N-linked glycoprotein and glycosylation site mapping. Clin. Proteom. 2019, 16, 35. [Google Scholar] [CrossRef] [PubMed]
- Abrahams, J.L.; Taherzadeh, G.; Jarvas, G.; Guttman, A.; Zhou, Y.; Campbell, M.P. Recent advances in glycoinformatic platforms for glycomics and glycoproteomics. Curr. Opin. Struct. Biol. 2020, 62, 56–69. [Google Scholar] [CrossRef]
- Egorova, K.S.; Toukach, P.V. Glycoinformatics: Bridging Isolated Islands in the Sea of Data. Angew. Chem. Int. Ed. Engl. 2018, 57, 14986–14990. [Google Scholar] [CrossRef]
- Apweiler, R.; Hermjakob, H.; Sharon, N. On the frequency of protein glycosylation, as deduced from analysis of the SWISS-PROT database. Biochim. Biophys. Acta 1999, 1473, 4–8. [Google Scholar] [CrossRef]
- Breitling, J.; Aebi, M. N-linked protein glycosylation in the endoplasmic reticulum. Cold Spring Harb. Perspect. Biol. 2013, 5, a013359. [Google Scholar] [CrossRef]
- Hanson, S.R.; Culyba, E.K.; Hsu, T.L.; Wong, C.H.; Kelly, J.W.; Powers, E.T. The core trisaccharide of an N-linked glycoprotein intrinsically accelerates folding and enhances stability. Proc. Natl. Acad. Sci. USA 2009, 106, 3131–3136. [Google Scholar] [CrossRef]
- Xu, C.; Ng, D.T. Glycosylation-directed quality control of protein folding. Nat. Rev. Mol. Cell Biol. 2015, 16, 742–752. [Google Scholar] [CrossRef]
- Ohtsubo, K.; Marth, J.D. Glycosylation in cellular mechanisms of health and disease. Cell 2006, 126, 855–867. [Google Scholar] [CrossRef]
- Wolfert, M.A.; Boons, G.J. Adaptive immune activation: Glycosylation does matter. Nat. Chem. Biol. 2013, 9, 776–784. [Google Scholar] [CrossRef]
- Dzobo, K.; Dandara, C. The Extracellular Matrix: Its Composition, Function, Remodeling, and Role in Tumorigenesis. Biomimetics 2023, 8, 146. [Google Scholar] [CrossRef]
- Pinho, S.S.; Alves, I.; Gaifem, J.; Rabinovich, G.A. Immune regulatory networks coordinated by glycans and glycan-binding proteins in autoimmunity and infection. Cell. Mol. Immunol. 2023, 20, 1101–1113. [Google Scholar] [CrossRef]
- Stowell, S.R.; Ju, T.; Cummings, R.D. Protein glycosylation in cancer. Annu. Rev. Pathol. 2015, 10, 473–510. [Google Scholar] [CrossRef]
- Taniguchi, N.; Kizuka, Y. Glycans and cancer: Role of N-glycans in cancer biomarker, progression and metastasis, and therapeutics. Adv. Cancer Res. 2015, 126, 11–51. [Google Scholar] [CrossRef] [PubMed]
- Chandler, K.B.; Costello, C.E.; Rahimi, N. Glycosylation in the Tumor Microenvironment: Implications for Tumor Angiogenesis and Metastasis. Cells 2019, 8, 544. [Google Scholar] [CrossRef] [PubMed]
- Costa, A.F.; Campos, D.; Reis, C.A.; Gomes, C. Targeting Glycosylation: A New Road for Cancer Drug Discovery. Trends Cancer 2020, 6, 757–766. [Google Scholar] [CrossRef] [PubMed]
- Mereiter, S.; Balmana, M.; Campos, D.; Gomes, J.; Reis, C.A. Glycosylation in the Era of Cancer-Targeted Therapy: Where Are We Heading? Cancer Cell 2019, 36, 6–16. [Google Scholar] [CrossRef]
- Thomas, D.; Rathinavel, A.K.; Radhakrishnan, P. Altered glycosylation in cancer: A promising target for biomarkers and therapeutics. Biochim. Biophys. Acta Rev. Cancer 2021, 1875, 188464. [Google Scholar] [CrossRef]
- Haukedal, H.; Freude, K.K. Implications of Glycosylation in Alzheimer’s Disease. Front. Neurosci. 2020, 14, 625348. [Google Scholar] [CrossRef]
- Pradeep, P.; Kang, H.; Lee, B. Glycosylation and behavioral symptoms in neurological disorders. Transl. Psychiatry 2023, 13, 154. [Google Scholar] [CrossRef]
- Zhao, J.; Lang, M. New insight into protein glycosylation in the development of Alzheimer’s disease. Cell Death Discov. 2023, 9, 314. [Google Scholar] [CrossRef]
- Conroy, L.R.; Hawkinson, T.R.; Young, L.E.A.; Gentry, M.S.; Sun, R.C. Emerging roles of N-linked glycosylation in brain physiology and disorders. Trends Endocrinol. Metab. 2021, 32, 980–993. [Google Scholar] [CrossRef] [PubMed]
- Paprocka, J.; Jezela-Stanek, A.; Tylki-Szymanska, A.; Grunewald, S. Congenital Disorders of Glycosylation from a Neurological Perspective. Brain Sci. 2021, 11, 88. [Google Scholar] [CrossRef] [PubMed]
- Groux-Degroote, S.; Cavdarli, S.; Uchimura, K.; Allain, F.; Delannoy, P. Glycosylation changes in inflammatory diseases. Adv. Protein Chem. Struct. Biol. 2020, 119, 111–156. [Google Scholar] [CrossRef]
- Kissel, T.; Toes, R.E.M.; Huizinga, T.W.J.; Wuhrer, M. Glycobiology of rheumatic diseases. Nat. Rev. Rheumatol. 2023, 19, 28–43. [Google Scholar] [CrossRef] [PubMed]
- Loke, I.; Kolarich, D.; Packer, N.H.; Thaysen-Andersen, M. Emerging roles of protein mannosylation in inflammation and infection. Mol. Aspects Med. 2016, 51, 31–55. [Google Scholar] [CrossRef] [PubMed]
- Ugonotti, J.; Chatterjee, S.; Thaysen-Andersen, M. Structural and functional diversity of neutrophil glycosylation in innate immunity and related disorders. Mol. Aspects Med. 2021, 79, 100882. [Google Scholar] [CrossRef]
- Lin, B.; Qing, X.; Liao, J.; Zhuo, K. Role of Protein Glycosylation in Host-Pathogen Interaction. Cells 2020, 9, 1022. [Google Scholar] [CrossRef]
- Wang, S.H.; Wu, T.J.; Lee, C.W.; Yu, J. Dissecting the conformation of glycans and their interactions with proteins. J. Biomed. Sci. 2020, 27, 93. [Google Scholar] [CrossRef]
- Varki, A.; Freeze, H.H. Glycans in Acquired Human Diseases. In Essentials of Glycobiology, 2nd ed.; Varki, A., Cummings, R.D., Esko, J.D., Freeze, H.H., Stanley, P., Bertozzi, C.R., Hart, G.W., Etzler, M.E., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2009. [Google Scholar]
- Zhou, J.Y.; Cobb, B.A. Glycans in Immunologic Health and Disease. Annu. Rev. Immunol. 2021, 39, 511–536. [Google Scholar] [CrossRef]
- Barb, A.W. Fc gamma receptor compositional heterogeneity: Considerations for immunotherapy development. J. Biol. Chem. 2021, 296, 100057. [Google Scholar] [CrossRef]
- Saporiti, S.; Parravicini, C.; Pergola, C.; Guerrini, U.; Rossi, M.; Centola, F.; Eberini, I. IgG1 conformational behavior: Elucidation of the N-glycosylation role via molecular dynamics. Biophys. J. 2021, 120, 5355–5370. [Google Scholar] [CrossRef]
- Subedi, G.P.; Hanson, Q.M.; Barb, A.W. Restricted motion of the conserved immunoglobulin G1 N-glycan is essential for efficient FcgammaRIIIa binding. Structure 2014, 22, 1478–1488. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Izadi, S.; Callahan, M.; Deperalta, G.; Wecksler, A.T. Antibody-receptor interactions mediate antibody-dependent cellular cytotoxicity. J. Biol. Chem. 2021, 297, 100826. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Liu, W.; Li, Y.; Ma, B.; Shang, S.; Tan, Z. Impact of N-Linked Glycosylation on Therapeutic Proteins. Molecules 2022, 27, 8859. [Google Scholar] [CrossRef] [PubMed]
- Pinho, S.S.; Reis, C.A. Glycosylation in cancer: Mechanisms and clinical implications. Nat. Rev. Cancer 2015, 15, 540–555. [Google Scholar] [CrossRef] [PubMed]
- Buffone, A.; Weaver, V.M. Don’t sugarcoat it: How glycocalyx composition influences cancer progression. J. Cell Biol. 2020, 219, e201910070. [Google Scholar] [CrossRef]
- Lee, H.S.; Qi, Y.; Im, W. Effects of N-glycosylation on protein conformation and dynamics: Protein Data Bank analysis and molecular dynamics simulation study. Sci. Rep. 2015, 5, 8926. [Google Scholar] [CrossRef]
- Mule, S.N.; Rosa-Fernandes, L.; Coutinho, J.V.P.; Gomes, V.M.; Macedo-da-Silva, J.; Santiago, V.F.; Quina, D.; de Oliveira, G.S.; Thaysen-Andersen, M.; Larsen, M.R.; et al. Systems-wide analysis of glycoprotein conformational changes by limited deglycosylation assay. J. Proteom. 2021, 248, 104355. [Google Scholar] [CrossRef] [PubMed]
- Papaleo, E.; Saladino, G.; Lambrughi, M.; Lindorff-Larsen, K.; Gervasio, F.L.; Nussinov, R. The Role of Protein Loops and Linkers in Conformational Dynamics and Allostery. Chem. Rev. 2016, 116, 6391–6423. [Google Scholar] [CrossRef]
- Cascella, M.; Rajnik, M.; Aleem, A.; Dulebohn, S.C.; Di Napoli, R. Features, Evaluation, and Treatment of Coronavirus (COVID-19). In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Casalino, L.; Gaieb, Z.; Goldsmith, J.A.; Hjorth, C.K.; Dommer, A.C.; Harbison, A.M.; Fogarty, C.A.; Barros, E.P.; Taylor, B.C.; McLellan, J.S.; et al. Beyond Shielding: The Roles of Glycans in the SARS-CoV-2 Spike Protein. ACS Cent. Sci. 2020, 6, 1722–1734. [Google Scholar] [CrossRef] [PubMed]
- Malaquias, M.A.S.; Gadotti, A.C.; Motta-Junior, J.D.S.; Martins, A.P.C.; Azevedo, M.L.V.; Benevides, A.P.K.; Cezar-Neto, P.; Panini do Carmo, L.A.; Zeni, R.C.; Raboni, S.M.; et al. The role of the lectin pathway of the complement system in SARS-CoV-2 lung injury. Transl. Res. 2021, 231, 55–63. [Google Scholar] [CrossRef]
- Jiang, S.; Hillyer, C.; Du, L. Neutralizing Antibodies against SARS-CoV-2 and Other Human Coronaviruses. Trends Immunol. 2020, 41, 355–359. [Google Scholar] [CrossRef]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef]
- Du, L.; He, Y.; Zhou, Y.; Liu, S.; Zheng, B.J.; Jiang, S. The spike protein of SARS-CoV—A target for vaccine and therapeutic development. Nat. Rev. Microbiol. 2009, 7, 226–236. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Gui, M.; Wang, X.; Xiang, Y. Cryo-EM structure of the SARS coronavirus spike glycoprotein in complex with its host cell receptor ACE2. PLoS Pathog. 2018, 14, e1007236. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.-H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef]
- Benton, D.J.; Wrobel, A.G.; Xu, P.; Roustan, C.; Martin, S.R.; Rosenthal, P.B.; Skehel, J.J.; Gamblin, S.J. Receptor binding and priming of the spike protein of SARS-CoV-2 for membrane fusion. Nature 2020, 588, 327–330. [Google Scholar] [CrossRef]
- Lu, M.; Uchil, P.D.; Li, W.; Zheng, D.; Terry, D.S.; Gorman, J.; Shi, W.; Zhang, B.; Zhou, T.; Ding, S.; et al. Real-Time Conformational Dynamics of SARS-CoV-2 Spikes on Virus Particles. Cell Host Microbe 2020, 28, 880–891.e8. [Google Scholar] [CrossRef]
- Walls, A.C.; Park, Y.J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 183, 1735. [Google Scholar] [CrossRef] [PubMed]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Parsons, L.M.; Jankowska, E.; Cipollo, J.F. Site-Specific Glycosylation Patterns of the SARS-CoV-2 Spike Protein Derived From Recombinant Protein and Viral WA1 and D614G Strains. Front. Chem. 2021, 9, 767448. [Google Scholar] [CrossRef]
- Cai, Y.; Zhang, J.; Xiao, T.; Peng, H.; Sterling, S.M.; Walsh, R.M., Jr.; Rawson, S.; Rits-Volloch, S.; Chen, B. Distinct conformational states of SARS-CoV-2 spike protein. Science 2020, 369, 1586–1592. [Google Scholar] [CrossRef]
- Fan, X.; Cao, D.; Kong, L.; Zhang, X. Cryo-EM analysis of the post-fusion structure of the SARS-CoV spike glycoprotein. Nat. Commun. 2020, 11, 3618. [Google Scholar] [CrossRef] [PubMed]
- Harbison, A.M.; Fogarty, C.A.; Phung, T.K.; Satheesan, A.; Schulz, B.L.; Fadda, E. Fine-tuning the spike: Role of the nature and topology of the glycan shield in the structure and dynamics of the SARS-CoV-2 S. Chem. Sci. 2022, 13, 386–395. [Google Scholar] [CrossRef]
- Ray, D.; Le, L.; Andricioaei, I. Distant residues modulate conformational opening in SARS-CoV-2 spike protein. Proc. Natl. Acad. Sci. USA 2021, 118, e2100943118. [Google Scholar] [CrossRef]
- Zimmerman, M.I.; Porter, J.R.; Ward, M.D.; Singh, S.; Vithani, N.; Meller, A.; Mallimadugula, U.L.; Kuhn, C.E.; Borowsky, J.H.; Wiewiora, R.P.; et al. SARS-CoV-2 simulations go exascale to predict dramatic spike opening and cryptic pockets across the proteome. Nat. Chem. 2021, 13, 651–659. [Google Scholar] [CrossRef] [PubMed]
- Fallon, L.; Belfon, K.A.A.; Raguette, L.; Wang, Y.; Stepanenko, D.; Cuomo, A.; Guerra, J.; Budhan, S.; Varghese, S.; Corbo, C.P.; et al. Free Energy Landscapes from SARS-CoV-2 Spike Glycoprotein Simulations Suggest that RBD Opening Can Be Modulated via Interactions in an Allosteric Pocket. J. Am. Chem. Soc. 2021, 143, 11349–11360. [Google Scholar] [CrossRef] [PubMed]
- Sztain, T.; Ahn, S.H.; Bogetti, A.T.; Casalino, L.; Goldsmith, J.A.; Seitz, E.; McCool, R.S.; Kearns, F.L.; Acosta-Reyes, F.; Maji, S.; et al. A glycan gate controls opening of the SARS-CoV-2 spike protein. Nat. Chem. 2021, 13, 963–968. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.W.; Jasnow, D.; Zuckerman, D.M. The “weighted ensemble” path sampling method is statistically exact for a broad class of stochastic processes and binning procedures. J. Chem. Phys. 2010, 132, 054107. [Google Scholar] [CrossRef]
- Pang, Y.T.; Acharya, A.; Lynch, D.L.; Pavlova, A.; Gumbart, J.C. SARS-CoV-2 spike opening dynamics and energetics reveal the individual roles of glycans and their collective impact. Commun. Biol. 2022, 5, 1170. [Google Scholar] [CrossRef]
- Dodero-Rojas, E.; Onuchic, J.N.; Whitford, P.C. Sterically confined rearrangements of SARS-CoV-2 Spike protein control cell invasion. eLife 2021, 10, e70362. [Google Scholar] [CrossRef] [PubMed]
- Geschwind, M.D. Prion Diseases. Contin. Lifelong Learn. Neurol. 2015, 21, 1612–1638. [Google Scholar] [CrossRef]
- Collinge, J. Mammalian prions and their wider relevance in neurodegenerative diseases. Nature 2016, 539, 217–226. [Google Scholar] [CrossRef]
- Johnson, R.T. Prion diseases. Lancet Neurol. 2005, 4, 635–642. [Google Scholar] [CrossRef] [PubMed]
- Westergard, L.; Christensen, H.M.; Harris, D.A. The cellular prion protein (PrPC): Its physiological function and role in disease. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2007, 1772, 629–644. [Google Scholar] [CrossRef]
- Ermonval, M.; Mouillet-Richard, S.; Codogno, P.; Kellermann, O.; Botti, J. Evolving views in prion glycosylation: Functional and pathological implications. Biochimie 2003, 85, 33–45. [Google Scholar] [CrossRef]
- Xiao, X.; Yuan, J.; Haik, S.; Cali, I.; Zhan, Y.; Moudjou, M.; Li, B.; Laplanche, J.L.; Laude, H.; Langeveld, J.; et al. Glycoform-selective prion formation in sporadic and familial forms of prion disease. PLoS ONE 2013, 8, e58786. [Google Scholar] [CrossRef]
- Mallucci, G.; Dickinson, A.; Linehan, J.; Klohn, P.C.; Brandner, S.; Collinge, J. Depleting neuronal PrP in prion infection prevents disease and reverses spongiosis. Science 2003, 302, 871–874. [Google Scholar] [CrossRef]
- Aguilar-Calvo, P.; Callender, J.A.; Sigurdson, C.J. Short and sweet: How glycans impact prion conversion, cofactor interactions, and cross-species transmission. PLOS Pathog. 2021, 17, e1009123. [Google Scholar] [CrossRef]
- Aguilar-Calvo, P.; Xiao, X.; Bett, C.; Erana, H.; Soldau, K.; Castilla, J.; Nilsson, K.P.; Surewicz, W.K.; Sigurdson, C.J. Post-translational modifications in PrP expand the conformational diversity of prions in vivo. Sci. Rep. 2017, 7, 43295. [Google Scholar] [CrossRef]
- Angers, R.C.; Kang, H.E.; Napier, D.; Browning, S.; Seward, T.; Mathiason, C.; Balachandran, A.; McKenzie, D.; Castilla, J.; Soto, C.; et al. Prion strain mutation determined by prion protein conformational compatibility and primary structure. Science 2010, 328, 1154–1158. [Google Scholar] [CrossRef] [PubMed]
- Nakic, N.; Tran, T.H.; Novokmet, M.; Andreoletti, O.; Lauc, G.; Legname, G. Site-specific analysis of N-glycans from different sheep prion strains. PLoS Pathog. 2021, 17, e1009232. [Google Scholar] [CrossRef]
- Sevillano, A.M.; Aguilar-Calvo, P.; Kurt, T.D.; Lawrence, J.A.; Soldau, K.; Nam, T.H.; Schumann, T.; Pizzo, D.P.; Nystrom, S.; Choudhury, B.; et al. Prion protein glycans reduce intracerebral fibril formation and spongiosis in prion disease. J. Clin. Investig. 2020, 130, 1350–1362. [Google Scholar] [CrossRef] [PubMed]
- Corsaro, A.; Thellung, S.; Villa, V.; Nizzari, M.; Florio, T. Role of prion protein aggregation in neurotoxicity. Int. J. Mol. Sci. 2012, 13, 8648–8669. [Google Scholar] [CrossRef]
- Poggiolini, I.; Saverioni, D.; Parchi, P. Prion protein misfolding, strains, and neurotoxicity: An update from studies on Mammalian prions. Int. J. Cell Biol. 2013, 2013, 910314. [Google Scholar] [CrossRef]
- Salamat, M.K.; Dron, M.; Chapuis, J.; Langevin, C.; Laude, H. Prion propagation in cells expressing PrP glycosylation mutants. J. Virol. 2011, 85, 3077–3085. [Google Scholar] [CrossRef]
- Cancellotti, E.; Mahal, S.P.; Somerville, R.; Diack, A.; Brown, D.; Piccardo, P.; Weissmann, C.; Manson, J.C. Post-translational changes to PrP alter transmissible spongiform encephalopathy strain properties. EMBO J. 2013, 32, 756–769. [Google Scholar] [CrossRef]
- Yi, C.W.; Wang, L.Q.; Huang, J.J.; Pan, K.; Chen, J.; Liang, Y. Glycosylation Significantly Inhibits the Aggregation of Human Prion Protein and Decreases Its Cytotoxicity. Sci. Rep. 2018, 8, 12603. [Google Scholar] [CrossRef]
- Schilling, K.M.; Jorwal, P.; Ubilla-Rodriguez, N.C.; Assafa, T.E.; Gatdula, J.R.P.; Vultaggio, J.S.; Harris, D.A.; Millhauser, G.L. N-glycosylation is a potent regulator of prion protein neurotoxicity. J. Biol. Chem. 2023, 299, 105101. [Google Scholar] [CrossRef]
- DeArmond, S.J.; Sanchez, H.; Yehiely, F.; Qiu, Y.; Ninchak-Casey, A.; Daggett, V.; Camerino, A.P.; Cayetano, J.; Rogers, M.; Groth, D.; et al. Selective neuronal targeting in prion disease. Neuron 1997, 19, 1337–1348. [Google Scholar] [CrossRef]
- Aguilar-Calvo, P.; Sevillano, A.M.; Bapat, J.; Soldau, K.; Sandoval, D.R.; Altmeppen, H.C.; Linsenmeier, L.; Pizzo, D.P.; Geschwind, M.D.; Sanchez, H.; et al. Shortening heparan sulfate chains prolongs survival and reduces parenchymal plaques in prion disease caused by mobile, ADAM10-cleaved prions. Acta Neuropathol. 2020, 139, 527–546. [Google Scholar] [CrossRef] [PubMed]
- Giachin, G.; Biljan, I.; Ilc, G.; Plavec, J.; Legname, G. Probing early misfolding events in prion protein mutants by NMR spectroscopy. Molecules 2013, 18, 9451–9476. [Google Scholar] [CrossRef] [PubMed]
- Riek, R.; Hornemann, S.; Wider, G.; Glockshuber, R.; Wuthrich, K. NMR characterization of the full-length recombinant murine prion protein, mPrP(23-231). FEBS Lett. 1997, 413, 282–288. [Google Scholar] [CrossRef] [PubMed]
- Zuegg, J.; Gready, J.E. Molecular dynamics simulation of human prion protein including both N-linked oligosaccharides and the GPI anchor. Glycobiology 2000, 10, 959–974. [Google Scholar] [CrossRef] [PubMed]
- Schilling, K.M.; Tao, L.; Wu, B.; Kiblen, J.T.M.; Ubilla-Rodriguez, N.C.; Pushie, M.J.; Britt, R.D.; Roseman, G.P.; Harris, D.A.; Millhauser, G.L. Both N-Terminal and C-Terminal Histidine Residues of the Prion Protein Are Essential for Copper Coordination and Neuroprotective Self-Regulation. J. Mol. Biol. 2020, 432, 4408–4425. [Google Scholar] [CrossRef] [PubMed]
- Evans, E.G.B.; Pushie, M.J.; Markham, K.A.; Lee, H.-W.; Millhauser, G.L. Interaction between Prion Protein’s Copper-Bound Octarepeat Domain and a Charged C-Terminal Pocket Suggests a Mechanism for N-Terminal Regulation. Structure 2016, 24, 1057–1067. [Google Scholar] [CrossRef]
- Spevacek, A.R.; Evans, E.G.B.; Miller, J.L.; Meyer, H.C.; Pelton, J.G.; Millhauser, G.L. Zinc Drives a Tertiary Fold in the Prion Protein with Familial Disease Mutation Sites at the Interface. Structure 2013, 21, 236–246. [Google Scholar] [CrossRef]
- Marzec, M.; Eletto, D.; Argon, Y. GRP94: An HSP90-like protein specialized for protein folding and quality control in the endoplasmic reticulum. Biochim. Biophys. Acta 2012, 1823, 774–787. [Google Scholar] [CrossRef] [PubMed]
- Ansa-Addo, E.A.; Thaxton, J.; Hong, F.; Wu, B.X.; Zhang, Y.; Fugle, C.W.; Metelli, A.; Riesenberg, B.; Williams, K.; Gewirth, D.T.; et al. Clients and Oncogenic Roles of Molecular Chaperone gp96/grp94. Curr. Top. Med. Chem. 2016, 16, 2765–2778. [Google Scholar] [CrossRef]
- Eletto, D.; Dersh, D.; Argon, Y. GRP94 in ER quality control and stress responses. Semin. Cell Dev. Biol. 2010, 21, 479–485. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.S. Glucose-regulated proteins in cancer: Molecular mechanisms and therapeutic potential. Nat. Rev. Cancer 2014, 14, 263–276. [Google Scholar] [CrossRef] [PubMed]
- Wiersma, V.R.; Michalak, M.; Abdullah, T.M.; Bremer, E.; Eggleton, P. Mechanisms of Translocation of ER Chaperones to the Cell Surface and Immunomodulatory Roles in Cancer and Autoimmunity. Front. Oncol. 2015, 5, 7. [Google Scholar] [CrossRef]
- Altmeyer, A.; Maki, R.G.; Feldweg, A.M.; Heike, M.; Protopopov, V.P.; Masur, S.K.; Srivastava, P.K. Tumor-specific cell surface expression of the-KDEL containing, endoplasmic reticular heat shock protein gp96. Int. J. Cancer 1996, 69, 340–349. [Google Scholar] [CrossRef]
- Booth, C.; Koch, G.L. Perturbation of cellular calcium induces secretion of luminal ER proteins. Cell 1989, 59, 729–737. [Google Scholar] [CrossRef]
- Martins, M.; Custodio, R.; Camejo, A.; Almeida, M.T.; Cabanes, D.; Sousa, S. Listeria monocytogenes triggers the cell surface expression of Gp96 protein and interacts with its N terminus to support cellular infection. J. Biol. Chem. 2012, 287, 43083–43093. [Google Scholar] [CrossRef]
- Rothan, H.A.; Zhong, Y.; Sanborn, M.A.; Teoh, T.C.; Ruan, J.; Yusof, R.; Hang, J.; Henderson, M.J.; Fang, S. Small molecule grp94 inhibitors block dengue and Zika virus replication. Antiviral Res. 2019, 171, 104590. [Google Scholar] [CrossRef] [PubMed]
- Sumitomo, T.; Nakata, M.; Nagase, S.; Takahara, Y.; Honda-Ogawa, M.; Mori, Y.; Akamatsu, Y.; Yamaguchi, M.; Okamoto, S.; Kawabata, S. GP96 Drives Exacerbation of Secondary Bacterial Pneumonia following Influenza A Virus Infection. mBio 2021, 12, e0326920. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Dai, J.; Zheng, H.; Stoilova, D.; Sun, S.; Li, Z. Cell surface expression of an endoplasmic reticulum resident heat shock protein gp96 triggers MyD88-dependent systemic autoimmune diseases. Proc. Natl. Acad. Sci. USA 2003, 100, 15824–15829. [Google Scholar] [CrossRef]
- Li, X.; Sun, L.; Hou, J.; Gui, M.; Ying, J.; Zhao, H.; Lv, N.; Meng, S. Cell membrane gp96 facilitates HER2 dimerization and serves as a novel target in breast cancer. Int. J. Cancer 2015, 137, 512–524. [Google Scholar] [CrossRef] [PubMed]
- Patel, P.D.; Yan, P.; Seidler, P.M.; Patel, H.J.; Sun, W.; Yang, C.; Que, N.S.; Taldone, T.; Finotti, P.; Stephani, R.A.; et al. Paralog-selective Hsp90 inhibitors define tumor-specific regulation of HER2. Nat. Chem. Biol. 2013, 9, 677–684. [Google Scholar] [CrossRef]
- Yan, P.; Patel, H.J.; Sharma, S.; Corben, A.; Wang, T.; Panchal, P.; Yang, C.; Sun, W.; Araujo, T.L.; Rodina, A.; et al. Molecular Stressors Engender Protein Connectivity Dysfunction through Aberrant N-Glycosylation of a Chaperone. Cell Rep. 2020, 31, 107840. [Google Scholar] [CrossRef]
- Chavany, C.; Mimnaugh, E.; Miller, P.; Bitton, R.; Nguyen, P.; Trepel, J.; Whitesell, L.; Schnur, R.; Moyer, J.; Neckers, L. p185erbB2 binds to GRP94 in vivo. Dissociation of the p185erbB2/GRP94 heterocomplex by benzoquinone ansamycins precedes depletion of p185erbB2. J. Biol. Chem. 1996, 271, 4974–4977. [Google Scholar] [CrossRef]
- Chaumonnot, K.; Masson, S.; Sikner, H.; Bouchard, A.; Baverel, V.; Bellaye, P.S.; Collin, B.; Garrido, C.; Kohli, E. The HSP GRP94 interacts with macrophage intracellular complement C3 and impacts M2 profile during ER stress. Cell Death Dis. 2021, 12, 114. [Google Scholar] [CrossRef]
- Ratna, A.; Lim, A.; Li, Z.; Argemi, J.; Bataller, R.; Chiosis, G.; Mandrekar, P. Myeloid Endoplasmic Reticulum Resident Chaperone GP96 Facilitates Inflammation and Steatosis in Alcohol-Associated Liver Disease. Hepatol. Commun. 2021, 5, 1165–1182. [Google Scholar] [CrossRef]
- Pagetta, A.; Tramentozzi, E.; Tibaldi, E.; Cendron, L.; Zanotti, G.; Brunati, A.M.; Vitadello, M.; Gorza, L.; Finotti, P. Structural insights into complexes of glucose-regulated Protein94 (Grp94) with human immunoglobulin G. relevance for Grp94-IgG complexes that form in vivo in pathological conditions. PLoS ONE 2014, 9, e86198. [Google Scholar] [CrossRef]
- Cherepanova, N.A.; Venev, S.V.; Leszyk, J.D.; Shaffer, S.A.; Gilmore, R. Quantitative glycoproteomics reveals new classes of STT3A- and STT3B-dependent N-glycosylation sites. J. Cell Biol. 2019, 218, 2782–2796. [Google Scholar] [CrossRef]
- Wearsch, P.A.; Nicchitta, C.V. Purification and partial molecular characterization of GRP94, an ER resident chaperone. Protein Expr. Purif. 1996, 7, 114–121. [Google Scholar] [CrossRef]
- Wen, P.; Chen, J.; Zuo, C.; Gao, X.; Fujita, M.; Yang, G. Proteome and Glycoproteome Analyses Reveal the Protein N-Linked Glycosylation Specificity of STT3A and STT3B. Cells 2022, 11, 2775. [Google Scholar] [CrossRef]
- Gottschalk, N.; Kimmig, R.; Lang, S.; Singh, M.; Brandau, S. Anti-epidermal growth factor receptor (EGFR) antibodies overcome resistance of ovarian cancer cells to targeted therapy and natural cytotoxicity. Int. J. Mol. Sci. 2012, 13, 12000–12016. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Zhang, H. Large-Scale Measurement of Absolute Protein Glycosylation Stoichiometry. Anal. Chem. 2015, 87, 6479–6482. [Google Scholar] [CrossRef] [PubMed]
- Castelli, M.; Yan, P.; Rodina, A.; Digwal, C.S.; Panchal, P.; Chiosis, G.; Moroni, E.; Colombo, G. How aberrant N-glycosylation can alter protein functionality and ligand binding: An atomistic view. Structure 2023, 31, 987–1004.e8. [Google Scholar] [CrossRef] [PubMed]
- Bouchard, A.; Sikner, H.; Baverel, V.; Garnier, A.R.; Monterrat, M.; Moreau, M.; Limagne, E.; Garrido, C.; Kohli, E.; Collin, B.; et al. The GRP94 Inhibitor PU-WS13 Decreases M2-like Macrophages in Murine TNBC Tumors: A Pharmaco-Imaging Study with (99m)Tc-Tilmanocept SPECT. Cells 2021, 10, 3393. [Google Scholar] [CrossRef] [PubMed]
- Rodina, A.; Xu, C.; Digwal, C.S.; Joshi, S.; Patel, Y.; Santhaseela, A.R.; Bay, S.; Merugu, S.; Alam, A.; Yan, P.; et al. Systems-level analyses of protein-protein interaction network dysfunctions via epichaperomics identify cancer-specific mechanisms of stress adaptation. Nat. Commun. 2023, 14, 3742. [Google Scholar] [CrossRef]
- Chiosis, G.; Digwal, C.S.; Trepel, J.B.; Neckers, L. Structural and functional complexity of HSP90 in cellular homeostasis and disease. Nat. Rev. Mol. Cell Biol. 2023, 24, 797–815. [Google Scholar] [CrossRef] [PubMed]
- Rodina, A.; Wang, T.; Yan, P.; Gomes, E.D.; Dunphy, M.P.; Pillarsetty, N.; Koren, J.; Gerecitano, J.F.; Taldone, T.; Zong, H.; et al. The epichaperome is an integrated chaperome network that facilitates tumour survival. Nature 2016, 538, 397–401. [Google Scholar] [CrossRef]
- Barouch, D.H. COVID-19 Vaccines—Immunity, Variants, Boosters. N. Engl. J. Med. 2022, 387, 1011–1020. [Google Scholar] [CrossRef]
- Corti, D.; Purcell, L.A.; Snell, G.; Veesler, D. Tackling COVID-19 with neutralizing monoclonal antibodies. Cell 2021, 184, 4593–4595. [Google Scholar] [CrossRef] [PubMed]
- Raybould, M.I.J.; Kovaltsuk, A.; Marks, C.; Deane, C.M. CoV-AbDab: The coronavirus antibody database. Bioinformatics 2021, 37, 734–735. [Google Scholar] [CrossRef]
- Voss, W.N.; Hou, Y.J.; Johnson, N.V.; Delidakis, G.; Kim, J.E.; Javanmardi, K.; Horton, A.P.; Bartzoka, F.; Paresi, C.J.; Tanno, Y.; et al. Prevalent, protective, and convergent IgG recognition of SARS-CoV-2 non-RBD spike epitopes. Science 2021, 372, 1108–1112. [Google Scholar] [CrossRef]
- Serapian, S.A.; Marchetti, F.; Triveri, A.; Morra, G.; Meli, M.; Moroni, E.; Sautto, G.A.; Rasola, A.; Colombo, G. The Answer Lies in the Energy: How Simple Atomistic Molecular Dynamics Simulations May Hold the Key to Epitope Prediction on the Fully Glycosylated SARS-CoV-2 Spike Protein. J. Phys. Chem. Lett. 2020, 11, 8084–8093. [Google Scholar] [CrossRef] [PubMed]
- Triveri, A.; Serapian, S.A.; Marchetti, F.; Doria, F.; Pavoni, S.; Cinquini, F.; Moroni, E.; Rasola, A.; Frigerio, F.; Colombo, G. SARS-CoV-2 Spike Protein Mutations and Escape from Antibodies: A Computational Model of Epitope Loss in Variants of Concern. J. Chem. Inf. Model. 2021, 61, 4687–4700. [Google Scholar] [CrossRef]
- Triveri, A.; Casali, E.; Frasnetti, E.; Doria, F.; Frigerio, F.; Cinquini, F.; Pavoni, S.; Moroni, E.; Marchetti, F.; Serapian, S.A.; et al. Conformational Behavior of SARS-CoV-2 Spike Protein Variants: Evolutionary Jumps in Sequence Reverberate in Structural Dynamic Differences. J. Chem. Theory Comput. 2023, 19, 2120–2134. [Google Scholar] [CrossRef]
- Mead, S.; Whitfield, J.; Poulter, M.; Shah, P.; Uphill, J.; Campbell, T.; Al-Dujaily, H.; Hummerich, H.; Beck, J.; Mein, C.A.; et al. A novel protective prion protein variant that colocalizes with kuru exposure. N. Engl. J. Med. 2009, 361, 2056–2065. [Google Scholar] [CrossRef] [PubMed]
- Asante, E.A.; Smidak, M.; Grimshaw, A.; Houghton, R.; Tomlinson, A.; Jeelani, A.; Jakubcova, T.; Hamdan, S.; Richard-Londt, A.; Linehan, J.M.; et al. A naturally occurring variant of the human prion protein completely prevents prion disease. Nature 2015, 522, 478–481. [Google Scholar] [CrossRef]
- Zheng, Z.; Zhang, M.; Wang, Y.; Ma, R.; Guo, C.; Feng, L.; Wu, J.; Yao, H.; Lin, D. Structural basis for the complete resistance of the human prion protein mutant G127V to prion disease. Sci. Rep. 2018, 8, 13211. [Google Scholar] [CrossRef]
- Kuwata, K. Logical design of medical chaperone for prion diseases. Curr. Top. Med. Chem. 2013, 13, 2432–2440. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, K.; Kamatari, Y.O.; Ono, F.; Shibata, H.; Fuse, T.; Elhelaly, A.E.; Fukuoka, M.; Kimura, T.; Hosokawa-Muto, J.; Ishikawa, T.; et al. A designer molecular chaperone against transmissible spongiform encephalopathy slows disease progression in mice and macaques. Nat. Biomed. Eng. 2019, 3, 206–219. [Google Scholar] [CrossRef] [PubMed]
- Lysek, D.A.; Schorn, C.; Nivon, L.G.; Esteve-Moya, V.; Christen, B.; Calzolai, L.; von Schroetter, C.; Fiorito, F.; Herrmann, T.; Guntert, P.; et al. Prion protein NMR structures of cats, dogs, pigs, and sheep. Proc. Natl. Acad. Sci. USA 2005, 102, 640–645. [Google Scholar] [CrossRef] [PubMed]
- Duan, X.; Iwanowycz, S.; Ngoi, S.; Hill, M.; Zhao, Q.; Liu, B. Molecular Chaperone GRP94/GP96 in Cancers: Oncogenesis and Therapeutic Target. Front. Oncol. 2021, 11, 629846. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.X.; Hong, F.; Zhang, Y.; Ansa-Addo, E.; Li, Z. GRP94/gp96 in Cancer: Biology, Structure, Immunology, and Drug Development. Adv. Cancer Res. 2016, 129, 165–190. [Google Scholar] [CrossRef]
- Gewirth, D.T. Paralog Specific Hsp90 Inhibitors—A Brief History and a Bright Future. Curr. Top. Med. Chem. 2016, 16, 2779–2791. [Google Scholar] [CrossRef]
- Patel, H.J.; Patel, P.D.; Ochiana, S.O.; Yan, P.; Sun, W.; Patel, M.R.; Shah, S.K.; Tramentozzi, E.; Brooks, J.; Bolaender, A.; et al. Structure-activity relationship in a purine-scaffold compound series with selectivity for the endoplasmic reticulum Hsp90 paralog Grp94. J. Med. Chem. 2015, 58, 3922–3943. [Google Scholar] [CrossRef]
- Shrestha, L.; Patel, H.J.; Chiosis, G. Chemical Tools to Investigate Mechanisms Associated with HSP90 and HSP70 in Disease. Cell Chem. Biol. 2016, 23, 158–172. [Google Scholar] [CrossRef]
- Sousa, S.; Brion, R.; Lintunen, M.; Kronqvist, P.; Sandholm, J.; Monkkonen, J.; Kellokumpu-Lehtinen, P.L.; Lauttia, S.; Tynninen, O.; Joensuu, H.; et al. Human breast cancer cells educate macrophages toward the M2 activation status. Breast Cancer Res. 2015, 17, 101. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pasala, C.; Sharma, S.; Roychowdhury, T.; Moroni, E.; Colombo, G.; Chiosis, G. N-Glycosylation as a Modulator of Protein Conformation and Assembly in Disease. Biomolecules 2024, 14, 282. https://doi.org/10.3390/biom14030282
Pasala C, Sharma S, Roychowdhury T, Moroni E, Colombo G, Chiosis G. N-Glycosylation as a Modulator of Protein Conformation and Assembly in Disease. Biomolecules. 2024; 14(3):282. https://doi.org/10.3390/biom14030282
Chicago/Turabian StylePasala, Chiranjeevi, Sahil Sharma, Tanaya Roychowdhury, Elisabetta Moroni, Giorgio Colombo, and Gabriela Chiosis. 2024. "N-Glycosylation as a Modulator of Protein Conformation and Assembly in Disease" Biomolecules 14, no. 3: 282. https://doi.org/10.3390/biom14030282
APA StylePasala, C., Sharma, S., Roychowdhury, T., Moroni, E., Colombo, G., & Chiosis, G. (2024). N-Glycosylation as a Modulator of Protein Conformation and Assembly in Disease. Biomolecules, 14(3), 282. https://doi.org/10.3390/biom14030282