Structural and Functional Insights into Human Nuclear Cyclophilins

Abstract
:1. Introduction
2. Background and Structures of the Nuclear Cyclophilins within Splicing Complexes
2.1. Peptidyl Prolyl Isomerase Isoform H
2.2. Peptidyl Prolyl Isomerase Isoform E
2.3. Peptidyl Prolyl Isomerase-Like Isoform 1
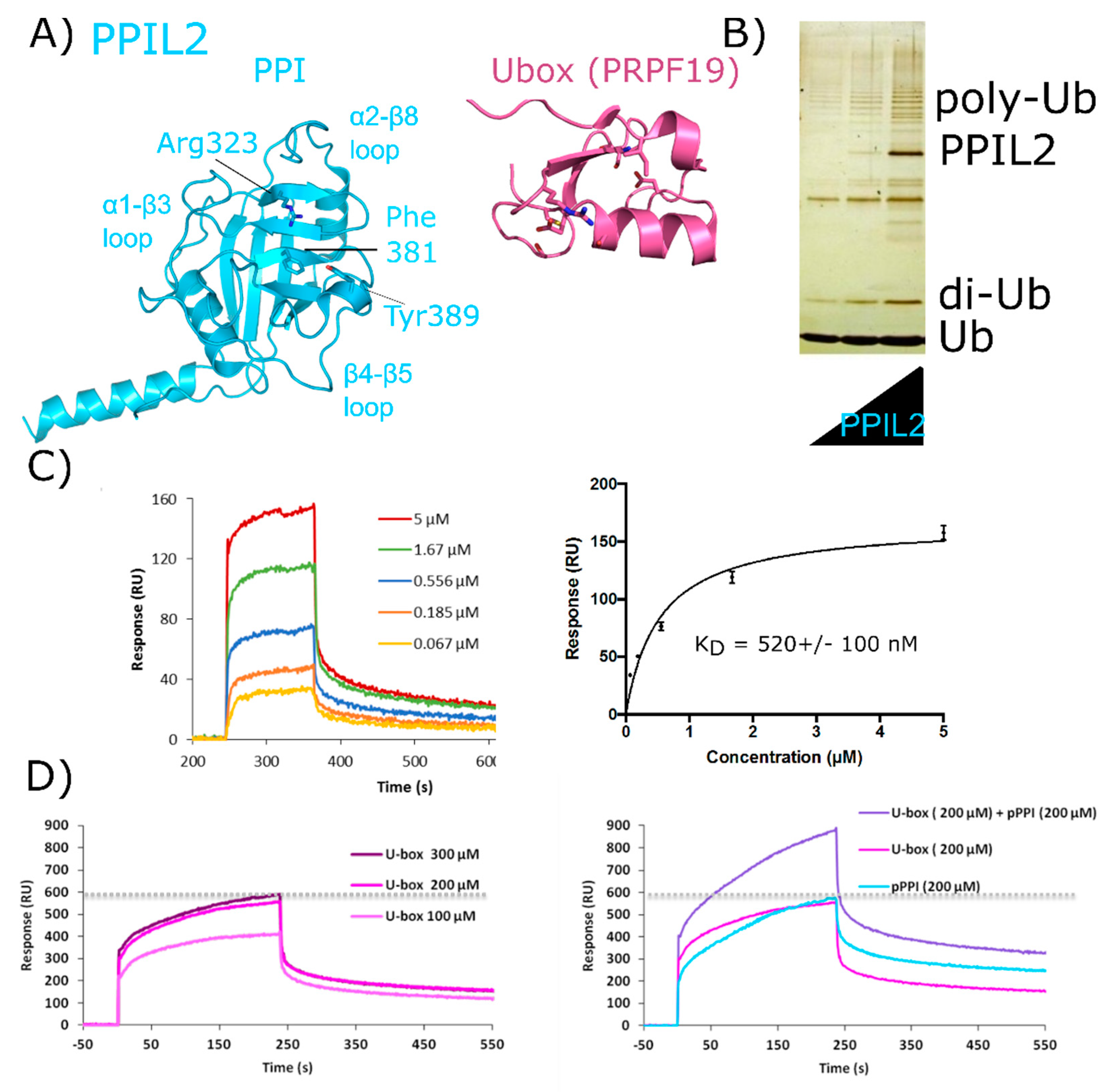
2.4. Peptidyl Prolyl Isomerase-Like Isoform 2
2.5. Peptidyl Prolyl Isomerase-Like Isoform 3
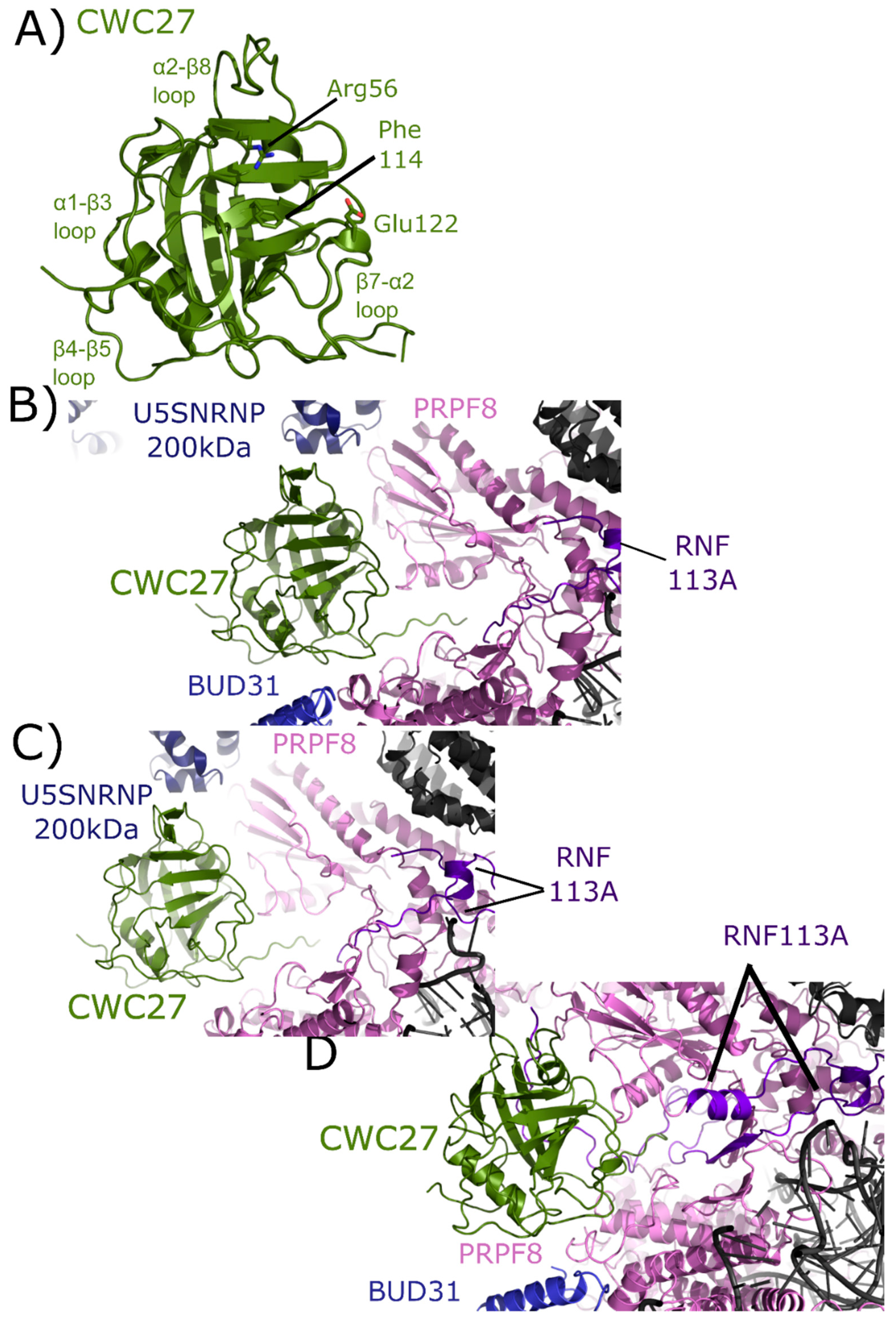
2.6. CWC27
2.7. WD40-Domain Containing Peptidyl Prolyl Isomerase 1
2.8. Peptidyl Prolyl Isomerase Isoform G
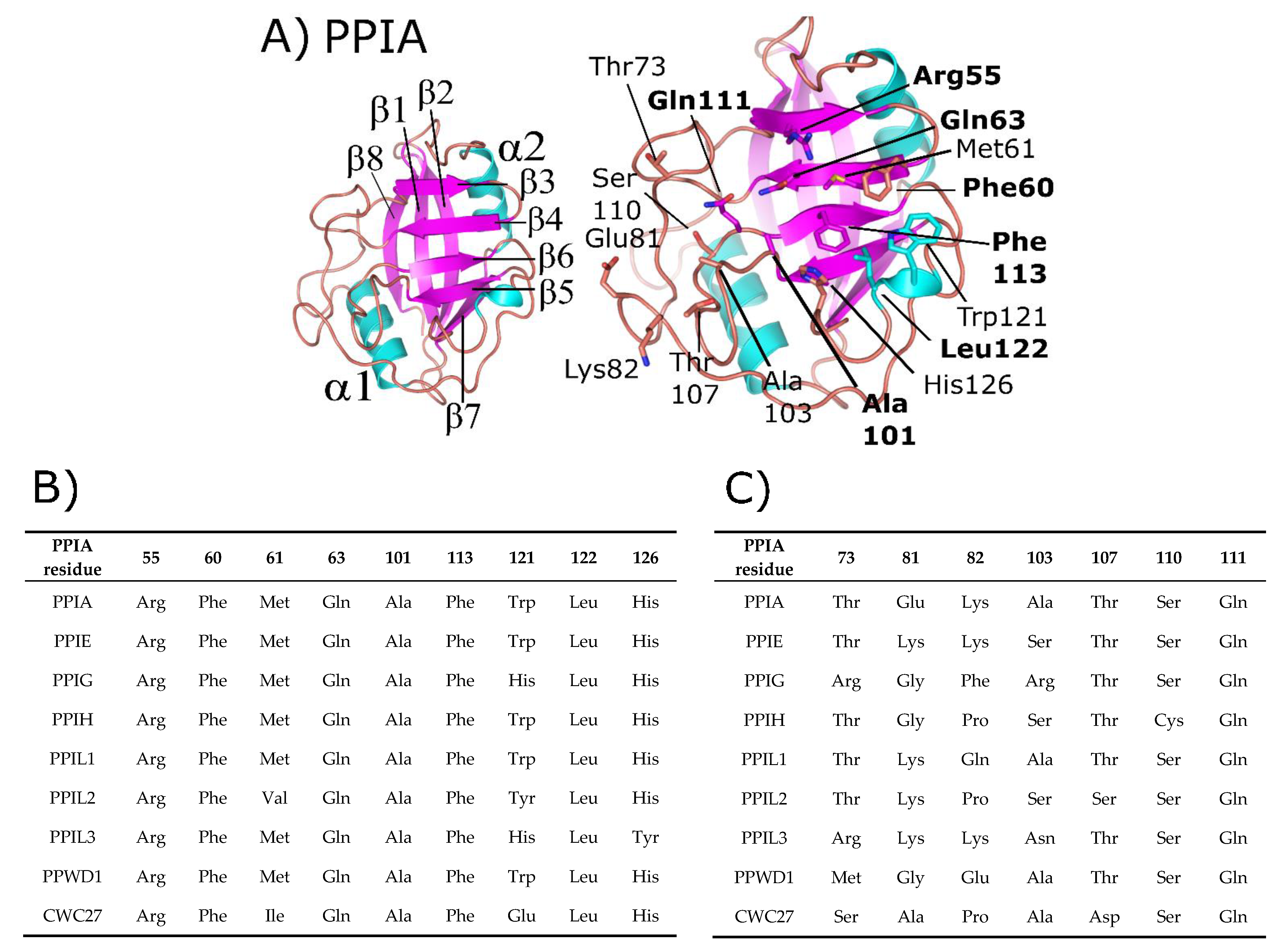
3. Structural Analysis of the Spliceophilins
4. Spliceophilin Function
5. Spliceophilins: Highly Druggable Modulators of Nuclear Function?
6. Conclusions and Final Thoughts
7. Accession Codes
Supplementary Materials
Funding
Acknowledgments
Conflicts of Interest
Appendix A
Appendix B
References
- Davis, T.L.; Walker, J.R.; Campagna-Slater, V.; Finerty, P.J.; Paramanathan, R.; Bernstein, G.; MacKenzie, F.; Tempel, W.; Ouyang, H.; Lee, W.H.; et al. Structural and biochemical characterization of the human cyclophilin family of peptidyl-prolyl isomerases. PLoS Boil. 2010, 8, e1000439. [Google Scholar] [CrossRef]
- Ferreira, P.A.; Orry, A. From Drosophila to humans: Reflections on the roles of the prolyl isomerases and chaperones, cyclophilins, in cell function and disease. J. Neurogenet. 2012, 26, 132–143. [Google Scholar] [CrossRef]
- Galat, A. Peptidylproline cis-trans-isomerases: Immunophilins. Eur. J. Biochem./FEBS 1993, 216, 689–707. [Google Scholar] [CrossRef]
- Kumari, S.; Roy, S.; Singh, P.; Singla-Pareek, S.L.; Pareek, A. Cyclophilins: Proteins in search of function. Plant Signal. Behav. 2013, 8, e22734. [Google Scholar] [CrossRef]
- Gordan, R.; Fefelova, N.; Gwathmey, J.K.; Xie, L.H. Involvement of mitochondrial permeability transition pore (mPTP) in cardiac arrhythmias: Evidence from cyclophilin D knockout mice. Cell Calcium 2016, 60, 363–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shore, E.R.; Awais, M.; Kershaw, N.M.; Gibson, R.R.; Pandalaneni, S.; Latawiec, D.; Wen, L.; Javed, M.A.; Criddle, D.N.; Berry, N.; et al. Small molecule inhibitors of cyclophilin D to protect mitochondrial function as a potential treatment for acute pancreatitis. J. Med. Chem. 2016, 59, 2596–2611. [Google Scholar] [CrossRef]
- Valasani, K.R.; Sun, Q.; Fang, D.; Zhang, Z.; Yu, Q.; Guo, Y.; Li, J.; Roy, A.; ShiDu Yan, S. Identification of a small molecule cyclophilin D inhibitor for rescuing Aβ-mediated mitochondrial dysfunction. ACS Med. Chem. Lett. 2016, 7, 294–299. [Google Scholar] [CrossRef] [PubMed]
- Ahmed-Belkacem, A.; Colliandre, L.; Ahnou, N.; Nevers, Q.; Gelin, M.; Bessin, Y.; Brillet, R.; Cala, O.; Douguet, D.; Bourguet, W.; et al. Fragment-based discovery of a new family of non-peptidic small-molecule cyclophilin inhibitors with potent antiviral activities. Nat. Commun. 2016, 7, 12777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dawar, F.U.; Tu, J.; Khattak, M.N.; Mei, J.; Lin, L. Cyclophilin A: A Key factor in virus replication and potential target for anti-viral therapy. Curr. Issues Mol. Biol. 2016, 21, 1–20. [Google Scholar] [PubMed]
- Yoshikawa, R.; Izumi, T.; Nakano, Y.; Yamada, E.; Moriwaki, M.; Misawa, N.; Ren, F.; Kobayashi, T.; Koyanagi, Y.; Sato, K. Small ruminant lentiviral Vif proteins commonly utilize cyclophilin A, an evolutionarily and structurally conserved protein, to degrade ovine and caprine APOBEC3 proteins. Microbiol. Immunol. 2016, 60, 427–436. [Google Scholar] [CrossRef] [PubMed]
- Kallen, J.; Mikol, V.; Taylor, P.; Walkinshaw, M.D. X-ray structures and analysis of 11 cyclosporin derivatives complexed with cyclophilin A1. J. Mol. Boil. 1998, 283, 435–449. [Google Scholar] [CrossRef]
- Kallen, J.; Spitzfaden, C.; Zurini, M.G.; Wider, G.; Widmer, H.; Wüthrich, K.; Walkinshaw, M.D. Structure of human cyclophilin and its binding site for cyclosporin A determined by X-ray crystallography and NMR spectroscopy. Nature 1991, 353, 276–279. [Google Scholar] [CrossRef] [PubMed]
- Kallen, J.; Walkinshaw, M.D. The X-ray structure of a tetrapeptide bound to the active site of human cyclophilin A. FEBS Lett. 1992, 300, 286–290. [Google Scholar] [CrossRef] [Green Version]
- Ke, H.M.; Zydowsky, L.D.; Liu, J.; Walsh, C.T. Crystal structure of recombinant human T-cell cyclophilin A at 2.5 A resolution. Proc. Natl. Acad. Sci. USA 1991, 88, 9483–9487. [Google Scholar] [CrossRef]
- Stegmann, C.M.; Luhrmann, R.; Wahl, M.C. The crystal structure of PPIL1 bound to cyclosporine A suggests a binding mode for a linear epitope of the SKIP protein. PLoS ONE 2010, 5, e10013. [Google Scholar] [CrossRef] [PubMed]
- Adams, B.M.; Coates, M.N.; Jackson, S.R.; Jurica, M.S.; Davis, T.L. Nuclear cyclophilins affect spliceosome assembly and function in vitro. Biochem. J. 2015, 469, 223–233. [Google Scholar] [CrossRef] [PubMed]
- Agafonov, D.E.; Deckert, J.; Wolf, E.; Odenwalder, P.; Bessonov, S.; Will, C.L.; Urlaub, H.; Luhrmann, R. Semiquantitative proteomic analysis of the human spliceosome via a novel two-dimensional gel electrophoresis method. Mol. Cell. Boil. 2011, 31, 2667–2682. [Google Scholar] [CrossRef] [PubMed]
- Cvitkovic, I.; Jurica, M.S. Spliceosome database: A tool for tracking components of the spliceosome. Nucleic Acids Res. 2013, 41, D132–D141. [Google Scholar] [CrossRef] [PubMed]
- Hegele, A.; Kamburov, A.; Grossmann, A.; Sourlis, C.; Wowro, S.; Weimann, M.; Will, C.L.; Pena, V.; Luhrmann, R.; Stelzl, U. Dynamic protein–protein interaction wiring of the human spliceosome. Mol. Cell 2012, 45, 567–580. [Google Scholar] [CrossRef]
- Jurica, M.S. Detailed close-ups and the big picture of spliceosomes. Curr. Opin. Struct. Biol. 2008, 18, 315–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wahl, M.C.; Will, C.L.; Luhrmann, R. The spliceosome: Design principles of a dynamic RNP machine. Cell 2009, 136, 701–718. [Google Scholar] [CrossRef] [PubMed]
- Will, C.L.; Luhrmann, R. Spliceosome structure and function. Cold Spring Harb. Perspect. Biol. 2011, 3. [Google Scholar] [CrossRef] [PubMed]
- Agafonov, D.E.; Kastner, B.; Dybkov, O.; Hofele, R.V.; Liu, W.T.; Urlaub, H.; Luhrmann, R.; Stark, H. Molecular architecture of the human U4/U6.U5 tri-snRNP. Science 2016, 351, 1416–1420. [Google Scholar] [CrossRef] [PubMed]
- Bertram, K.; Agafonov, D.E.; Dybkov, O.; Haselbach, D.; Leelaram, M.N.; Will, C.L.; Urlaub, H.; Kastner, B.; Luhrmann, R.; Stark, H. Cryo-EM structure of a pre-catalytic human spliceosome primed for activation. Cell 2017, 170, 701–713.e711. [Google Scholar] [CrossRef] [PubMed]
- Haselbach, D.; Komarov, I.; Agafonov, D.E.; Hartmuth, K.; Graf, B.; Dybkov, O.; Urlaub, H.; Kastner, B.; Luhrmann, R.; Stark, H. Structure and conformational dynamics of the human spliceosomal Bact complex. Cell 2018, 172, 454–464.e411. [Google Scholar] [CrossRef]
- Zhang, X.; Yan, C.; Zhan, X.; Li, L.; Lei, J.; Shi, Y. Structure of the human activated spliceosome in three conformational states. Cell Res. 2018, 28, 307–322. [Google Scholar] [CrossRef] [Green Version]
- Bertram, K.; Agafonov, D.E.; Liu, W.T.; Dybkov, O.; Will, C.L.; Hartmuth, K.; Urlaub, H.; Kastner, B.; Stark, H.; Luhrmann, R. Cryo-EM structure of a human spliceosome activated for step 2 of splicing. Nature 2017, 542, 318–323. [Google Scholar] [CrossRef]
- Zhan, X.; Yan, C.; Zhang, X.; Lei, J.; Shi, Y. Structure of a human catalytic step I spliceosome. Science 2018, 359, 537–545. [Google Scholar] [CrossRef]
- Zhang, X.; Yan, C.; Hang, J.; Finci, L.I.; Lei, J.; Shi, Y. An atomic structure of the human spliceosome. Cell 2017, 169, 918–929.e914. [Google Scholar] [CrossRef]
- Davis, T.L.; Walker, J.R.; Ouyang, H.; MacKenzie, F.; Butler-Cole, C.; Newman, E.M.; Eisenmesser, E.Z.; Dhe-Paganon, S. The crystal structure of human WD40 repeat-containing peptidylprolyl isomerase (PPWD1). FEBS J. 2008, 275, 2283–2295. [Google Scholar] [CrossRef] [Green Version]
- Gustafson, C.L.; Parsley, N.C.; Asimgil, H.; Lee, H.W.; Ahlbach, C.; Michael, A.K.; Xu, H.; Williams, O.L.; Davis, T.L.; Liu, A.C.; et al. A slow conformational switch in the BMAL1 transactivation domain modulates circadian rhythms. Mol. Cell 2017, 66, 447–457.e447. [Google Scholar] [CrossRef] [PubMed]
- Rajiv, C.; Jackson, S.R.; Cocklin, S.; Eisenmesser, E.Z.; Davis, T.L. The spliceosomal proteins PPIH and PRPF4 exhibit bi-partite binding. Biochem. J. 2017, 474, 3689–3704. [Google Scholar] [CrossRef] [PubMed]
- Skruzný, M.; Ambrozková, M.; Fuková, I.; Martínková, K.; Blahůsková, A.; Hamplová, L.; Půta, F.; Folk, P. Cyclophilins of a novel subfamily interact with SNW/SKIP coregulator in Dictyostelium discoideum and Schizosaccharomyces pombe. Biochim. Biophys. Acta 2001, 1521, 146–151. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, S.; Zhang, J.; Huang, X.; Xu, C.; Wang, W.; Liu, Z.; Wu, J.; Shi, Y. A large intrinsically disordered region in SKIP and its disorder-order transition induced by PPIL1 binding revealed by NMR. J. Boil. Chem. 2009, 285, 4951–4963. [Google Scholar] [CrossRef] [PubMed]
- Xu, C. Solution structure of human peptidyl prolyl isomerase-like protein 1 and insights into its interaction with SKIP. J. Boil. Chem. 2006, 281, 15900–15908. [Google Scholar] [CrossRef]
- Galej, W.P. Structural studies of the spliceosome: Past, present and future perspectives. Biochem. Soc. Trans. 2018. [Google Scholar] [CrossRef]
- Vander Kooi, C.W.; Ohi, M.D.; Rosenberg, J.A.; Oldham, M.L.; Newcomer, M.E.; Gould, K.L.; Chazin, W.J. The Prp19 U-box crystal structure suggests a common dimeric architecture for a class of oligomeric E3 ubiquitin ligases. Biochemistry 2006, 45, 121–130. [Google Scholar] [CrossRef]
- Horowitz, D.S.; Lee, E.J.; Mabon, S.A.; Misteli, T. A cyclophilin functions in pre-mRNA splicing. EMBO J. 2002, 21, 470–480. [Google Scholar] [CrossRef] [Green Version]
- Teigelkamp, S.; Achsel, T.; Mundt, C.; Göthel, S.F.; Cronshagen, U.; Lane, W.S.; Marahiel, M.; Lührmann, R. The 20kD protein of human [U4/U6.U5] tri-snRNPs is a novel cyclophilin that forms a complex with the U4/U6-specific 60 kD and 90 kD proteins. RNA 1998, 4, 127–141. [Google Scholar]
- Ingelfinger, D.; Gothel, S.F.; Marahiel, M.A.; Reidt, U.; Ficner, R.; Luhrmann, R.; Achsel, T. Two protein-protein interaction sites on the spliceosome-associated human cyclophilin CypH. Nucleic Acids Res. 2003, 31, 4791–4796. [Google Scholar] [CrossRef] [Green Version]
- Reidt, U.; Wahl, M.C.; Fasshauer, D.; Horowitz, D.S.; Lührmann, R.; Ficner, R. Crystal structure of a complex between human spliceosomal cyclophilin H and a U4/U6 snRNP-60K peptide. J. Mol. Boil. 2003, 331, 45–56. [Google Scholar] [CrossRef]
- Vidovic, I.; Nottrott, S.; Hartmuth, K.; Lührmann, R.; Ficner, R. Crystal structure of the spliceosomal 15.5 kD protein bound to a U4 snRNA fragment. Mol. Cell 2000, 6, 1331–1342. [Google Scholar] [CrossRef]
- Structural Genomics Consortium (SGC). Available online: www.thesgc.org/ (accessed on 30 October 2018).
- Wang, Y.; Han, R.; Zhang, W.; Yuan, Y.; Zhang, X.; Long, Y.; Mi, H. Human CyP33 binds specifically to mRNA and binding stimulates PPIase activity of hCyP33. FEBS Lett. 2008, 582, 835–839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joint Center for Structural Genomics. Available online: http://www.jcsg.org/ (accessed on 30 October 2018).
- RIKEN. Available online: http://www.riken.jp/en/ (accessed on 30 October 2018).
- Wang, Z.; Song, J.; Milne, T.A.; Wang, G.G.; Li, H.; Allis, C.D.; Patel, D.J. Pro Isomerization in MLL1 PHD3-Bromo Cassette Connects H3K4me Readout to CyP33 and HDAC-Mediated Repression. Cell 2010, 141, 1183–1194. [Google Scholar] [CrossRef] [Green Version]
- Hom, R.A.; Chang, P.Y.; Roy, S.; Musselman, C.A.; Glass, K.C.; Selezneva, A.I.; Gozani, O.; Ismagilov, R.F.; Cleary, M.L.; Kutateladze, T.G. Molecular mechanism of MLL PHD3 and RNA recognition by the Cyp33 RRM domain. J. Mol. Boil. 2010, 400, 145–154. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Osmers, U.; Raman, G.; Schwantes, R.H.; Diaz, M.O.; Bushweller, J.H. The PHD3 domain of MLL acts as a CYP33-regulated switch between MLL-mediated activation and repression. Biochemistry 2010, 49, 6576–6586. [Google Scholar] [CrossRef] [PubMed]
- Kuraoka, I.; Ito, S.; Wada, T.; Hayashida, M.; Lee, L.; Saijo, M.; Nakatsu, Y.; Matsumoto, M.; Matsunaga, T.; Handa, H.; et al. Isolation of XAB2 complex involved in pre-mRNA splicing, transcription, and transcription-coupled repair. J. Boil. Chem. 2007, 283, 940–950. [Google Scholar] [CrossRef]
- De, I.; Bessonov, S.; Hofele, R.; dos Santos, K.; Will, C.L.; Urlaub, H.; Luhrmann, R.; Pena, V. The RNA helicase Aquarius exhibits structural adaptations mediating its recruitment to spliceosomes. Nat. Struct. Mol. Boil. 2015, 22, 138–144. [Google Scholar] [CrossRef]
- Huang, L.L.; Zhao, X.M.; Huang, C.Q.; Yu, L.; Xia, Z.X. Structure of recombinant human cyclophilin J, a novel member of the cyclophilin family. Acta Crystallogr. D Biol. Crystallogr. 2005, 61, 316–321. [Google Scholar] [CrossRef]
- Hatakeyama, S.; Nakayama, K.I. U-box proteins as a new family of ubiquitin ligases. Biochem. Biophys. Res. Commun. 2003, 302, 635–645. [Google Scholar] [CrossRef]
- Davis, T.L.; Dhe-Paganon, S. Testing of E1-E2-E3 proteins to find ubiquitin modifier pairs. Structural Genomics Consortium: Toronto, ON, Canada.
- Korneta, I.; Magnus, M.; Bujnicki, J.M. Structural bioinformatics of the human spliceosomal proteome. Nucleic Acids Res. 2012, 40, 7046–7065. [Google Scholar] [CrossRef] [PubMed]
- Makarova, O.V.; Makarov, E.M.; Luhrmann, R. The 65 and 110 kDa SR-related proteins of the U4/U6.U5 tri-snRNP are essential for the assembly of mature spliceosomes. EMBO J. 2001, 20, 2553–2563. [Google Scholar] [CrossRef] [PubMed]
- Mishra, S.K.; Ammon, T.; Popowicz, G.M.; Krajewski, M.; Nagel, R.J.; Ares, M.; Holak, T.A.; Jentsch, S. Role of the ubiquitin-like protein Hub1 in splice-site usage and alternative splicing. Nature 2011, 474, 173–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohi, M.D.; Vander Kooi, C.W.; Rosenberg, J.A.; Chazin, W.J.; Gould, K.L. Structural insights into the U-box, a domain associated with multi-ubiquitination. Nat. Struct. Biol. 2003, 10, 250–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schimmel, J.; Balog, C.I.; Deelder, A.M.; Drijfhout, J.W.; Hensbergen, P.J.; Vertegaal, A.C. Positively charged amino acids flanking a sumoylation consensus tetramer on the 110kDa tri-snRNP component SART1 enhance sumoylation efficiency. J. Proteom. 2010, 73, 1523–1534. [Google Scholar] [CrossRef] [PubMed]
- UniprotKB. Available online: www.uniprot.org (accessed on 30 October 2018).
- Rajiv, C. A Study of Two Multi-Domain Spliceosomal Proteins: Peptidyl Prolyl Isomerase-Like 2 and Zinc Finger 830. Master’s thesis, Department of Biochemistry and Molecular Biology, Drexel University, Philadelphia, PA, USA, 2016. [Google Scholar]
- Chen, J.; Liefke, R.; Jiang, L.; Wang, J.; Huang, C.; Gong, Z.; Schiene-Fischer, C.; Yu, L. Biochemical features of recombinant human cyclophilin J. Anticancer. Res 2016, 36, 1175–1180. [Google Scholar] [PubMed]
- Ulrich, A.; Wahl, M.C. Structure and evolution of the spliceosomal peptidyl-prolyl cis-trans isomerase Cwc27. Acta Crystallogr. D Biol. Crystallogr. 2014, 70, 3110–3123. [Google Scholar] [CrossRef]
- Stegmann, C.M.; Seeliger, D.; Sheldrick, G.M.; de Groot, B.L.; Wahl, M.C. The thermodynamic influence of trapped water molecules on a protein–ligand interaction. Angew. Chem. Int. Ed. Engl. 2009, 48, 5207–5210. [Google Scholar] [CrossRef]
- Nguyen, T.H.; Galej, W.P.; Bai, X.C.; Oubridge, C.; Newman, A.J.; Scheres, S.H.; Nagai, K. Cryo-EM structure of the yeast U4/U6.U5 tri-snRNP at 3.7 Å resolution. Nature 2016, 530, 298–302. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, T.H.; Galej, W.P.; Bai, X.C.; Savva, C.G.; Newman, A.J.; Scheres, S.H.; Nagai, K. The architecture of the spliceosomal U4/U6.U5 tri-snRNP. Nature 2015, 523, 47–52. [Google Scholar] [CrossRef]
- Papasaikas, P.; Tejedor, J.R.; Vigevani, L.; Valcarcel, J. Functional splicing network reveals extensive regulatory potential of the core spliceosomal machinery. Mol. Cell 2015, 57, 7–22. [Google Scholar] [CrossRef] [PubMed]
- Tejedor, J.R.; Papasaikas, P.; Valcarcel, J. Genome-wide identification of Fas/CD95 alternative splicing regulators reveals links with iron homeostasis. Mol. Cell 2015, 57, 23–38. [Google Scholar] [CrossRef] [PubMed]
- Armah, N.M.; Jackson, S.R.; Adams, B.M.; Thorpe, J.; Trinh, A.; Marinock, J.M.; Davis, T.L. Development of a functional bioassay for the nuclear cyclophilin PPIH. Department of Biochemistry and Molecular Biology, Drexel University: Philadelphia, PA, USA, 2018. [Google Scholar]
- Javadov, S.; Kuznetsov, A. Mitochondrial permeability transition and cell death: The role of cyclophilin D. Front. Physiol. 2013, 4, 76. [Google Scholar] [CrossRef] [PubMed]
- Daum, S.; Schumann, M.; Mathea, S.; Aumüller, T.; Balsley, M.A.; Constant, S.L.; de Lacroix, B.F.a.; Kruska, F.; Braun, M.; Schiene-Fischer, C. Isoform-specific inhibition of cyclophilins. Biochemistry 2009, 48, 6268–6277. [Google Scholar] [CrossRef] [PubMed]
- Wear, M.A.; Nowicki, M.W.; Blackburn, E.A.; McNae, I.W.; Walkinshaw, M.D. Thermo-kinetic analysis space expansion for cyclophilin-ligand interactions—Identification of a new nonpeptide inhibitor using BiacoreTM T200. FEBS Open Bio 2017, 7, 533–549. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | UniProt Accession No. | # aa | Domain 1 | Boundary | Domain 2 | Boundary | Cryo-EM Structures |
|---|---|---|---|---|---|---|---|
| PPIA | P62937 | 165 | PPI | 1–165 | - | - | - |
| PPIE | Q9UNP9 | 301 | RRM | 7–80 | PPI | 143–299 | 5MQF, 5YZG, 5Z56, 5Z57 |
| PPIG | Q13427 | 754 | PPI | 11–176 | SR/RS repeats | 540–639 | 5YZG |
| PPIH | O43447 | 177 | PPI | 1–177 | - | - | 5O9Z |
| PPIL1 | Q9Y3C6 | 166 | PPI | 1–166 | - | - | 5MQF, 5XJC, 5YZG, 5Z56, 5Z57, 6FF4 |
| PPIL2 | Q13356 | 527 | U-BOX | 42–101 | PPI | 281–433 | - |
| PPIL3 | Q9H2H8 | 161 | PPI | 1–161 | - | - | |
| PPWD1 | Q96BP3 | 646 | WD40 | 80–453 | PPI | 490–645 | 5YZG |
| CWC27 | Q6UX04 | 472 | PPI | 14–166 | coiled-coil | 306–351 | 5Z56, 5Z58, 6FF4 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rajiv, C.; Davis, T.L. Structural and Functional Insights into Human Nuclear Cyclophilins. Biomolecules 2018, 8, 161. https://doi.org/10.3390/biom8040161
Rajiv C, Davis TL. Structural and Functional Insights into Human Nuclear Cyclophilins. Biomolecules. 2018; 8(4):161. https://doi.org/10.3390/biom8040161
Chicago/Turabian StyleRajiv, Caroline, and Tara L. Davis. 2018. "Structural and Functional Insights into Human Nuclear Cyclophilins" Biomolecules 8, no. 4: 161. https://doi.org/10.3390/biom8040161
APA StyleRajiv, C., & Davis, T. L. (2018). Structural and Functional Insights into Human Nuclear Cyclophilins. Biomolecules, 8(4), 161. https://doi.org/10.3390/biom8040161