Cyclophilin D, Somehow a Master Regulator of Mitochondrial Function



Abstract
:1. Introduction
2. Mitochondrial Function and the Mitochondrial Permeability Transition Pore
3. Cyclophilin D Regulates the Mitochondrial Permeability Transition Pore
4. Cyclophilin D Regulates Mitochondrial Function
5. Cyclophilin D’s Binding Partners
6. Cyclophilin D’s Enzymatic Activity
7. Physiologic Regulation of Cyclophilin D
7.1. Acetylation
7.2. Oxidation/Nitrosation/Glutathionylation
7.3. Phosphorylation
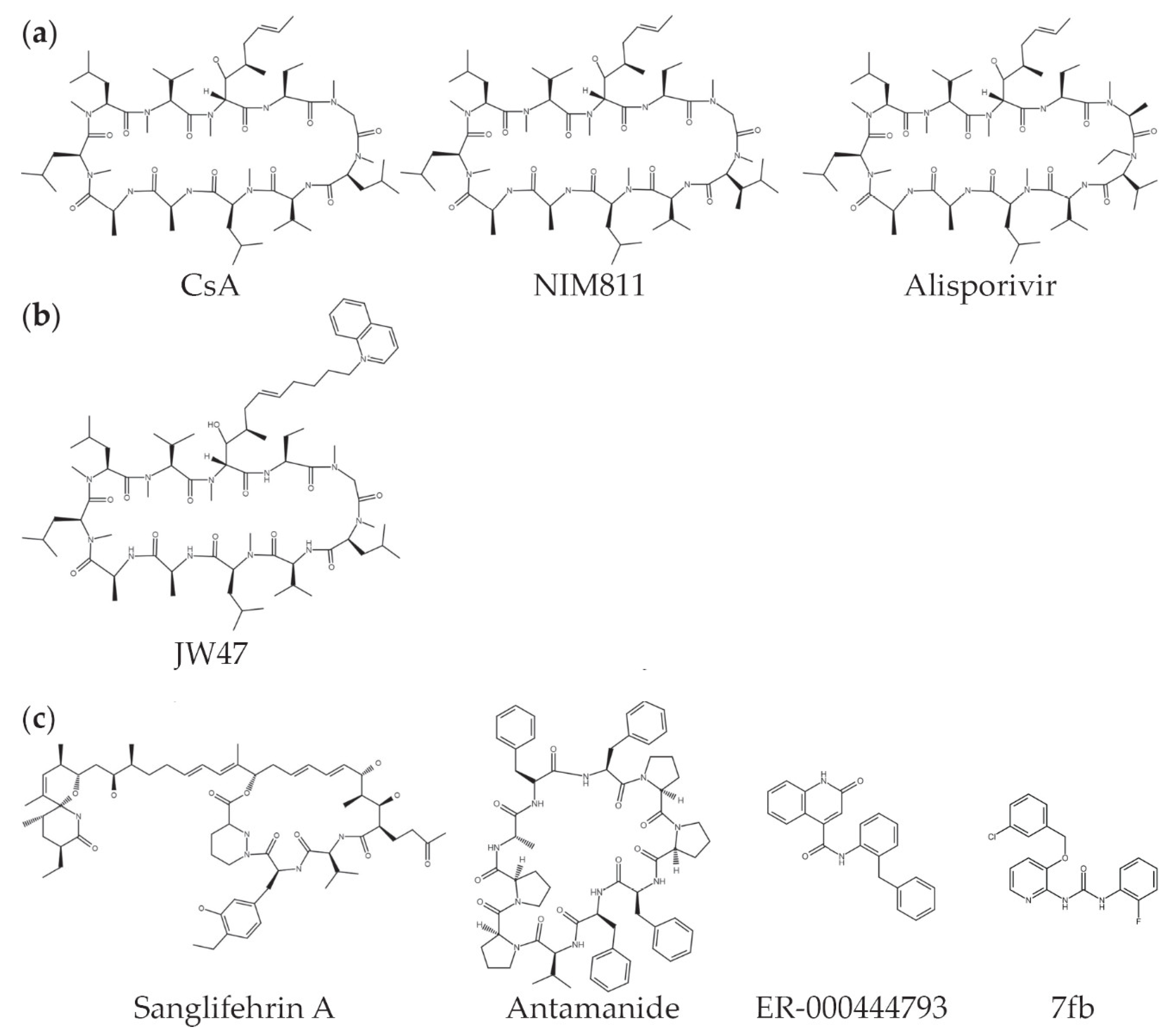
8. Therapeutic Potential of Targeting Cyclophilin D
9. Conclusions
10. Patents
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Halestrap, A.P.; Davidson, A.M. Inhibition of Ca2+-induced large-amplitude swelling of liver and heart mitochondria by cyclosporin is probably caused by the inhibitor binding to mitochondrial-matrix peptidyl-prolyl cis-trans isomerase and preventing it interacting with the adenine nucleotide translocase. Biochem. J. 1990, 268, 153–160. [Google Scholar] [PubMed]
- McGuinness, O.; Yafei, N.; Costi, A.; Crompton, M. The presence of two classes of high-affinity cyclosporin A binding sites in mitochondria. Evidence that the minor component is involved in the opening of an inner-membrane Ca2+-dependent pore. Eur. J. Biochem. 1990, 194, 671–679. [Google Scholar] [CrossRef] [PubMed]
- Alam, M.R.; Baetz, D.; Ovize, M. Cyclophilin D and myocardial ischemia-reperfusion injury: A fresh perspective. J. Mol. Cell. Cardiol. 2015, 78, 80–89. [Google Scholar] [CrossRef] [PubMed]
- Elrod, J.W.; Molkentin, J.D. Physiologic functions of cyclophilin D and the mitochondrial permeability transition pore. Circ. J. 2013, 77, 1111–1122. [Google Scholar] [CrossRef]
- Giorgio, V.; Soriano, M.E.; Basso, E.; Bisetto, E.; Lippe, G.; Forte, M.A.; Bernardi, P. Cyclophilin D in mitochondrial pathophysiology. Biochim. Biophys. Acta 2010, 1797, 1113–1118. [Google Scholar] [CrossRef] [Green Version]
- Gutierrez-Aguilar, M.; Baines, C.P. Structural mechanisms of cyclophilin D-dependent control of the mitochondrial permeability transition pore. Biochim. Biophys. Acta 2015, 1850, 2041–2047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Javadov, S.; Kuznetsov, A. Mitochondrial permeability transition and cell death: The role of cyclophilin D. Front. Physiol. 2013, 4, 76. [Google Scholar] [CrossRef]
- Stelzer, G.; Rosen, N.; Plaschkes, I.; Zimmerman, S.; Twik, M.; Fishilevich, S.; Stein, T.I.; Nudel, R.; Lieder, I.; Mazor, Y.; et al. The GeneCards Suite: From Gene Data Mining to Disease Genome Sequence Analyses. Curr. Protoc. Bioinform. 2016, 54, 1.30.1–1.30.33. [Google Scholar] [CrossRef]
- GeneCards. Available online: www.genecards.org (accessed on 10 December 2018).
- Acin-Perez, R.; Enriquez, J.A. The function of the respiratory supercomplexes: The plasticity model. Biochim. Biophys. Acta 2014, 1837, 444–450. [Google Scholar] [CrossRef]
- Beutner, G.; Alavian, K.N.; Jonas, E.A.; Porter, G.A., Jr. The Mitochondrial Permeability Transition Pore and ATP Synthase. In Pharmacology of Mitochondria; Singh, H., Sheu, S.S., Eds.; Springer: Berlin/Heidelberg, Germany, 2017; pp. 21–46. [Google Scholar]
- Chen, C.; Ko, Y.; Delannoy, M.; Ludtke, S.J.; Chiu, W.; Pedersen, P.L. Mitochondrial ATP synthasome: Three-dimensional structure by electron microscopy of the ATP synthase in complex formation with carriers for Pi and ADP/ATP. J. Biol. Chem. 2004, 279, 31761–31768. [Google Scholar] [CrossRef]
- Genova, M.L.; Lenaz, G. Functional role of mitochondrial respiratory supercomplexes. Biochim. Biophys. Acta 2014, 1837, 427–443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milenkovic, D.; Blaza, J.N.; Larsson, N.G.; Hirst, J. The Enigma of the Respiratory Chain Supercomplex. Cell Metab. 2017, 25, 765–776. [Google Scholar] [CrossRef] [PubMed]
- Saks, V.; Kuznetsov, A.V.; Gonzalez-Granillo, M.; Tepp, K.; Timohhina, N.; Karu-Varikmaa, M.; Kaambre, T.; Dos Santos, P.; Boucher, F.; Guzun, R. Intracellular Energetic Units regulate metabolism in cardiac cells. J. Mol. Cell. Cardiol. 2012, 52, 419–436. [Google Scholar] [CrossRef] [PubMed]
- Chouchani, E.T.; Kazak, L.; Jedrychowski, M.P.; Lu, G.Z.; Erickson, B.K.; Szpyt, J.; Pierce, K.A.; Laznik-Bogoslavski, D.; Vetrivelan, R.; Clish, C.B.; et al. Mitochondrial ROS regulate thermogenic energy expenditure and sulfenylation of UCP1. Nature 2016, 532, 112–116. [Google Scholar] [CrossRef] [Green Version]
- Jonas, E.A.; Porter, G.A., Jr.; Beutner, G.; Mnatsakanyan, N.; Alavian, K.N. Cell death disguised: The mitochondrial permeability transition pore as the c-subunit of the F(1)F(O) ATP synthase. Pharmacol. Res. 2015, 99, 382–392. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, P. The mitochondrial permeability transition pore: A mystery solved? Front. Physiol. 2013, 4, 95. [Google Scholar] [CrossRef] [PubMed]
- Biasutto, L.; Azzolini, M.; Szabo, I.; Zoratti, M. The mitochondrial permeability transition pore in AD 2016: An update. Biochim. Biophys. Acta 2016, 1863, 2515–2530. [Google Scholar] [CrossRef] [PubMed]
- Haworth, R.A.; Hunter, D.R. The Ca2+-induced membrane transition in mitochondria. II. Nature of the Ca2+ trigger site. Arch. Biochem. Biophys. 1979, 195, 460–467. [Google Scholar] [CrossRef]
- Hunter, D.R.; Haworth, R.A. The Ca2+-induced membrane transition in mitochondria. I. The protective mechanisms. Arch. Biochem. Biophys. 1979, 195, 453–459. [Google Scholar] [CrossRef]
- Hunter, D.R.; Haworth, R.A. The Ca2+-induced membrane transition in mitochondria. III. Transitional Ca2+ release. Arch. Biochem. Biophys. 1979, 195, 468–477. [Google Scholar] [CrossRef]
- Ichas, F.; Mazat, J.P. From calcium signaling to cell death: Two conformations for the mitochondrial permeability transition pore. Switching from low- to high-conductance state. Biochim. Biophys. Acta 1998, 1366, 33–50. [Google Scholar] [CrossRef]
- Baines, C.P. The mitochondrial permeability transition pore and the cardiac necrotic program. Pediatr. Cardiol. 2011, 32, 258–262. [Google Scholar] [CrossRef] [PubMed]
- Brenner, C.; Moulin, M. Physiological roles of the permeability transition pore. Circ. Res. 2012, 111, 1237–1247. [Google Scholar] [CrossRef]
- Hom, J.R.; Quintanilla, R.A.; Hoffman, D.L.; de Mesy Bentley, K.L.; Molkentin, J.D.; Sheu, S.S.; Porter, G.A., Jr. The permeability transition pore controls cardiac mitochondrial maturation and myocyte differentiation. Dev. Cell 2011, 21, 469–478. [Google Scholar] [CrossRef]
- Lingan, J.V.; Alanzalon, R.E.; Porter, G.A., Jr. Preventing permeability transition pore opening increases mitochondrial maturation, myocyte differentiation and cardiac function in the neonatal mouse heart. Pediatr. Res. 2017, 81, 932–941. [Google Scholar] [CrossRef] [PubMed]
- Mnatsakanyan, N.; Beutner, G.; Porter, G.A.; Alavian, K.N.; Jonas, E.A. Physiological roles of the mitochondrial permeability transition pore. J. Bioenerg. Biomembr. 2017, 49, 13–25. [Google Scholar] [CrossRef]
- Baines, C.P.; Gutierrez-Aguilar, M. The still uncertain identity of the channel-forming unit(s) of the mitochondrial permeability transition pore. Cell Calcium 2018, 73, 121–130. [Google Scholar] [CrossRef]
- Baines, C.P.; Kaiser, R.A.; Sheiko, T.; Craigen, W.J.; Molkentin, J.D. Voltage-dependent anion channels are dispensable for mitochondrial-dependent cell death. Nat. Cell Biol. 2007, 9, 550–555. [Google Scholar] [CrossRef] [Green Version]
- Gutierrez-Aguilar, M.; Douglas, D.L.; Gibson, A.K.; Domeier, T.L.; Molkentin, J.D.; Baines, C.P. Genetic manipulation of the cardiac mitochondrial phosphate carrier does not affect permeability transition. J. Mol. Cell. Cardiol. 2014, 72, 316–325. [Google Scholar] [CrossRef] [Green Version]
- Kokoszka, J.E.; Waymire, K.G.; Levy, S.E.; Sligh, J.E.; Cai, J.; Jones, D.P.; MacGregor, G.R.; Wallace, D.C. The ADP/ATP translocator is not essential for the mitochondrial permeability transition pore. Nature 2004, 427, 461–465. [Google Scholar] [CrossRef] [Green Version]
- Krauskopf, A.; Eriksson, O.; Craigen, W.J.; Forte, M.A.; Bernardi, P. Properties of the permeability transition in VDAC1(-/-) mitochondria. Biochim. Biophys. Acta 2006, 1757, 590–595. [Google Scholar] [CrossRef] [PubMed]
- Kwong, J.Q.; Davis, J.; Baines, C.P.; Sargent, M.A.; Karch, J.; Wang, X.; Huang, T.; Molkentin, J.D. Genetic deletion of the mitochondrial phosphate carrier desensitizes the mitochondrial permeability transition pore and causes cardiomyopathy. Cell Death Differ. 2014, 21, 1209–1217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sileikyte, J.; Blachly-Dyson, E.; Sewell, R.; Carpi, A.; Menabo, R.; Di Lisa, F.; Ricchelli, F.; Bernardi, P.; Forte, M. Regulation of the Mitochondrial Permeability Transition Pore by the Outer Membrane does not Involve the Peripheral Benzodiazepine Receptor (TSPO). J. Biol. Chem. 2014, 289, 13769–13781. [Google Scholar] [CrossRef] [PubMed]
- Vyssokikh, M.Y.; Brdiczka, D. The function of complexes between the outer mitochondrial membrane pore (VDAC) and the adenine nucleotide translocase in regulation of energy metabolism and apoptosis. Acta Biochim. Pol. 2003, 50, 389–404. [Google Scholar]
- He, L.; Lemasters, J.J. Regulated and unregulated mitochondrial permeability transition pores: A new paradigm of pore structure and function? FEBS Lett. 2002, 512, 1–7. [Google Scholar] [CrossRef]
- Elustondo, P.A.; Nichols, M.; Negoda, A.; Thirumaran, A.; Zakharian, E.; Robertson, G.S.; Pavlov, E.V. Mitochondrial permeability transition pore induction is linked to formation of the complex of ATPase C-subunit, polyhydroxybutyrate and inorganic polyphosphate. Cell Death Discov. 2016, 2, 16070. [Google Scholar] [CrossRef] [Green Version]
- Seidlmayer, L.K.; Juettner, V.V.; Kettlewell, S.; Pavlov, E.V.; Blatter, L.A.; Dedkova, E.N. Distinct mPTP activation mechanisms in ischaemia-reperfusion: Contributions of Ca2+, ROS, pH, and inorganic polyphosphate. Cardiovasc. Res. 2015, 106, 237–248. [Google Scholar] [CrossRef]
- Fournier, N.; Ducet, G.; Crevat, A. Action of cyclosporine on mitochondrial calcium fluxes. J. Bioenerg. Biomembr. 1987, 19, 297–303. [Google Scholar] [CrossRef]
- Jung, K.; Pergande, M. Influence of cyclosporin A on the respiration of isolated rat kidney mitochondria. FEBS Lett. 1985, 183, 167–169. [Google Scholar] [CrossRef] [Green Version]
- Mihatsch, M.J.; Olivieri, W.; Marbet, U.; Thiel, G.; Harder, F.; Zollinger, H.U. Giant mitochondria in renal tubular cells and cyclosporin A. Lancet 1981, 1, 1162–1163. [Google Scholar] [CrossRef]
- Broekemeier, K.M.; Dempsey, M.E.; Pfeiffer, D.R. Cyclosporin A is a potent inhibitor of the inner membrane permeability transition in liver mitochondria. J. Biol. Chem. 1989, 264, 7826–7830. [Google Scholar] [PubMed]
- Crompton, M.; Ellinger, H.; Costi, A. Inhibition by cyclosporin A of a Ca2+-dependent pore in heart mitochondria activated by inorganic phosphate and oxidative stress. Biochem. J. 1988, 255, 357–360. [Google Scholar] [PubMed]
- Tanveer, A.; Virji, S.; Andreeva, L.; Totty, N.F.; Hsuan, J.J.; Ward, J.M.; Crompton, M. Involvement of cyclophilin D in the activation of a mitochondrial pore by Ca2+ and oxidant stress. Eur. J. Biochem. 1996, 238, 166–172. [Google Scholar] [CrossRef] [PubMed]
- Baines, C.P.; Kaiser, R.A.; Purcell, N.H.; Blair, N.S.; Osinska, H.; Hambleton, M.A.; Brunskill, E.W.; Sayen, M.R.; Gottlieb, R.A.; Dorn, G.W.; et al. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature 2005, 434, 658–662. [Google Scholar] [CrossRef]
- Basso, E.; Fante, L.; Fowlkes, J.; Petronilli, V.; Forte, M.A.; Bernardi, P. Properties of the permeability transition pore in mitochondria devoid of Cyclophilin D. J. Biol. Chem. 2005, 280, 18558–18561. [Google Scholar] [CrossRef]
- Nakagawa, T.; Shimizu, S.; Watanabe, T.; Yamaguchi, O.; Otsu, K.; Yamagata, H.; Inohara, H.; Kubo, T.; Tsujimoto, Y. Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature 2005, 434, 652–658. [Google Scholar] [CrossRef] [PubMed]
- Schinzel, A.C.; Takeuchi, O.; Huang, Z.; Fisher, J.K.; Zhou, Z.; Rubens, J.; Hetz, C.; Danial, N.N.; Moskowitz, M.A.; Korsmeyer, S.J. Cyclophilin D is a component of mitochondrial permeability transition and mediates neuronal cell death after focal cerebral ischemia. Proc. Natl. Acad. Sci. USA 2005, 102, 12005–12010. [Google Scholar] [CrossRef] [Green Version]
- Basso, E.; Petronilli, V.; Forte, M.A.; Bernardi, P. Phosphate is essential for inhibition of the mitochondrial permeability transition pore by cyclosporin A and by cyclophilin D ablation. J. Biol. Chem. 2008, 283, 26307–26311. [Google Scholar] [CrossRef]
- Eliseev, R.A.; Filippov, G.; Velos, J.; VanWinkle, B.; Goldman, A.; Rosier, R.N.; Gunter, T.E. Role of cyclophilin D in the resistance of brain mitochondria to the permeability transition. Neurobiol. Aging 2007, 28, 1532–1542. [Google Scholar] [CrossRef]
- Beutner, G.; Alanzalon, R.E.; Porter, G.A., Jr. Cyclophilin D regulates the dynamic assembly of mitochondrial ATP synthase into synthasomes. Sci. Rep. 2017, 7, 14488. [Google Scholar] [CrossRef]
- Berman, S.B.; Watkins, S.C.; Hastings, T.G. Quantitative biochemical and ultrastructural comparison of mitochondrial permeability transition in isolated brain and liver mitochondria: Evidence for reduced sensitivity of brain mitochondria. Exp. Neurol. 2000, 164, 415–425. [Google Scholar] [CrossRef] [PubMed]
- Palmer, J.W.; Pfeiffer, D.R. The control of Ca2+ release from heart mitochondria. J. Biol. Chem. 1981, 256, 6742–6750. [Google Scholar] [PubMed]
- Zhu, J.; Rebecchi, M.J.; Glass, P.S.; Brink, P.R.; Liu, L. Interactions of GSK-3β with mitochondrial permeability transition pore modulators during preconditioning: Age-associated differences. J. Gerontol. A Biol. Sci. Med. Sci. 2013, 68, 395–403. [Google Scholar] [CrossRef]
- Gauba, E.; Guo, L.; Du, H. Cyclophilin D Promotes Brain Mitochondrial F1FO ATP Synthase Dysfunction in Aging Mice. J. Alzheimers Dis. 2017, 55, 1351–1362. [Google Scholar] [CrossRef]
- Giorgio, V.; Bisetto, E.; Soriano, M.E.; Dabbeni-Sala, F.; Basso, E.; Petronilli, V.; Forte, M.A.; Bernardi, P.; Lippe, G. Cyclophilin D modulates mitochondrial F0F1-ATP synthase by interacting with the lateral stalk of the complex. J. Biol. Chem. 2009, 284, 33982–33988. [Google Scholar] [CrossRef] [PubMed]
- Chinopoulos, C.; Konrad, C.; Kiss, G.; Metelkin, E.; Torocsik, B.; Zhang, S.F.; Starkov, A.A. Modulation of F0F1-ATP synthase activity by cyclophilin D regulates matrix adenine nucleotide levels. FEBS J. 2011, 278, 1112–1125. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Chauvin, C.; De Paulis, D.; De Oliveira, F.; Gharib, A.; Vial, G.; Lablanche, S.; Leverve, X.; Bernardi, P.; Ovize, M.; et al. Inhibition of complex I regulates the mitochondrial permeability transition through a phosphate-sensitive inhibitory site masked by cyclophilin D. Biochim. Biophys. Acta 2012, 1817, 1628–1634. [Google Scholar] [CrossRef] [Green Version]
- Teixeira, G.; Abrial, M.; Portier, K.; Chiari, P.; Couture-Lepetit, E.; Tourneur, Y.; Ovize, M.; Gharib, A. Synergistic protective effect of cyclosporin A and rotenone against hypoxia-reoxygenation in cardiomyocytes. J. Mol. Cell. Cardiol. 2013, 56, 55–62. [Google Scholar] [CrossRef]
- Etzler, J.C.; Bollo, M.; Holstein, D.; Deng, J.J.; Perez, V.; Lin, D.T.; Richardson, A.; Bai, Y.; Lechleiter, J.D. Cyclophilin D over-expression increases mitochondrial complex III activity and accelerates supercomplex formation. Arch. Biochem. Biophys. 2017, 613, 61–68. [Google Scholar] [CrossRef] [Green Version]
- Beutner, G.; Eliseev, R.A.; Porter, G.A., Jr. Initiation of electron transport chain activity in the embryonic heart coincides with the activation of mitochondrial complex 1 and the formation of supercomplexes. PLoS ONE 2014, 9, e113330. [Google Scholar] [CrossRef]
- Beutner, G.; Porter, G.A., Jr. Unpublished data not yet submitted for publication. 2018. [Google Scholar]
- Luvisetto, S.; Basso, E.; Petronilli, V.; Bernardi, P.; Forte, M. Enhancement of anxiety, facilitation of avoidance behavior, and occurrence of adult-onset obesity in mice lacking mitochondrial cyclophilin D. Neuroscience 2008, 155, 585–596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elrod, J.W.; Wong, R.; Mishra, S.; Vagnozzi, R.J.; Sakthievel, B.; Goonasekera, S.A.; Karch, J.; Gabel, S.; Farber, J.; Force, T.; et al. Cyclophilin D controls mitochondrial pore-dependent Ca2+ exchange, metabolic flexibility, and propensity for heart failure in mice. J. Clin. Investig. 2010, 120, 3680–3687. [Google Scholar] [CrossRef] [PubMed]
- Tavecchio, M.; Lisanti, S.; Bennett, M.J.; Languino, L.R.; Altieri, D.C. Deletion of Cyclophilin D Impairs beta-Oxidation and Promotes Glucose Metabolism. Sci. Rep. 2015, 5, 15981. [Google Scholar] [CrossRef] [PubMed]
- Menazza, S.; Wong, R.; Nguyen, T.; Wang, G.; Gucek, M.; Murphy, E. CypD(-/-) hearts have altered levels of proteins involved in Krebs cycle, branch chain amino acid degradation and pyruvate metabolism. J. Mol. Cell. Cardiol. 2013, 56, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.T.; Wong, R.; Menazza, S.; Sun, J.; Chen, Y.; Wang, G.; Gucek, M.; Steenbergen, C.; Sack, M.N.; Murphy, E. Cyclophilin D modulates mitochondrial acetylome. Circ. Res. 2013, 113, 1308–1319. [Google Scholar] [CrossRef] [PubMed]
- Crompton, M.; Virji, S.; Ward, J.M. Cyclophilin-D binds strongly to complexes of the voltage-dependent anion channel and the adenine nucleotide translocase to form the permeability transition pore. Eur. J. Biochem. 1998, 258, 729–735. [Google Scholar] [CrossRef] [PubMed]
- Woodfield, K.; Ruck, A.; Brdiczka, D.; Halestrap, A.P. Direct demonstration of a specific interaction between cyclophilin-D and the adenine nucleotide translocase confirms their role in the mitochondrial permeability transition. Biochem. J. 1998, 336(Pt. 2), 287–290. [Google Scholar] [CrossRef] [Green Version]
- Vyssokikh, M.Y.; Katz, A.; Rueck, A.; Wuensch, C.; Dorner, A.; Zorov, D.B.; Brdiczka, D. Adenine nucleotide translocator isoforms 1 and 2 are differently distributed in the mitochondrial inner membrane and have distinct affinities to cyclophilin D. Biochem. J. 2001, 358, 349–358. [Google Scholar] [CrossRef] [PubMed]
- Leung, A.W.; Varanyuwatana, P.; Halestrap, A.P. The mitochondrial phosphate carrier interacts with cyclophilin D and may play a key role in the permeability transition. J. Biol. Chem. 2008, 283, 26312–26323. [Google Scholar] [CrossRef]
- Beutner, G.; Ruck, A.; Riede, B.; Brdiczka, D. Complexes between porin, hexokinase, mitochondrial creatine kinase and adenylate translocator display properties of the permeability transition pore. Implication for regulation of permeability transition by the kinases. Biochim. Biophys. Acta 1998, 1368, 7–18. [Google Scholar] [CrossRef]
- Beutner, G.; Ruck, A.; Riede, B.; Welte, W.; Brdiczka, D. Complexes between kinases, mitochondrial porin and adenylate translocator in rat brain resemble the permeability transition pore. FEBS Lett. 1996, 396, 189–195. [Google Scholar] [CrossRef] [Green Version]
- Antoniel, M.; Jones, K.; Antonucci, S.; Spolaore, B.; Fogolari, F.; Petronilli, V.; Giorgio, V.; Carraro, M.; Di Lisa, F.; Forte, M.; et al. The unique histidine in OSCP subunit of F-ATP synthase mediates inhibition of the permeability transition pore by acidic pH. EMBO Rep. 2018, 19, 257–268. [Google Scholar] [CrossRef] [PubMed]
- Giorgio, V.; von Stockum, S.; Antoniel, M.; Fabbro, A.; Fogolari, F.; Forte, M.; Glick, G.D.; Petronilli, V.; Zoratti, M.; Szabo, I.; et al. Dimers of mitochondrial ATP synthase form the permeability transition pore. Proc. Natl. Acad. Sci. USA 2013, 110, 5887–5892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, J.; Carroll, J.; Ding, S.; Fearnley, I.M.; Walker, J.E. Permeability transition in human mitochondria persists in the absence of peripheral stalk subunits of ATP synthase. Proc. Natl. Acad. Sci. USA 2017. [Google Scholar] [CrossRef]
- Wu, Y.T.; Lee, H.C.; Liao, C.C.; Wei, Y.H. Regulation of mitochondrial F(o)F(1)ATPase activity by Sirt3-catalyzed deacetylation and its deficiency in human cells harboring 4977bp deletion of mitochondrial DNA. Biochim. Biophys. Acta 2013, 1832, 216–227. [Google Scholar] [CrossRef] [PubMed]
- Burstein, S.R.; Kim, H.J.; Fels, J.A.; Qian, L.; Zhang, S.; Zhou, P.; Starkov, A.A.; Iadecola, C.; Manfredi, G. Estrogen receptor β modulates permeability transition in brain mitochondria. Biochim. Biophys. Acta 2018, 1859, 423–433. [Google Scholar] [CrossRef] [PubMed]
- Shanmughapriya, S.; Rajan, S.; Hoffman, N.E.; Higgins, A.M.; Tomar, D.; Nemani, N.; Hines, K.J.; Smith, D.J.; Eguchi, A.; Vallem, S.; et al. SPG7 Is an Essential and Conserved Component of the Mitochondrial Permeability Transition Pore. Mol. Cell 2015, 60, 47–62. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, P.; Forte, M. Commentary: SPG7 is an essential and conserved component of the mitochondrial permeability transition pore. Front. Physiol. 2015, 6, 320. [Google Scholar] [CrossRef]
- Alavian, K.N.; Beutner, G.; Lazrove, E.; Sacchetti, S.; Park, H.A.; Licznerski, P.; Li, H.; Nabili, P.; Hockensmith, K.; Graham, M.; et al. An uncoupling channel within the c-subunit ring of the F1FO ATP synthase is the mitochondrial permeability transition pore. Proc. Natl. Acad. Sci. USA 2014, 111, 10580–10585. [Google Scholar] [CrossRef] [Green Version]
- Azarashvili, T.; Odinokova, I.; Bakunts, A.; Ternovsky, V.; Krestinina, O.; Tyynela, J.; Saris, N.E. Potential role of subunit c of F0F1-ATPase and subunit c of storage body in the mitochondrial permeability transition. Effect of the phosphorylation status of subunit c on pore opening. Cell Calcium 2014, 55, 69–77. [Google Scholar] [CrossRef]
- Bonora, M.; Bononi, A.; De Marchi, E.; Giorgi, C.; Lebiedzinska, M.; Marchi, S.; Patergnani, S.; Rimessi, A.; Suski, J.M.; Wojtala, A.; et al. Role of the c subunit of the FO ATP synthase in mitochondrial permeability transition. Cell Cycle 2013, 12, 674–683. [Google Scholar] [CrossRef] [PubMed]
- Bonora, M.; Morganti, C.; Morciano, G.; Pedriali, G.; Lebiedzinska-Arciszewska, M.; Aquila, G.; Giorgi, C.; Rizzo, P.; Campo, G.; Ferrari, R.; et al. Mitochondrial permeability transition involves dissociation of F1FO ATP synthase dimers and C-ring conformation. EMBO Rep. 2017, 18, 1077–1089. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Ford, H.C.; Carroll, J.; Ding, S.; Fearnley, I.M.; Walker, J.E. Persistence of the mitochondrial permeability transition in the absence of subunit c of human ATP synthase. Proc. Natl. Acad. Sci. USA 2017, 114, 3409–3414. [Google Scholar] [CrossRef] [Green Version]
- Amodeo, G.F.; Torregrosa, M.E.S.; Pavlov, E.V. From ATP synthase dimers to C-ring conformational changes: Unified model of the mitochondrial permeability transition pore. Cell Death Dis. 2017, 8, 1. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, P.; Di Lisa, F. The mitochondrial permeability transition pore: Molecular nature and role as a target in cardioprotection. J. Mol. Cell. Cardiol. 2015, 78, 100–106. [Google Scholar] [CrossRef]
- Bonora, M.; Pinton, P. Shedding light on molecular mechanisms and identity of mPTP. Mitochondrion 2015, 21, 11. [Google Scholar] [CrossRef]
- Chinopoulos, C. ATP synthase complex and the mitochondrial permeability transition pore: Poles of attraction. EMBO Rep. 2017, 18, 1041–1042. [Google Scholar] [CrossRef]
- Gerle, C. On the structural possibility of pore-forming mitochondrial FF ATP synthase. Biochim. Biophys. Acta 2016, 1857, 1191–1196. [Google Scholar] [CrossRef]
- Halestrap, A.P.; Richardson, A.P. The mitochondrial permeability transition: A current perspective on its identity and role in ischaemia/reperfusion injury. J. Mol. Cell. Cardiol. 2015, 78, 129–141. [Google Scholar] [CrossRef]
- Karch, J.; Molkentin, J.D. Identifying the components of the elusive mitochondrial permeability transition pore. Proc. Natl. Acad. Sci. USA 2014, 111, 10396–10397. [Google Scholar] [CrossRef] [Green Version]
- Perez, M.J.; Quintanilla, R.A. Development or disease: Duality of the mitochondrial permeability transition pore. Dev. Biol. 2017, 426, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Barreto-Torres, G.; Hernandez, J.S.; Jang, S.; Rodriguez-Munoz, A.R.; Torres-Ramos, C.A.; Basnakian, A.G.; Javadov, S. The beneficial effects of AMP kinase activation against oxidative stress are associated with prevention of PPARα-cyclophilin D interaction in cardiomyocytes. Am. J. Physiol. Heart Circ. Physiol. 2015, 308, H749–H758. [Google Scholar] [CrossRef]
- Rasola, A.; Sciacovelli, M.; Chiara, F.; Pantic, B.; Brusilow, W.S.; Bernardi, P. Activation of mitochondrial ERK protects cancer cells from death through inhibition of the permeability transition. Proc. Natl. Acad. Sci. USA 2010, 107, 726–731. [Google Scholar] [CrossRef] [PubMed]
- Xi, J.; Wang, H.; Mueller, R.A.; Norfleet, E.A.; Xu, Z. Mechanism for resveratrol-induced cardioprotection against reperfusion injury involves glycogen synthase kinase 3β and mitochondrial permeability transition pore. Eur. J. Pharmacol. 2009, 604, 111–116. [Google Scholar] [CrossRef] [Green Version]
- Nishihara, M.; Miura, T.; Miki, T.; Tanno, M.; Yano, T.; Naitoh, K.; Ohori, K.; Hotta, H.; Terashima, Y.; Shimamoto, K. Modulation of the mitochondrial permeability transition pore complex in GSK-3β-mediated myocardial protection. J. Mol. Cell. Cardiol. 2007, 43, 564–570. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.H.; Li, D.L.; Yang, F.; Wu, Z.; Zhao, Y.Y.; Jiang, Y. Gemcitabine-induced pancreatic cancer cell death is associated with MST1/cyclophilin D mitochondrial complexation. Biochimie 2014, 103, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, J.C.; Siegelin, M.D.; Dohi, T.; Altieri, D.C. Heat shock protein 60 regulation of the mitochondrial permeability transition pore in tumor cells. Cancer Res. 2010, 70, 8988–8993. [Google Scholar] [CrossRef]
- Kang, B.H.; Plescia, J.; Dohi, T.; Rosa, J.; Doxsey, S.J.; Altieri, D.C. Regulation of tumor cell mitochondrial homeostasis by an organelle-specific Hsp90 chaperone network. Cell 2007, 131, 257–270. [Google Scholar] [CrossRef]
- Sinha, D.; D’Silva, P. Chaperoning mitochondrial permeability transition: Regulation of transition pore complex by a J-protein, DnaJC15. Cell Death Dis. 2014, 5, e1101. [Google Scholar] [CrossRef]
- Chen, M.B.; Jiang, Q.; Liu, Y.Y.; Zhang, Y.; He, B.S.; Wei, M.X.; Lu, J.W.; Ji, Y.; Lu, P.H. C6 ceramide dramatically increases vincristine sensitivity both in vivo and in vitro, involving AMP-activated protein kinase-p53 signaling. Carcinogenesis 2015, 36, 1061–1070. [Google Scholar] [CrossRef]
- Wolff, S.; Erster, S.; Palacios, G.; Moll, U.M. p53’s mitochondrial translocation and MOMP action is independent of Puma and Bax and severely disrupts mitochondrial membrane integrity. Cell Res. 2008, 18, 733–744. [Google Scholar] [CrossRef] [PubMed]
- Lebedev, I.; Nemajerova, A.; Foda, Z.H.; Kornaj, M.; Tong, M.; Moll, U.M.; Seeliger, M.A. A Novel In Vitro CypD-Mediated p53 Aggregation Assay Suggests a Model for Mitochondrial Permeability Transition by Chaperone Systems. J. Mol. Biol. 2016, 428, 4154–4167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.H.; Zhuang, J.; Li, J.; Hwang, P.M. p53 as guardian of the mitochondrial genome. FEBS Lett. 2016, 590, 924–934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eliseev, R.A.; Malecki, J.; Lester, T.; Zhang, Y.; Humphrey, J.; Gunter, T.E. Cyclophilin D interacts with Bcl2 and exerts an anti-apoptotic effect. J. Biol. Chem. 2009, 284, 9692–9699. [Google Scholar] [CrossRef] [PubMed]
- Boengler, K.; Hilfiker-Kleiner, D.; Heusch, G.; Schulz, R. Inhibition of permeability transition pore opening by mitochondrial STAT3 and its role in myocardial ischemia/reperfusion. Basic Res. Cardiol. 2010, 105, 771–785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meier, J.A.; Hyun, M.; Cantwell, M.; Raza, A.; Mertens, C.; Raje, V.; Sisler, J.; Tracy, E.; Torres-Odio, S.; Gispert, S.; et al. Stress-induced dynamic regulation of mitochondrial STAT3 and its association with cyclophilin D reduce mitochondrial ROS production. Sci. Signal. 2017, 10. [Google Scholar] [CrossRef] [PubMed]
- Youcef, G.; Belaidi, E.; Waeckel, L.; Fazal, L.; Clemessy, M.; Vincent, M.P.; Zadigue, G.; Richer, C.; Alhenc-Gelas, F.; Ovize, M.; et al. Tissue kallikrein is required for the cardioprotective effect of cyclosporin A in myocardial ischemia in the mouse. Biochem. Pharmacol. 2015, 94, 22–29. [Google Scholar] [CrossRef] [PubMed]
- McGee, A.M.; Baines, C.P. Complement 1q-binding protein inhibits the mitochondrial permeability transition pore and protects against oxidative stress-induced death. Biochem. J. 2011, 433, 119–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baburina, Y.; Azarashvili, T.; Grachev, D.; Krestinina, O.; Galvita, A.; Stricker, R.; Reiser, G. Mitochondrial 2’,3’-cyclic nucleotide 3’-phosphodiesterase (CNP) interacts with mPTP modulators and functional complexes (I-V) coupled with release of apoptotic factors. Neurochem. Int. 2015, 90, 46–55. [Google Scholar] [CrossRef] [PubMed]
- Matouschek, A.; Rospert, S.; Schmid, K.; Glick, B.S.; Schatz, G. Cyclophilin catalyzes protein folding in yeast mitochondria. Proc. Natl. Acad. Sci. USA 1995, 92, 6319–6323. [Google Scholar] [CrossRef] [PubMed]
- Rassow, J.; Mohrs, K.; Koidl, S.; Barthelmess, I.B.; Pfanner, N.; Tropschug, M. Cyclophilin 20 is involved in mitochondrial protein folding in cooperation with molecular chaperones Hsp70 and Hsp60. Mol. Cell. Boil. 1995, 15, 2654–2662. [Google Scholar] [CrossRef]
- Pestana, C.R.; Silva, C.H.; Uyemura, S.A.; Santos, A.C.; Curti, C. Impact of adenosine nucleotide translocase (ANT) proline isomerization on Ca2+-induced cysteine relative mobility/mitochondrial permeability transition pore. J. Bioenerg. Biomembr. 2010, 42, 329–335. [Google Scholar] [CrossRef] [PubMed]
- Lin, D.T.; Lechleiter, J.D. Mitochondrial targeted cyclophilin D protects cells from cell death by peptidyl prolyl isomerization. J. Biol. Chem. 2002, 277, 31134–31141. [Google Scholar] [CrossRef] [PubMed]
- Scorrano, L.; Nicolli, A.; Basso, E.; Petronilli, V.; Bernardi, P. Two modes of activation of the permeability transition pore: The role of mitochondrial cyclophilin. Mol. Cell. Biochem. 1997, 174, 181–184. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Heitman, J. The cyclophilins. Genome Boil. 2005, 6, 226. [Google Scholar] [CrossRef]
- Zydowsky, L.D.; Etzkorn, F.A.; Chang, H.Y.; Ferguson, S.B.; Stolz, L.A.; Ho, S.I.; Walsh, C.T. Active site mutants of human cyclophilin A separate peptidyl-prolyl isomerase activity from cyclosporin A binding and calcineurin inhibition. Protein Sci. 1992, 1, 1092–1099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischer, G.; Bang, H.; Mech, C. Determination of enzymatic catalysis for the cis-trans-isomerization of peptide binding in proline-containing peptides. Biomed. Biochim. Acta 1984, 43, 1101–1111. [Google Scholar] [PubMed]
- Fischer, G.; Bang, H.; Berger, E.; Schellenberger, A. Conformational specificity of chymotrypsin toward proline-containing substrates. Biochim. Biophys. Acta 1984, 791, 87–97. [Google Scholar] [CrossRef]
- Kofron, J.L.; Kuzmic, P.; Kishore, V.; Colon-Bonilla, E.; Rich, D.H. Determination of kinetic constants for peptidyl prolyl cis-trans isomerases by an improved spectrophotometric assay. Biochemistry 1991, 30, 6127–6134. [Google Scholar] [CrossRef]
- Liu, J.; Albers, M.W.; Chen, C.M.; Schreiber, S.L.; Walsh, C.T. Cloning, expression, and purification of human cyclophilin in Escherichia coli and assessment of the catalytic role of cysteines by site-directed mutagenesis. Proc. Natl. Acad. Sci. USA 1990, 87, 2304–2308. [Google Scholar] [CrossRef]
- Wagner, G.R.; Payne, R.M. Widespread and enzyme-independent Nepsilon-acetylation and Nepsilon-succinylation of proteins in the chemical conditions of the mitochondrial matrix. J. Biol. Chem. 2013, 288, 29036–29045. [Google Scholar] [CrossRef] [PubMed]
- Scott, I.; Webster, B.R.; Li, J.H.; Sack, M.N. Identification of a molecular component of the mitochondrial acetyltransferase programme: A novel role for GCN5L1. Biochem. J. 2012, 443, 655–661. [Google Scholar] [CrossRef] [PubMed]
- Thapa, D.; Wu, K.; Stoner, M.W.; Xie, B.; Zhang, M.; Manning, J.R.; Lu, Z.; Li, J.H.; Chen, Y.; Gucek, M.; et al. The protein acetylase GCN5L1 modulates hepatic fatty acid oxidation activity via acetylation of the mitochondrial β-oxidation enzyme HADHA. J. Biol. Chem. 2018, 293, 17676–17684. [Google Scholar] [CrossRef] [PubMed]
- Webster, B.R.; Scott, I.; Han, K.; Li, J.H.; Lu, Z.; Stevens, M.V.; Malide, D.; Chen, Y.; Samsel, L.; Connelly, P.S.; et al. Restricted mitochondrial protein acetylation initiates mitochondrial autophagy. J. Cell. Sci. 2013, 126, 4843–4849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lammers, M.; Neumann, H.; Chin, J.W.; James, L.C. Acetylation regulates cyclophilin A catalysis, immunosuppression and HIV isomerization. Nat. Chem. Boil. 2010, 6, 331–337. [Google Scholar] [CrossRef] [PubMed]
- Hafner, A.V.; Dai, J.; Gomes, A.P.; Xiao, C.Y.; Palmeira, C.M.; Rosenzweig, A.; Sinclair, D.A. Regulation of the mPTP by SIRT3-mediated deacetylation of CypD at lysine 166 suppresses age-related cardiac hypertrophy. Aging (Albany NY) 2010, 2, 914–923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parodi-Rullan, R.; Barreto-Torres, G.; Ruiz, L.; Casasnovas, J.; Javadov, S. Direct renin inhibition exerts an anti-hypertrophic effect associated with improved mitochondrial function in post-infarction heart failure in diabetic rats. Cell. Physiol. Biochem. 2012, 29, 841–850. [Google Scholar] [CrossRef] [PubMed]
- Bochaton, T.; Crola-Da-Silva, C.; Pillot, B.; Villedieu, C.; Ferreras, L.; Alam, M.R.; Thibault, H.; Strina, M.; Gharib, A.; Ovize, M.; et al. Inhibition of myocardial reperfusion injury by ischemic postconditioning requires sirtuin 3-mediated deacetylation of cyclophilin D. J. Mol. Cell. Cardiol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Jahandiez, V.; Cour, M.; Bochaton, T.; Abrial, M.; Loufouat, J.; Gharib, A.; Varennes, A.; Ovize, M.; Argaud, L. Fast therapeutic hypothermia prevents post-cardiac arrest syndrome through cyclophilin D-mediated mitochondrial permeability transition inhibition. Basic Res. Cardiol. 2017, 112, 35. [Google Scholar] [CrossRef]
- Teodoro, J.S.; Varela, A.T.; Duarte, F.V.; Gomes, A.P.; Palmeira, C.M.; Rolo, A.P. Indirubin and NAD(+) prevent mitochondrial ischaemia/reperfusion damage in fatty livers. Eur. J. Clin. Investig. 2018, 48, e12932. [Google Scholar] [CrossRef]
- Linard, D.; Kandlbinder, A.; Degand, H.; Morsomme, P.; Dietz, K.J.; Knoops, B. Redox characterization of human cyclophilin D: Identification of a new mammalian mitochondrial redox sensor? Arch. Biochem. Biophys. 2009, 491, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Kohr, M.J.; Aponte, A.M.; Sun, J.; Wang, G.; Murphy, E.; Gucek, M.; Steenbergen, C. Characterization of potential S-nitrosylation sites in the myocardium. Am. J. Physiol. Heart Circ. Physiol. 2011, 300, H1327–H1335. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.T.; Stevens, M.V.; Kohr, M.; Steenbergen, C.; Sack, M.N.; Murphy, E. Cysteine 203 of cyclophilin D is critical for cyclophilin D activation of the mitochondrial permeability transition pore. J. Biol. Chem. 2011, 286, 40184–40192. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, G.; Fernandez, C.; Montecinos, L.; Domenech, R.J.; Donoso, P. Preconditioning tachycardia decreases the activity of the mitochondrial permeability transition pore in the dog heart. Biochem. Biophys. Res. Commun. 2011, 410, 916–921. [Google Scholar] [CrossRef]
- Ghosh, J.C.; Siegelin, M.D.; Vaira, V.; Faversani, A.; Tavecchio, M.; Chae, Y.C.; Lisanti, S.; Rampini, P.; Giroda, M.; Caino, M.C.; et al. Adaptive mitochondrial reprogramming and resistance to PI3K therapy. J. Natl. Cancer Inst. 2015, 107. [Google Scholar] [CrossRef]
- Parks, R.J.; Menazza, S.; Holmstrom, K.M.; Amanakis, G.; Fergusson, M.; Ma, H.; Aponte, A.M.; Bernardi, P.; Finkel, T.; Murphy, E. Cyclophilin D-mediated regulation of the permeability transition pore is altered in mice lacking the mitochondrial calcium uniporter. Cardiovasc. Res. 2018. [Google Scholar] [CrossRef]
- Griffiths, E.J.; Halestrap, A.P. Protection by Cyclosporin A of ischemia/reperfusion-induced damage in isolated rat hearts. J. Mol. Cell. Cardiol. 1993, 25, 1461–1469. [Google Scholar] [CrossRef]
- Hausenloy, D.J.; Lim, S.Y.; Ong, S.G.; Davidson, S.M.; Yellon, D.M. Mitochondrial cyclophilin-D as a critical mediator of ischaemic preconditioning. Cardiovasc. Res. 2010, 88, 67–74. [Google Scholar] [CrossRef] [Green Version]
- Pottecher, J.; Guillot, M.; Belaidi, E.; Charles, A.L.; Lejay, A.; Gharib, A.; Diemunsch, P.; Geny, B. Cyclosporine A normalizes mitochondrial coupling, reactive oxygen species production, and inflammation and partially restores skeletal muscle maximal oxidative capacity in experimental aortic cross-clamping. J. Vasc. Surg. 2013, 57, 1100.e2–1108.e2. [Google Scholar] [CrossRef]
- Li, J.; Yan, Z.; Fang, Q. A Mechanism Study Underlying the Protective Effects of Cyclosporine-A on Lung Ischemia-Reperfusion Injury. Pharmacology 2017, 100, 83–90. [Google Scholar] [CrossRef]
- Hokari, M.; Kuroda, S.; Iwasaki, Y. Pretreatment with the ciclosporin derivative NIM811 reduces delayed neuronal death in the hippocampus after transient forebrain ischaemia. J. Pharm. Pharmacol. 2010, 62, 485–490. [Google Scholar] [CrossRef] [PubMed]
- Devalaraja-Narashimha, K.; Diener, A.M.; Padanilam, B.J. Cyclophilin D gene ablation protects mice from ischemic renal injury. Am. J. Physiol. Ren. Physiol. 2009, 297, F749–F759. [Google Scholar] [CrossRef] [PubMed]
- Piot, C.; Croisille, P.; Staat, P.; Thibault, H.; Rioufol, G.; Mewton, N.; Elbelghiti, R.; Cung, T.T.; Bonnefoy, E.; Angoulvant, D.; et al. Effect of cyclosporine on reperfusion injury in acute myocardial infarction. N. Engl. J. Med. 2008, 359, 473–481. [Google Scholar] [CrossRef] [PubMed]
- Cung, T.T.; Morel, O.; Cayla, G.; Rioufol, G.; Garcia-Dorado, D.; Angoulvant, D.; Bonnefoy-Cudraz, E.; Guerin, P.; Elbaz, M.; Delarche, N.; et al. Cyclosporine before PCI in Patients with Acute Myocardial Infarction. N. Engl. J. Med. 2015, 373, 1021–1031. [Google Scholar] [CrossRef] [Green Version]
- Bernardi, P.; Di Lisa, F. Cyclosporine before PCI in Acute Myocardial Infarction. N. Engl. J. Med. 2016, 374, 89–90. [Google Scholar] [CrossRef] [PubMed]
- Hausenloy, D.J.; Yellon, D.M. Targeting Myocardial Reperfusion Injury–The Search Continues. N. Engl. J. Med. 2015, 373, 1073–1075. [Google Scholar] [CrossRef] [PubMed]
- Hansson, M.J.; Mattiasson, G.; Mansson, R.; Karlsson, J.; Keep, M.F.; Waldmeier, P.; Ruegg, U.T.; Dumont, J.M.; Besseghir, K.; Elmer, E. The nonimmunosuppressive cyclosporin analogs NIM811 and UNIL025 display nanomolar potencies on permeability transition in brain-derived mitochondria. J. Bioenerg. Biomembr. 2004, 36, 407–413. [Google Scholar] [CrossRef]
- Khaspekov, L.; Friberg, H.; Halestrap, A.; Viktorov, I.; Wieloch, T. Cyclosporin A and its nonimmunosuppressive analogue N-Me-Val-4-cyclosporin A mitigate glucose/oxygen deprivation-induced damage to rat cultured hippocampal neurons. Eur. J. Neurosci. 1999, 11, 3194–3198. [Google Scholar] [CrossRef] [PubMed]
- Waldmeier, P.C.; Feldtrauer, J.J.; Qian, T.; Lemasters, J.J. Inhibition of the mitochondrial permeability transition by the nonimmunosuppressive cyclosporin derivative NIM811. Mol. Pharmacol. 2002, 62, 22–29. [Google Scholar] [CrossRef]
- Dube, H.; Selwood, D.; Malouitre, S.; Capano, M.; Simone, M.I.; Crompton, M. A mitochondrial-targeted cyclosporin A with high binding affinity for cyclophilin D yields improved cytoprotection of cardiomyocytes. Biochem. J. 2012, 441, 901–907. [Google Scholar] [CrossRef] [Green Version]
- Malouitre, S.; Dube, H.; Selwood, D.; Crompton, M. Mitochondrial targeting of cyclosporin A enables selective inhibition of cyclophilin-D and enhanced cytoprotection after glucose and oxygen deprivation. Biochem. J. 2009, 425, 137–148. [Google Scholar] [CrossRef] [PubMed]
- Warne, J.; Pryce, G.; Hill, J.M.; Shi, X.; Lenneras, F.; Puentes, F.; Kip, M.; Hilditch, L.; Walker, P.; Simone, M.I.; et al. Selective Inhibition of the Mitochondrial Permeability Transition Pore Protects against Neurodegeneration in Experimental Multiple Sclerosis. J. Biol. Chem. 2016, 291, 4356–4373. [Google Scholar] [CrossRef] [PubMed]
- Azzolin, L.; Antolini, N.; Calderan, A.; Ruzza, P.; Sciacovelli, M.; Marin, O.; Mammi, S.; Bernardi, P.; Rasola, A. Antamanide, a derivative of Amanita phalloides, is a novel inhibitor of the mitochondrial permeability transition pore. PLoS ONE 2011, 6, e16280. [Google Scholar] [CrossRef] [PubMed]
- Briston, T.; Lewis, S.; Koglin, M.; Mistry, K.; Shen, Y.; Hartopp, N.; Katsumata, R.; Fukumoto, H.; Duchen, M.R.; Szabadkai, G.; et al. Identification of ER-000444793, a Cyclophilin D-independent inhibitor of mitochondrial permeability transition, using a high-throughput screen in cryopreserved mitochondria. Sci. Rep. 2016, 6, 37798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clarke, S.J.; McStay, G.P.; Halestrap, A.P. Sanglifehrin A acts as a potent inhibitor of the mitochondrial permeability transition and reperfusion injury of the heart by binding to cyclophilin-D at a different site from cyclosporin A. J. Biol. Chem. 2002, 277, 34793–34799. [Google Scholar] [CrossRef]
- Elkamhawy, A.; Park, J.E.; Hassan, A.H.E.; Pae, A.N.; Lee, J.; Park, B.G.; Roh, E.J. Synthesis and evaluation of 2-(3-arylureido)pyridines and 2-(3-arylureido)pyrazines as potential modulators of Aβ-induced mitochondrial dysfunction in Alzheimer’s disease. Eur. J. Med. Chem. 2018, 144, 529–543. [Google Scholar] [CrossRef] [PubMed]
- Sileikyte, J.; Forte, M. Shutting down the pore: The search for small molecule inhibitors of the mitochondrial permeability transition. Biochim. Biophys. Acta 2016, 1857, 1197–1202. [Google Scholar] [CrossRef]
- Shore, E.R.; Awais, M.; Kershaw, N.M.; Gibson, R.R.; Pandalaneni, S.; Latawiec, D.; Wen, L.; Javed, M.A.; Criddle, D.N.; Berry, N.; et al. Small Molecule Inhibitors of Cyclophilin D To Protect Mitochondrial Function as a Potential Treatment for Acute Pancreatitis. J. Med. Chem. 2016, 59, 2596–2611. [Google Scholar] [CrossRef]
- Fischer Scientific. Available online: https://www.fishersci.com/us/en/search/chemical/substructure (accessed on 10 December 2018).
- Hopkins, S.; Gallay, P. Cyclophilin inhibitors: An emerging class of therapeutics for the treatment of chronic hepatitis C infection. Viruses 2012, 4, 2558–2577. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Abbreviation | Full Name | Gene Name 1 |
|---|---|---|
| AMPK | AMP kinase | |
| ANT | adenine nucleotide translocator | Slc25a4-6 |
| ATP5PB | ATP synthase peripheral stalk-membrane subunit B | Atp5pb |
| ATP5PD | ATP synthase peripheral stalk-membrane subunit D | Atp5pd |
| BCL2 | B-cell lymphoma 2 | Bcl2 |
| C1QBP | complement 1q-binding protein | C1qbp |
| CNP | 2′,3′-Cyclic Nucleotide 3’ phosphodiesterase | Cnp |
| CsA | cyclosporin A | |
| CyPD | cyclophilin D | Ppif |
| DNAJC15 | DnaJ heat shock protein family (Hsp40) member C15 | Dnajc15 |
| ERK | mitogen-activated protein kinase 1 | Mapk1 |
| ETC | electron transport chain | |
| GSK-3β | glycogen synthase kinase-3β | Gsk3b |
| HK | hexokinase | Hk1-4 |
| HSP60 | heat shock protein 60 | Hsp60 |
| HSP70 | heat shock protein 70 | Hsp70 |
| HSP90 | heat shock protein 90 | Hsp90 |
| IMM | inner mitochondrial membrane | |
| IRI | ischemia–reperfusion injury | |
| kDa | kilodaltons | |
| MST1 | mammalian sterile 20-like kinase 1 | Stk4 |
| mtCK | mitochondrial creatine kinase | Ckmt1,2 |
| OMM | outer mitochondrial membrane | |
| OSCP | oligomycin sensitivity conferral protein | Atp5po |
| OXPHOS | oxidative phosphorylation | |
| PiC | mitochondrial phosphate carrier | Slc25a3 |
| PMSF | phenylmethylsulfonyl fluoride | |
| PPARα | peroxisome proliferator-activated receptor-α | Ppara |
| PPIase | peptidyl-prolyl, cis-trans isomerase | |
| PTP | mitochondrial permeability transition pore | |
| SIRT3 | sirtuin 3 | Sirt3 |
| SPG7 | spastic paraplegia 7 | Spg7 |
| STAT3 | signal transducer and activator of transcription 3 | Stat3 |
| TRAP-1 | tumor necrosis factor type 1 receptor-associated protein | Trap1 |
| tBID | BH3 interacting domain death agonist | Bid |
| TSPO | translocator protein of 18 kDa | Tspo |
| VDAC | voltage-dependent anion channel | Vdac1-3 |
| Protein | Binding to CyPD 1 | References |
|---|---|---|
| ANT | Direct | [1,69,70,71] |
| ATP5PB | Possibly direct | [57,75] |
| ATP5PD | Possibly direct | [57,75] |
| BCL2 | Direct | [107] |
| C1QBP | Direct | [111] |
| CNP | Indirect | [112] |
| DNAJC15 | Direct | [102] |
| ERK | Direct | [96] |
| GSK-3β | Direct | [55,96,97] |
| HK | Indirect | [73,74] |
| Hsp60 | Direct | [100,101] |
| Hsp70 | Direct | [102] |
| Hsp90 | Direct | [100,101] |
| MST1 | Direct | [99] |
| mtCK | Indirect | [73,74] |
| OCSP | Direct | [56,57,75] |
| p53 | Direct | [103,104] |
| PiC | Direct | [72] |
| PPARα | Direct | [95] |
| SIRT3 | Indirect | [78] |
| SPG7 | Direct | [80] |
| STAT3 | Direct | [108,109] |
| Tissue kallikrein | Possibly direct | [110] |
| TRAP-1 | Direct | [100,101,102] |
| VDAC | Indirect | [69,80] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Porter, G.A., Jr.; Beutner, G. Cyclophilin D, Somehow a Master Regulator of Mitochondrial Function. Biomolecules 2018, 8, 176. https://doi.org/10.3390/biom8040176
Porter GA Jr., Beutner G. Cyclophilin D, Somehow a Master Regulator of Mitochondrial Function. Biomolecules. 2018; 8(4):176. https://doi.org/10.3390/biom8040176
Chicago/Turabian StylePorter, George A., Jr., and Gisela Beutner. 2018. "Cyclophilin D, Somehow a Master Regulator of Mitochondrial Function" Biomolecules 8, no. 4: 176. https://doi.org/10.3390/biom8040176
APA StylePorter, G. A., Jr., & Beutner, G. (2018). Cyclophilin D, Somehow a Master Regulator of Mitochondrial Function. Biomolecules, 8(4), 176. https://doi.org/10.3390/biom8040176