Generation of a Mouse Model Lacking the Non-Homologous End-Joining Factor Mri/Cyren

 ,
,  and
and 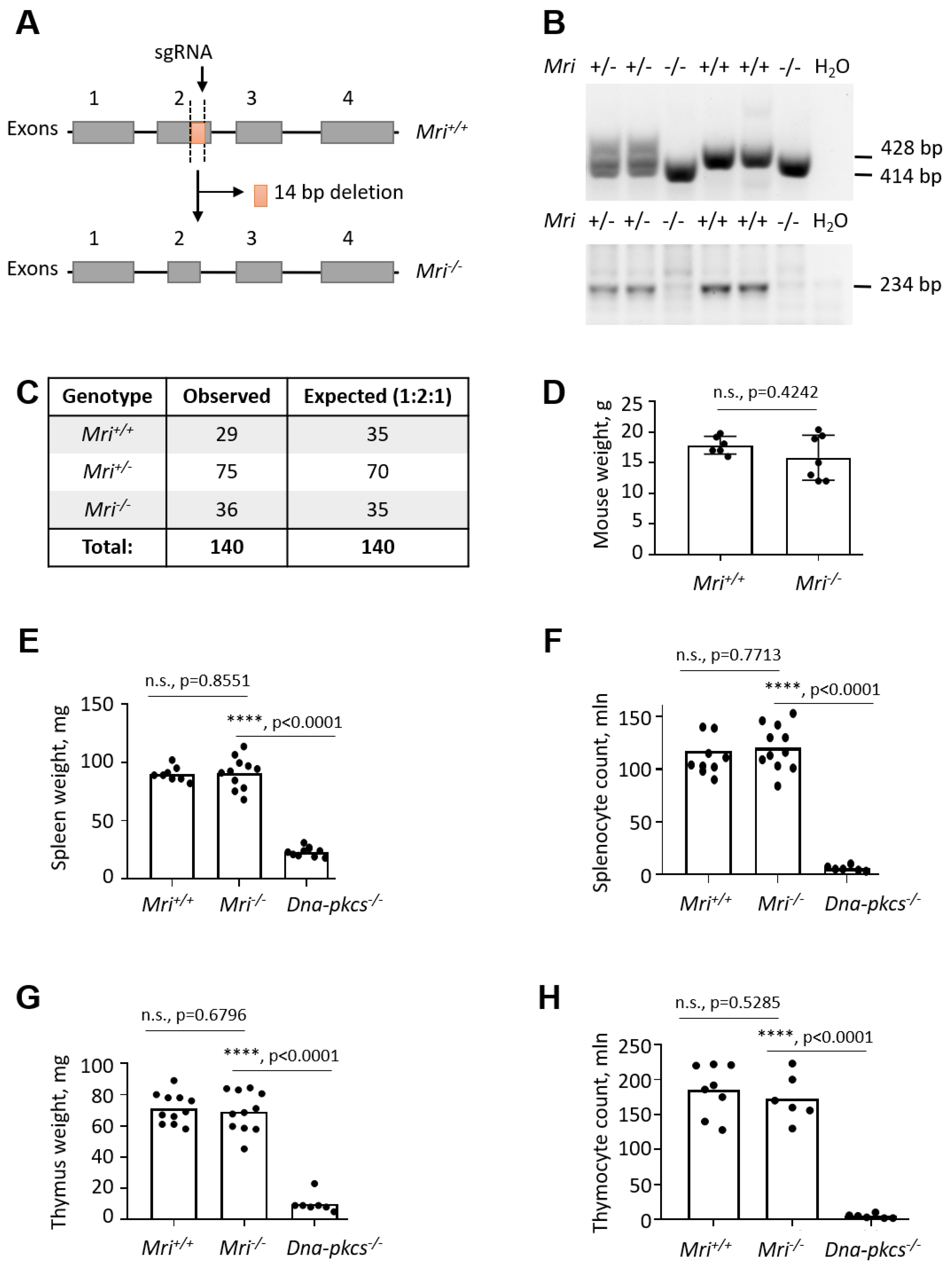{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Mouse Models
2.2. Generation of Mri+/− Mice
2.3. Mouse Genotyping
2.4. Fluorescence-Activated Cell Sorting, Splenocyte, and Thymocyte Count
2.5. Class Switch Recombination
2.6. Double Strand Break Sensitivity Assay
2.7. Brain Isolation and Neural Stem Progenitor Cell Culture
2.8. Neural Stem Progenitor Cell Proliferation and Self-Renewal Assays
2.9. Antibodies
3. Results
3.1. Generation of Mri−/− Mice
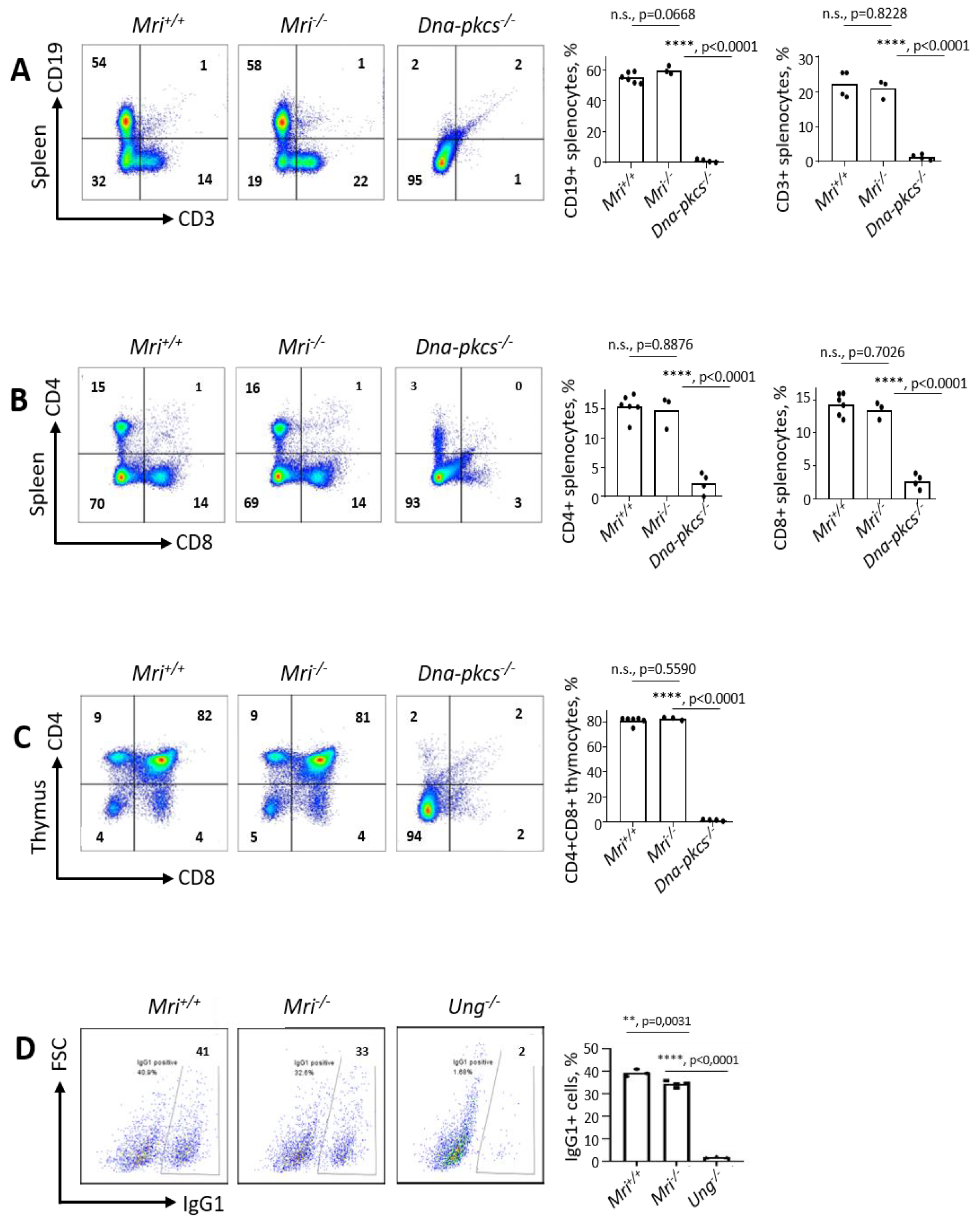
3.2. Mri−/− Mice Develop Normal Spleens and Thymi
3.3. Class Switch Recombination to IgG1 Is Reduced in Mri−/− Mice
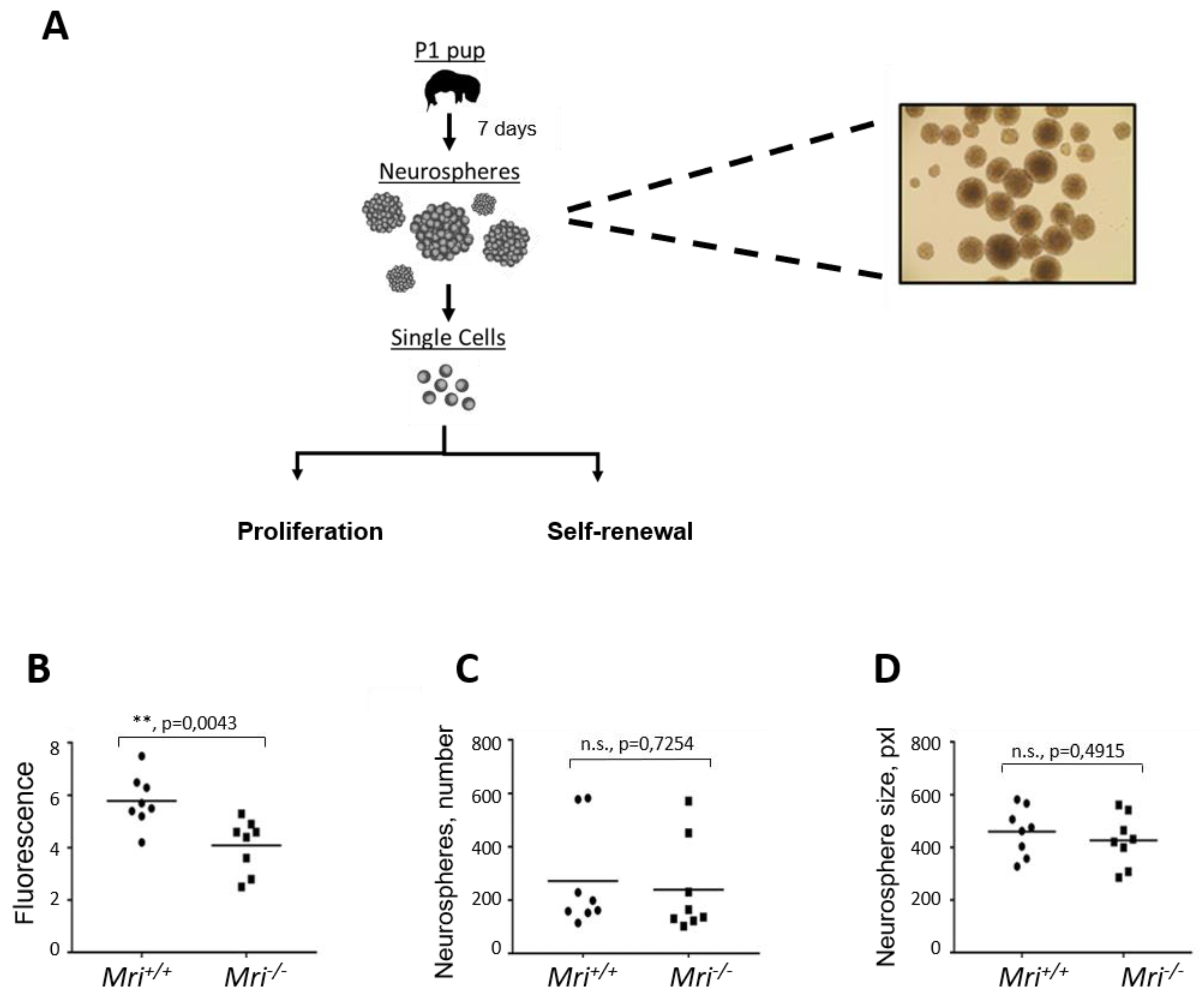
3.4. Lack of Mri Results in the Reduced Proliferation Rate of Neuronal Stem Progenitor Cells
3.5. Normal Self-Renewal Capacity of Mri-Deficient Neuronal Stem Progenitor Cells
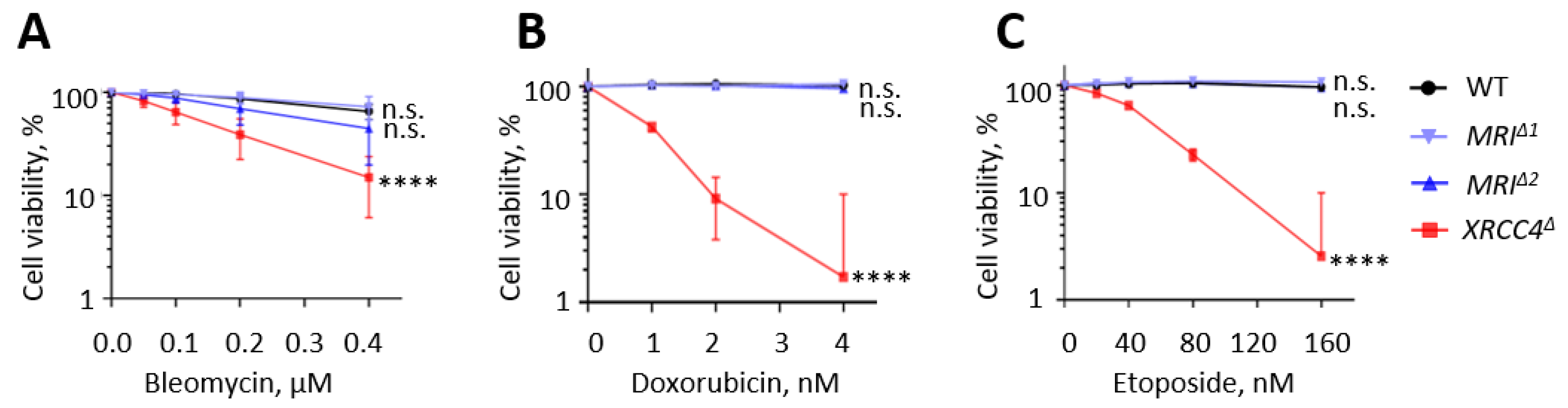
3.6. Human HAP1 Cells Lacking Mri Possess Normal Levels of Sensitivity to DNA Double-Strand Breaks
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ATM | Ataxia-telangiectasia mutated |
| CSR | Class switch recombination |
| DDR | DNA damage response |
| DNA-PKcs | DNA-dependent protein kinase |
| DSBs | DNA double-strand breaks |
| GFAP | Glial fibrillary acid protein |
| HAP1 | A near-haploid human cell line derived from KBM-7 cell line |
| IL-4 | Interleukin 4 |
| Lig4 | DNA ligase IV |
| LPS | Lipopolysaccharides |
| Mri | Modulator of retroviral infection |
| NHEJ | Non-homologous end-joining |
| NSPC | Neuronal stem progenitor cells |
| PAXX | Paralogue of XRCC4 and XLF |
| PCR | Polymerase chain reaction |
| XLF | XRCC4-like factor |
| XRCC4 | X-ray repair cross-complementing protein 4 |
References
- Kumar, V.; Alt, F.W.; Oksenych, V. Functional overlaps between XLF and the ATM-dependent DNA double strand break response. DNA Repair (Amst) 2014, 16, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Pannunzio, N.R.; Watanabe, G.; Lieber, M.R. Nonhomologous DNA end-joining for repair of DNA double-strand breaks. J. Biol. Chem. 2018, 293, 10512–10523. [Google Scholar] [CrossRef] [PubMed]
- Scully, R.; Panday, A.; Elango, R.; Willis, N.A. DNA double-strand break repair-pathway choice in somatic mammalian cells. Nat. Rev. Mol. Cell Biol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Alt, F.W.; Cheng, H.L.; Brush, J.W.; Goff, P.H.; Murphy, M.M.; Franco, S.; Zhang, Y.; Zha, S. Lymphocyte-specific compensation for XLF/cernunnos end-joining functions in V(D)J recombination. Mol. Cell 2008, 31, 631–640. [Google Scholar] [CrossRef]
- Vera, G.; Rivera-Munoz, P.; Abramowski, V.; Malivert, L.; Lim, A.; Bole-Feysot, C.; Martin, C.; Florkin, B.; Latour, S.; Revy, P.; et al. Cernunnos deficiency reduces thymocyte life span and alters the T cell repertoire in mice and humans. Mol. Cell Biol. 2013, 33, 701–711. [Google Scholar] [CrossRef]
- Zha, S.; Guo, C.; Boboila, C.; Oksenych, V.; Cheng, H.L.; Zhang, Y.; Wesemann, D.R.; Yuen, G.; Patel, H.; Goff, P.H.; et al. ATM damage response and XLF repair factor are functionally redundant in joining DNA breaks. Nature 2011, 469, 250–254. [Google Scholar] [CrossRef]
- Oksenych, V.; Alt, F.W.; Kumar, V.; Schwer, B.; Wesemann, D.R.; Hansen, E.; Patel, H.; Su, A.; Guo, C. Functional redundancy between repair factor XLF and damage response mediator 53BP1 in V(D)J recombination and DNA repair. Proc. Natl. Acad. Sci. USA 2012, 109, 2455–2460. [Google Scholar] [CrossRef]
- Abramowski, V.; Etienne, O.; Elsaid, R.; Yang, J.; Berland, A.; Kermasson, L.; Roch, B.; Musilli, S.; Moussu, J.P.; Lipson-Ruffert, K.; et al. PAXX and Xlf interplay revealed by impaired CNS development and immunodeficiency of double KO mice. Cell Death Differ. 2018, 25, 444–452. [Google Scholar] [CrossRef]
- Balmus, G.; Barros, A.C.; Wijnhoven, P.W.; Lescale, C.; Hasse, H.L.; Boroviak, K.; le Sage, C.; Doe, B.; Speak, A.O.; Galli, A.; et al. Synthetic lethality between PAXX and XLF in mammalian development. Genes Dev. 2016, 30, 2152–2157. [Google Scholar] [CrossRef]
- Castaneda-Zegarra, S.; Xing, M.; Gago-Fuentes, R.; Saeterstad, S.; Oksenych, V. Synthetic lethality between DNA repair factors Xlf and Paxx is rescued by inactivation of Trp53. DNA Repair (Amst) 2019, 73, 164–169. [Google Scholar] [CrossRef]
- Gago-Fuentes, R.; Xing, M.; Saeterstad, S.; Sarno, A.; Dewan, A.; Beck, C.; Bradamante, S.; Bjoras, M.; Oksenych, V. Normal development of mice lacking PAXX, the paralogue of XRCC4 and XLF. FEBS Open Bio 2018, 8, 426–434. [Google Scholar] [CrossRef]
- Liu, X.; Shao, Z.; Jiang, W.; Lee, B.J.; Zha, S. PAXX promotes KU accumulation at DNA breaks and is essential for end-joining in XLF-deficient mice. Nat. Commun. 2017, 8, 13816. [Google Scholar] [CrossRef]
- Frank, K.M.; Sharpless, N.E.; Gao, Y.; Sekiguchi, J.M.; Ferguson, D.O.; Zhu, C.; Manis, J.P.; Horner, J.; DePinho, R.A.; Alt, F.W. DNA ligase IV deficiency in mice leads to defective neurogenesis and embryonic lethality via the p53 pathway. Mol. Cell 2000, 5, 993–1002. [Google Scholar] [CrossRef]
- Gao, Y.; Ferguson, D.O.; Xie, W.; Manis, J.P.; Sekiguchi, J.; Frank, K.M.; Chaudhuri, J.; Horner, J.; DePinho, R.A.; Alt, F.W. Interplay of p53 and DNA-repair protein XRCC4 in tumorigenesis, genomic stability and development. Nature 2000, 404, 897–900. [Google Scholar] [CrossRef] [PubMed]
- Boboila, C.; Jankovic, M.; Yan, C.T.; Wang, J.H.; Wesemann, D.R.; Zhang, T.; Fazeli, A.; Feldman, L.; Nussenzweig, A.; Nussenzweig, M.; et al. Alternative end-joining catalyzes robust IgH locus deletions and translocations in the combined absence of ligase 4 and Ku70. Proc. Natl. Acad. Sci. USA 2010, 107, 3034–3039. [Google Scholar] [CrossRef]
- Boboila, C.; Yan, C.; Wesemann, D.R.; Jankovic, M.; Wang, J.H.; Manis, J.; Nussenzweig, A.; Nussenzweig, M.; Alt, F.W. Alternative end-joining catalyzes class switch recombination in the absence of both Ku70 and DNA ligase 4. J. Exp. Med. 2010, 207, 417–427. [Google Scholar] [CrossRef]
- Karanjawala, Z.E.; Adachi, N.; Irvine, R.A.; Oh, E.K.; Shibata, D.; Schwarz, K.; Hsieh, C.L.; Lieber, M.R. The embryonic lethality in DNA ligase IV-deficient mice is rescued by deletion of Ku: Implications for unifying the heterogeneous phenotypes of NHEJ mutants. DNA Repair (Amst) 2002, 1, 1017–1026. [Google Scholar] [CrossRef]
- Oksenych, V.; Kumar, V.; Liu, X.; Guo, C.; Schwer, B.; Zha, S.; Alt, F.W. Functional redundancy between the XLF and DNA-PKcs DNA repair factors in V(D)J recombination and nonhomologous DNA end joining. Proc. Natl. Acad. Sci. USA 2013, 110, 2234–2239. [Google Scholar] [CrossRef]
- Xing, M.; Bjoras, M.; Daniel, J.A.; Alt, F.W.; Oksenych, V. Synthetic lethality between murine DNA repair factors XLF and DNA-PKcs is rescued by inactivation of Ku70. DNA Repair (Amst) 2017, 57, 133–138. [Google Scholar] [CrossRef]
- Hung, P.J.; Johnson, B.; Chen, B.R.; Byrum, A.K.; Bredemeyer, A.L.; Yewdell, W.T.; Johnson, T.E.; Lee, B.J.; Deivasigamani, S.; Hindi, I.; et al. MRI Is a DNA Damage Response Adaptor during Classical Non-homologous End Joining. Mol. Cell 2018, 71, 332–342.e8. [Google Scholar] [CrossRef]
- Lescale, C.; Abramowski, V.; Bedora-Faure, M.; Murigneux, V.; Vera, G.; Roth, D.B.; Revy, P.; de Villartay, J.P.; Deriano, L. RAG2 and XLF/Cernunnos interplay reveals a novel role for the RAG complex in DNA repair. Nat. Commun. 2016, 7, 10529. [Google Scholar] [CrossRef]
- Chen, B.R.; Quinet, A.; Byrum, A.K.; Jackson, J.; Berti, M.; Thangavel, S.; Bredemeyer, A.L.; Hindi, I.; Mosammaparast, N.; Tyler, J.K.; et al. XLF and H2AX function in series to promote replication fork stability. J. Cell Biol. 2019, 218, 2113–2123. [Google Scholar] [CrossRef]
- Liu, X.; Jiang, W.; Dubois, R.L.; Yamamoto, K.; Wolner, Z.; Zha, S. Overlapping functions between XLF repair protein and 53BP1 DNA damage response factor in end joining and lymphocyte development. Proc. Natl. Acad. Sci. USA 2012, 109, 3903–3908. [Google Scholar] [CrossRef] [PubMed]
- Alt, F.W.; Zhang, Y.; Meng, F.L.; Guo, C.; Schwer, B. Mechanisms of programmed DNA lesions and genomic instability in the immune system. Cell 2013, 152, 417–429. [Google Scholar] [CrossRef]
- Yeap, L.S.; Meng, F.L. Cis- and trans-factors affecting AID targeting and mutagenic outcomes in antibody diversification. Adv. Immunol. 2019, 141, 51–103. [Google Scholar] [CrossRef]
- Alt, F.W.; Schwer, B. DNA double-strand breaks as drivers of neural genomic change, function, and disease. DNA Repair (Amst) 2018, 71, 158–163. [Google Scholar] [CrossRef]
- Agarwal, S.; Harada, J.; Schreifels, J.; Lech, P.; Nikolai, B.; Yamaguchi, T.; Chanda, S.K.; Somia, N.V. Isolation, characterization, and genetic complementation of a cellular mutant resistant to retroviral infection. Proc. Natl. Acad. Sci. USA 2006, 103, 15933–15938. [Google Scholar] [CrossRef]
- Slavoff, S.A.; Heo, J.; Budnik, B.A.; Hanakahi, L.A.; Saghatelian, A. A human short open reading frame (sORF)-encoded polypeptide that stimulates DNA end joining. J. Biol. Chem. 2014, 289, 10950–10957. [Google Scholar] [CrossRef]
- Grundy, G.J.; Rulten, S.L.; Arribas-Bosacoma, R.; Davidson, K.; Kozik, Z.; Oliver, A.W.; Pearl, L.H.; Caldecott, K.W. The Ku-binding motif is a conserved module for recruitment and stimulation of non-homologous end-joining proteins. Nat. Commun. 2016, 7, 11242. [Google Scholar] [CrossRef]
- Arnoult, N.; Correia, A.; Ma, J.; Merlo, A.; Garcia-Gomez, S.; Maric, M.; Tognetti, M.; Benner, C.W.; Boulton, S.J.; Saghatelian, A.; et al. Regulation of DNA repair pathway choice in S and G2 phases by the NHEJ inhibitor CYREN. Nature 2017, 549, 548–552. [Google Scholar] [CrossRef]
- Nilsen, H.; Rosewell, I.; Robins, P.; Skjelbred, C.F.; Andersen, S.; Slupphaug, G.; Daly, G.; Krokan, H.E.; Lindahl, T.; Barnes, D.E. Uracil-DNA glycosylase (UNG)-deficient mice reveal a primary role of the enzyme during DNA replication. Mol. Cell 2000, 5, 1059–1065. [Google Scholar] [CrossRef]
- Dewan, A.; Xing, M.; Lundbaek, M.B.; Gago-Fuentes, R.; Beck, C.; Aas, P.A.; Liabakk, N.B.; Saeterstad, S.; Chau, K.T.P.; Kavli, B.M.; et al. Robust DNA repair in PAXX-deficient mammalian cells. FEBS Open Bio 2018, 8, 442–448. [Google Scholar] [CrossRef]
- Xing, M.; Oksenych, V. Genetic interaction between DNA repair factors PAXX, XLF, XRCC4 and DNA-PKcs in human cells. FEBS Open Bio 2019. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Osenbroch, P.; Skinnes, R.; Esbensen, Y.; Bjoras, M.; Eide, L. Mitochondrial DNA integrity is essential for mitochondrial maturation during differentiation of neural stem cells. Stem Cells 2010, 28, 2195–2204. [Google Scholar] [CrossRef]
- Rooney, S.; Sekiguchi, J.; Zhu, C.; Cheng, H.L.; Manis, J.; Whitlow, S.; DeVido, J.; Foy, D.; Chaudhuri, J.; Lombard, D.; et al. Leaky Scid phenotype associated with defective V(D)J coding end processing in Artemis-deficient mice. Mol. Cell 2002, 10, 1379–1390. [Google Scholar] [CrossRef]
- Gao, Y.; Chaudhuri, J.; Zhu, C.; Davidson, L.; Weaver, D.T.; Alt, F.W. A targeted DNA-PKcs-null mutation reveals DNA-PK-independent functions for KU in V(D)J recombination. Immunity 1998, 9, 367–376. [Google Scholar] [CrossRef] [Green Version]
- Gu, Y.; Seidl, K.J.; Rathbun, G.A.; Zhu, C.; Manis, J.P.; van der Stoep, N.; Davidson, L.; Cheng, H.L.; Sekiguchi, J.M.; Frank, K.; et al. Growth retardation and leaky SCID phenotype of Ku70-deficient mice. Immunity 1997, 7, 653–665. [Google Scholar] [CrossRef] [Green Version]
- Nussenzweig, A.; Chen, C.; da Costa Soares, V.; Sanchez, M.; Sokol, K.; Nussenzweig, M.C.; Li, G.C. Requirement for Ku80 in growth and immunoglobulin V(D)J recombination. Nature 1996, 382, 551–555. [Google Scholar] [CrossRef]
- Gao, Y.; Sun, Y.; Frank, K.M.; Dikkes, P.; Fujiwara, Y.; Seidl, K.J.; Sekiguchi, J.M.; Rathbun, G.A.; Swat, W.; Wang, J.; et al. A critical role for DNA end-joining proteins in both lymphogenesis and neurogenesis. Cell 1998, 95, 891–902. [Google Scholar] [CrossRef] [Green Version]
- Frank, K.M.; Sekiguchi, J.M.; Seidl, K.J.; Swat, W.; Rathbun, G.A.; Cheng, H.L.; Davidson, L.; Kangaloo, L.; Alt, F.W. Late embryonic lethality and impaired V(D)J recombination in mice lacking DNA ligase IV. Nature 1998, 396, 173–177. [Google Scholar] [CrossRef]
- Sekiguchi, J.; Ferguson, D.O.; Chen, H.T.; Yang, E.M.; Earle, J.; Frank, K.; Whitlow, S.; Gu, Y.; Xu, Y.; Nussenzweig, A.; et al. Genetic interactions between ATM and the nonhomologous end-joining factors in genomic stability and development. Proc. Natl. Acad. Sci. USA 2001, 98, 3243–3248. [Google Scholar] [CrossRef] [PubMed] [Green Version]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Castañeda-Zegarra, S.; Huse, C.; Røsand, Ø.; Sarno, A.; Xing, M.; Gago-Fuentes, R.; Zhang, Q.; Alirezaylavasani, A.; Werner, J.; Ji, P.; et al. Generation of a Mouse Model Lacking the Non-Homologous End-Joining Factor Mri/Cyren. Biomolecules 2019, 9, 798. https://doi.org/10.3390/biom9120798
Castañeda-Zegarra S, Huse C, Røsand Ø, Sarno A, Xing M, Gago-Fuentes R, Zhang Q, Alirezaylavasani A, Werner J, Ji P, et al. Generation of a Mouse Model Lacking the Non-Homologous End-Joining Factor Mri/Cyren. Biomolecules. 2019; 9(12):798. https://doi.org/10.3390/biom9120798
Chicago/Turabian StyleCastañeda-Zegarra, Sergio, Camilla Huse, Øystein Røsand, Antonio Sarno, Mengtan Xing, Raquel Gago-Fuentes, Qindong Zhang, Amin Alirezaylavasani, Julia Werner, Ping Ji, and et al. 2019. "Generation of a Mouse Model Lacking the Non-Homologous End-Joining Factor Mri/Cyren" Biomolecules 9, no. 12: 798. https://doi.org/10.3390/biom9120798
APA StyleCastañeda-Zegarra, S., Huse, C., Røsand, Ø., Sarno, A., Xing, M., Gago-Fuentes, R., Zhang, Q., Alirezaylavasani, A., Werner, J., Ji, P., Liabakk, N. -B., Wang, W., Bjørås, M., & Oksenych, V. (2019). Generation of a Mouse Model Lacking the Non-Homologous End-Joining Factor Mri/Cyren. Biomolecules, 9(12), 798. https://doi.org/10.3390/biom9120798