Novel Variants of Angiotensin Converting Enzyme-2 of Shorter Molecular Size to Target the Kidney Renin Angiotensin System

Abstract
:1. Introduction
2. Methods
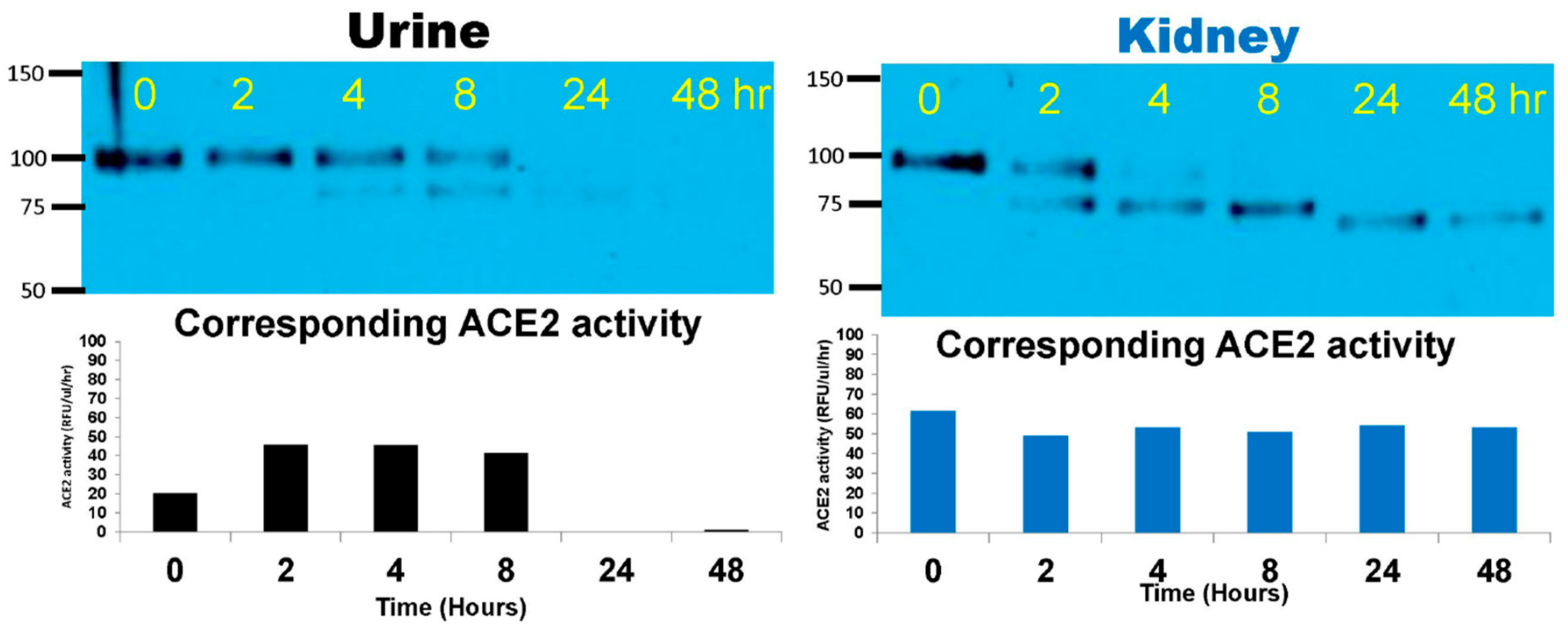
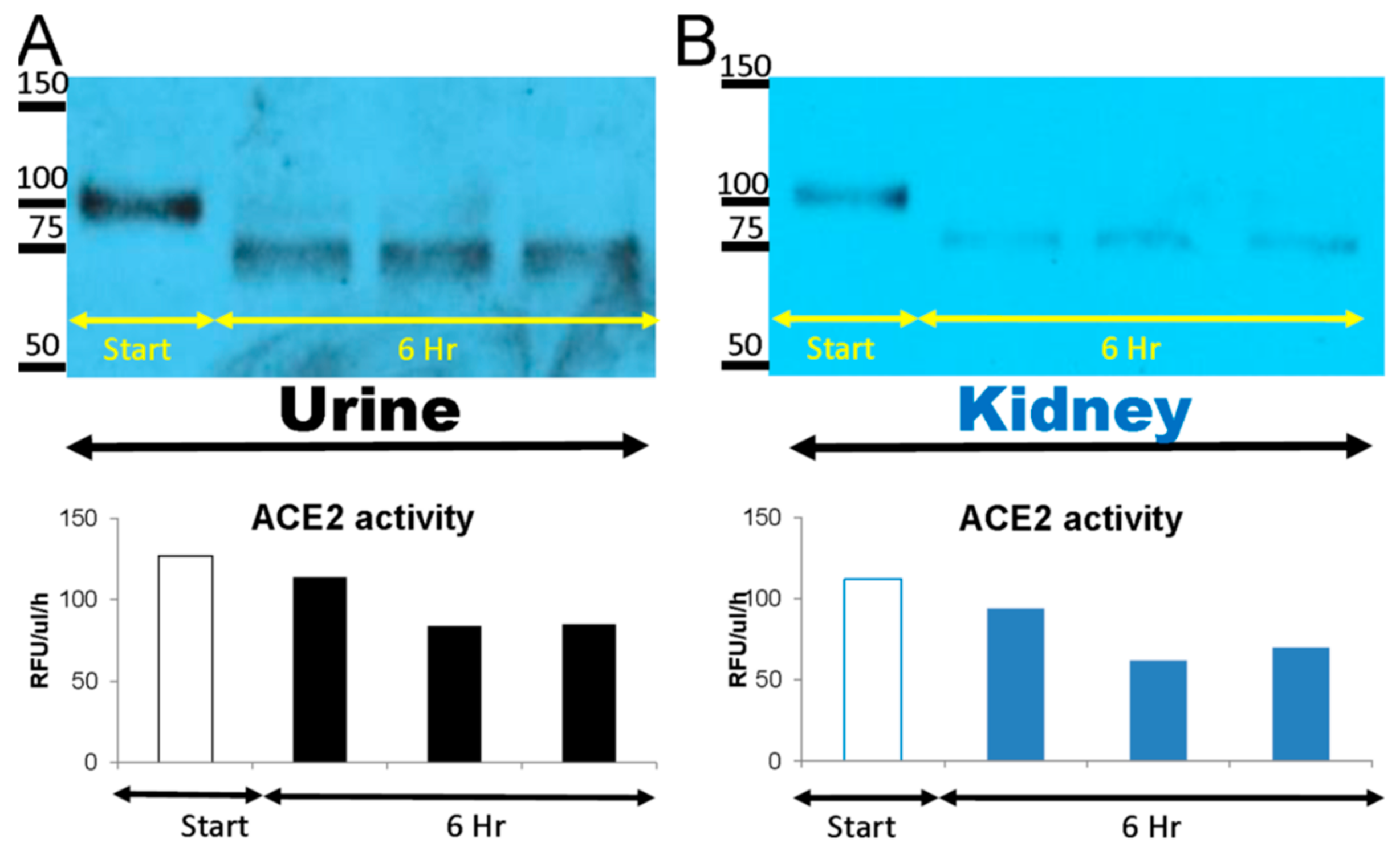
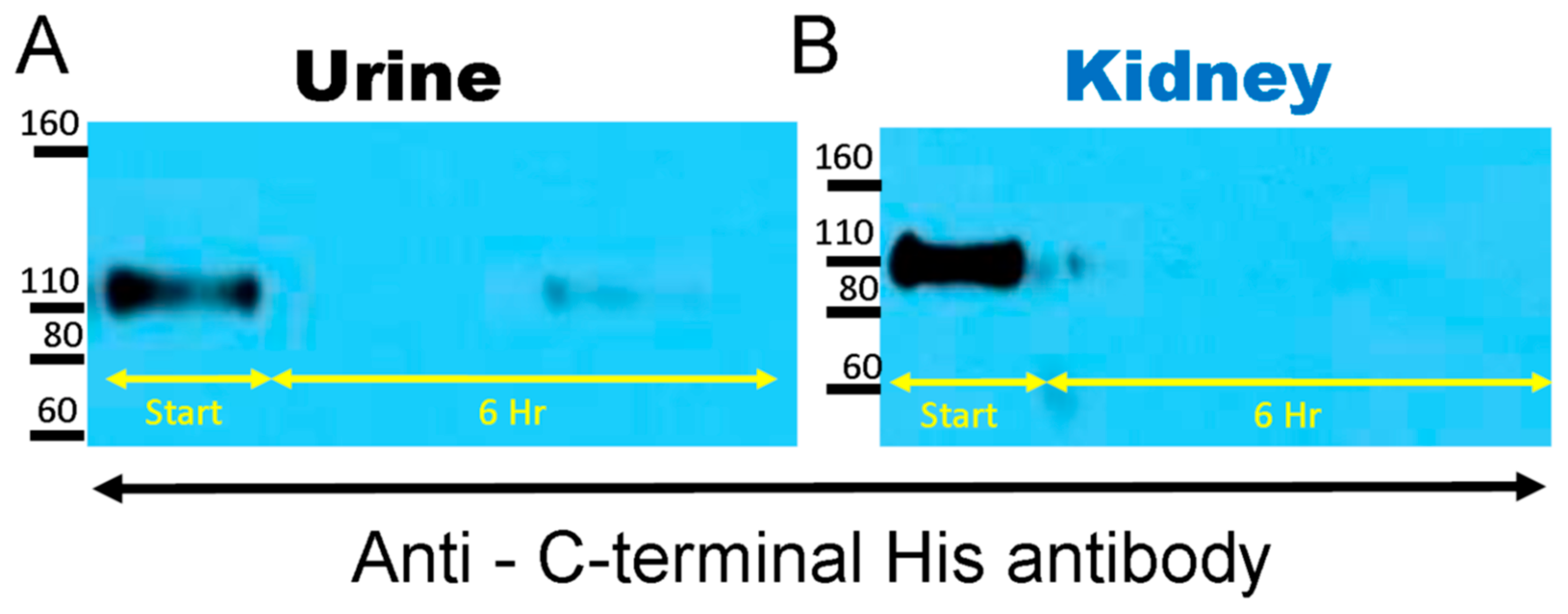
2.1. Degradation of Native rACE2 in Urine and Kidney Lysates from ACE2KO Mice
2.2. Design of Short ACE2 Variants
2.3. Production and Purification of Selected Short ACE2 Variants
2.4. Enzyme Activity
2.5. Acute Blood Pressure Response Ising Ang II-Induced Hypertension Mouse Model
2.6. Recovery of Urinary ACE2 after Recombinant ACE2 Infusion to ACE2-Deficient mice and the Effect of Blocking Tubular Reabsorption with L-lysine
2.7. Pharmacokinetics Analysis
2.8. Statistical Analyses
3. Results
3.1. Degradation of Native rACE2 to Shorter Enzymatically Active ACE2 Truncates
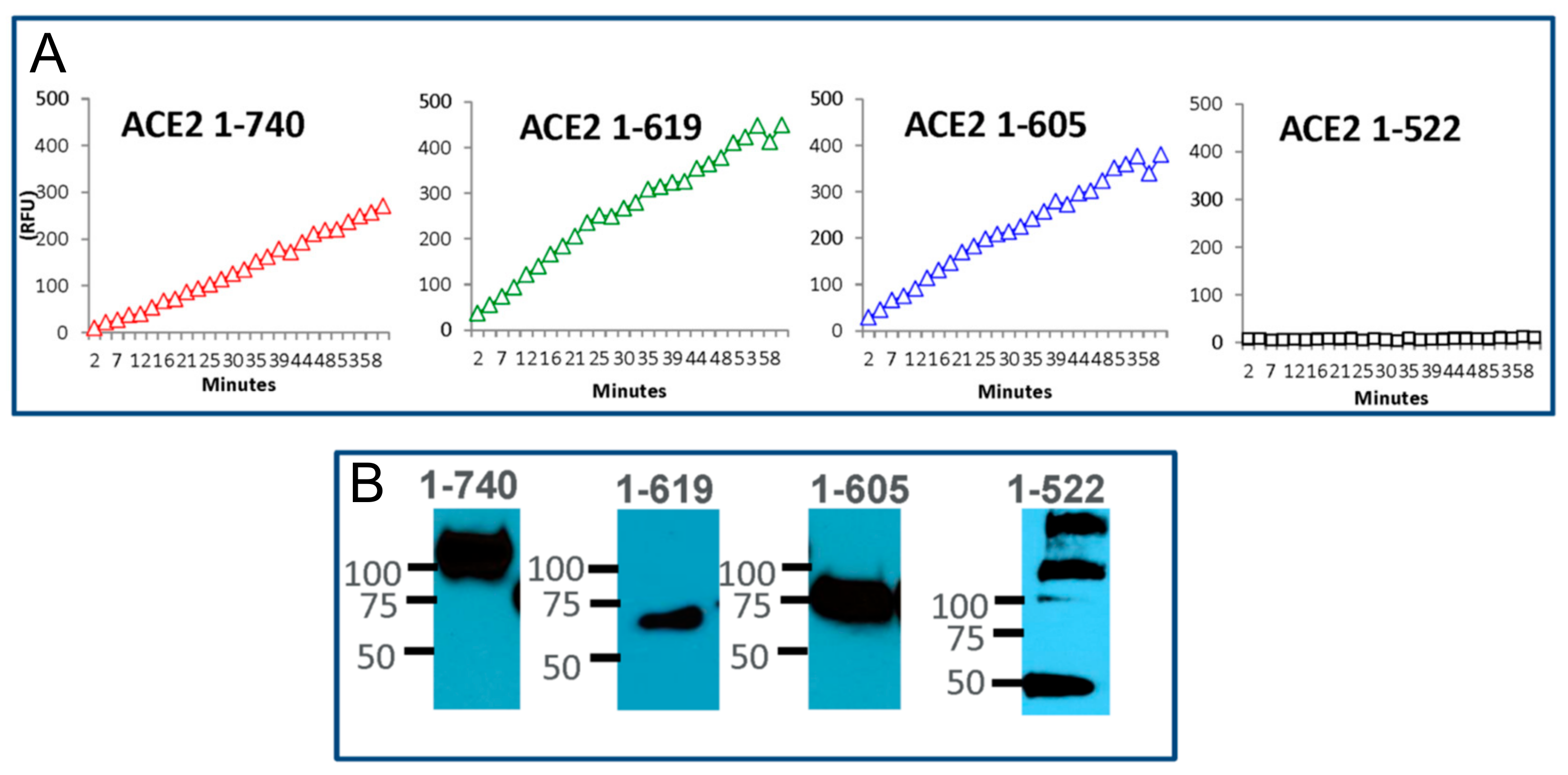
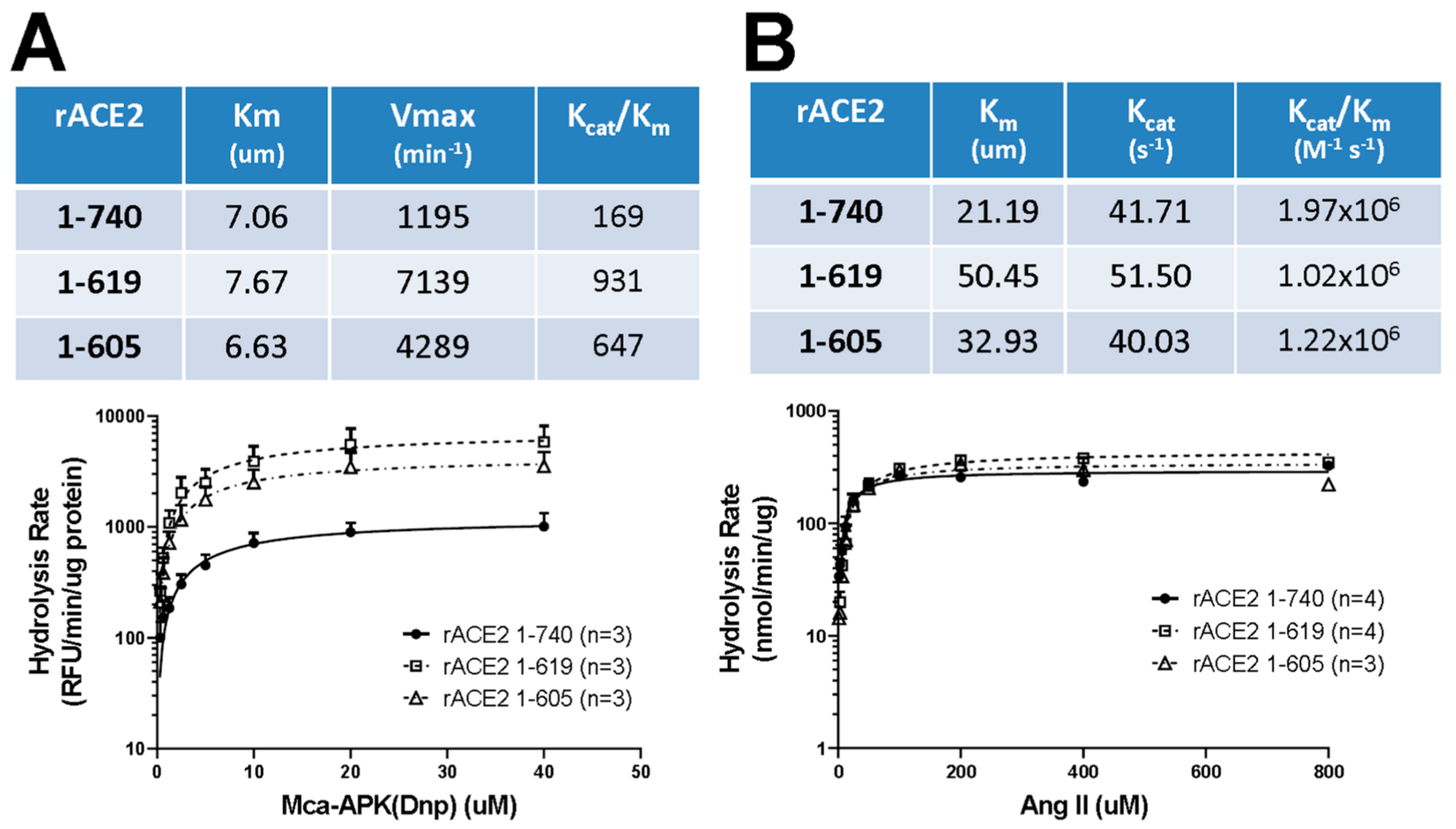
3.2. Design of Small ACE2 Variants and Demonstration of Their Enzyme Activity and Molecular Size
3.3. Enzyme Activities and Kinetic Constants of Purified Short Recombinant ACE2 Variants Using Angiotensin II as a Substrate
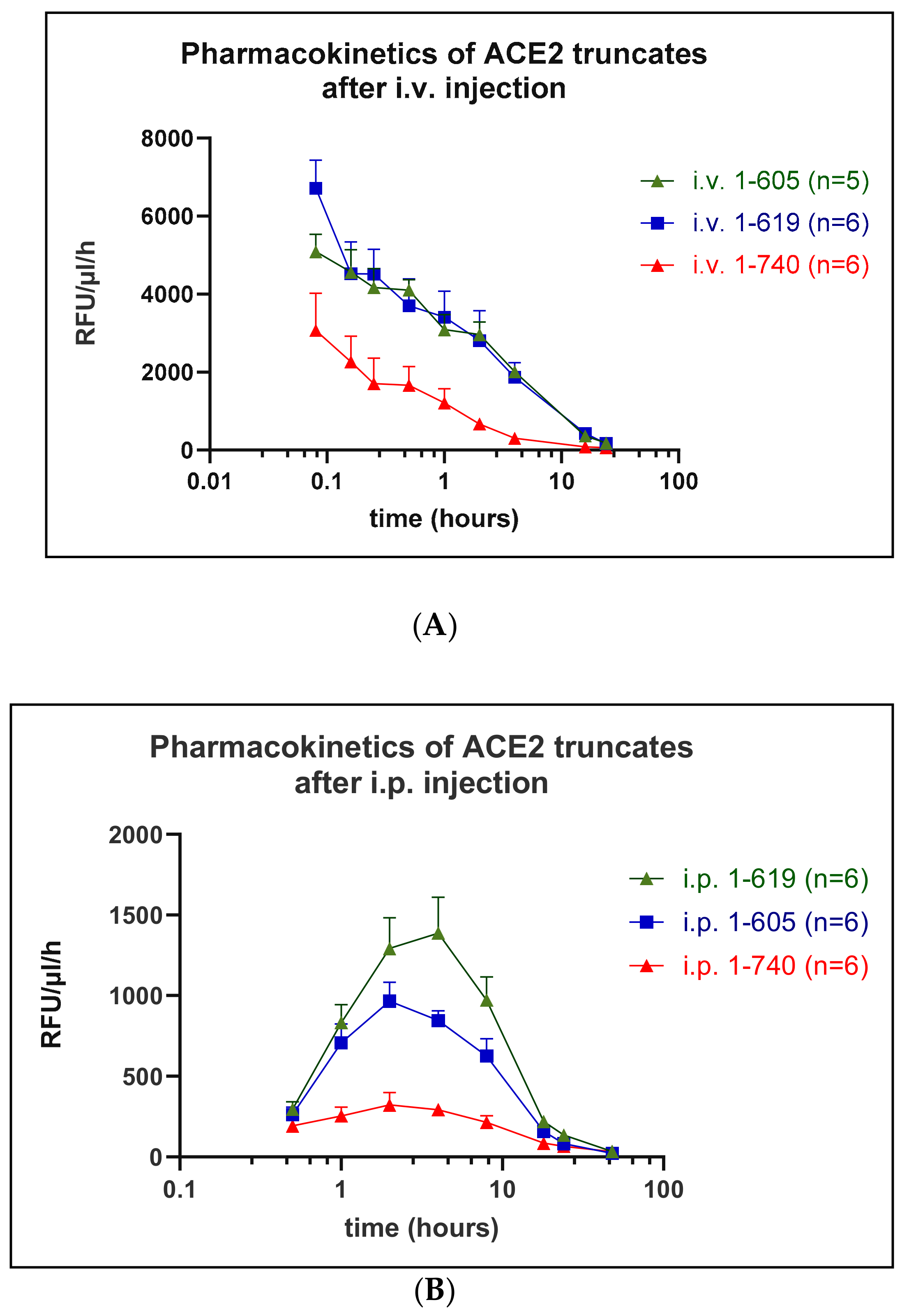
3.4. Pharmacokinetics
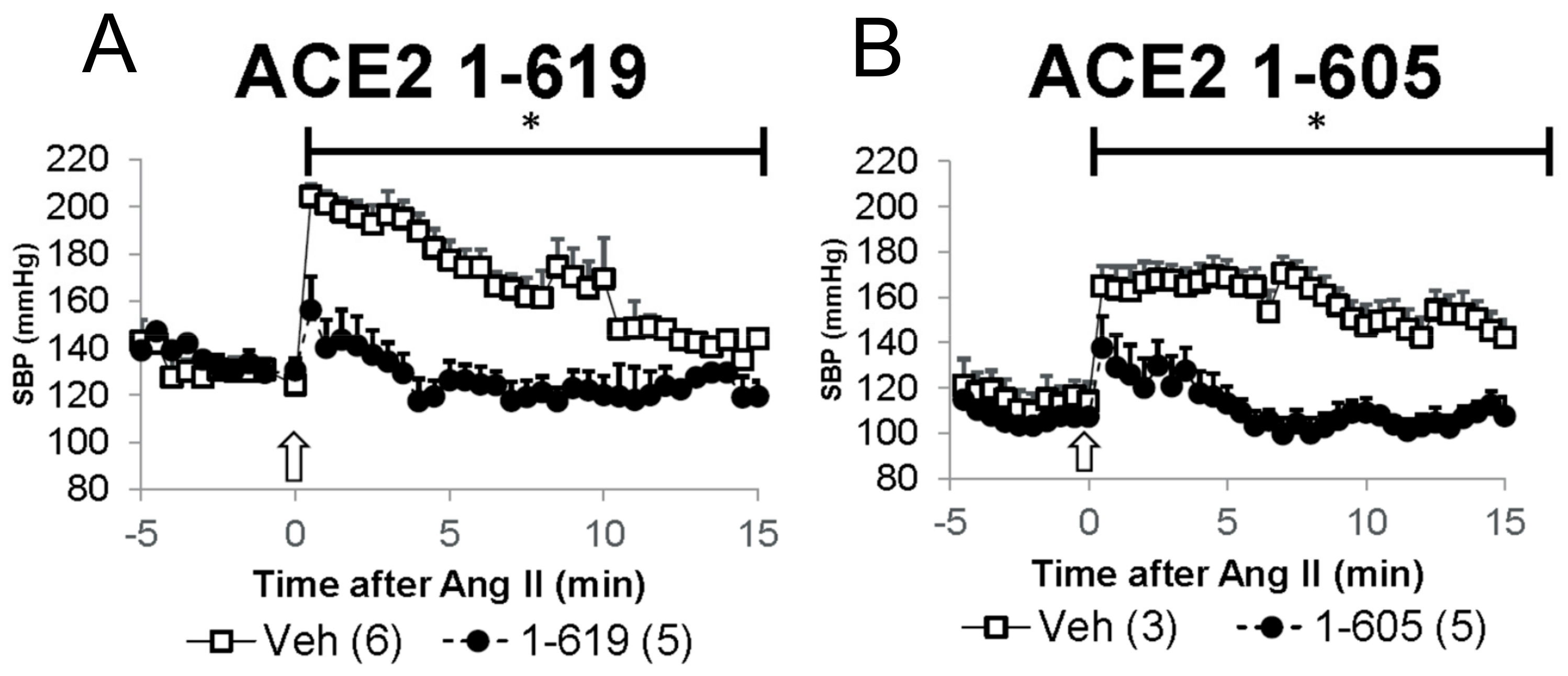
3.5. Demonstration of In Vivo Activity as Reflected by Blood Pressure-Lowering Effect of Short rACE2 Variants during Acute Ang II Infusion in Mice
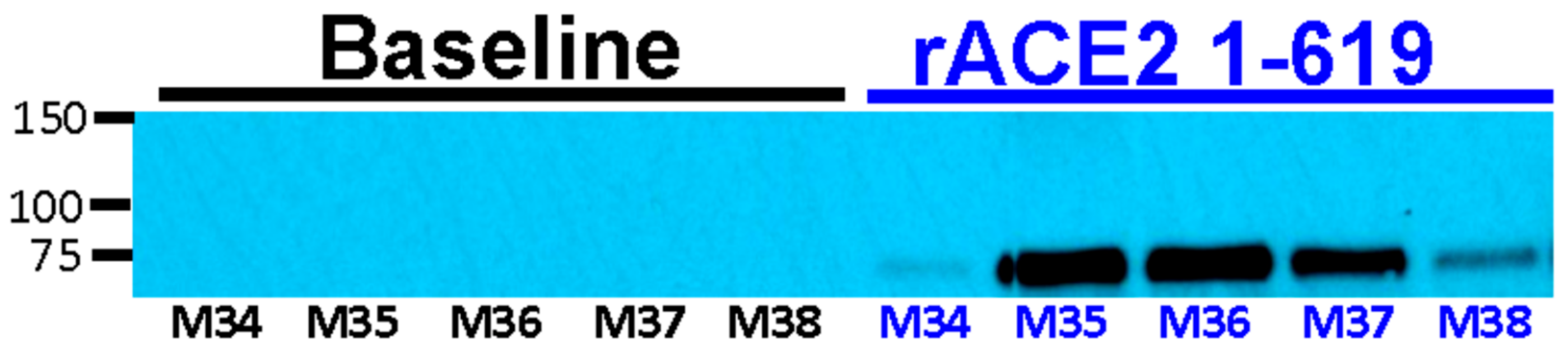
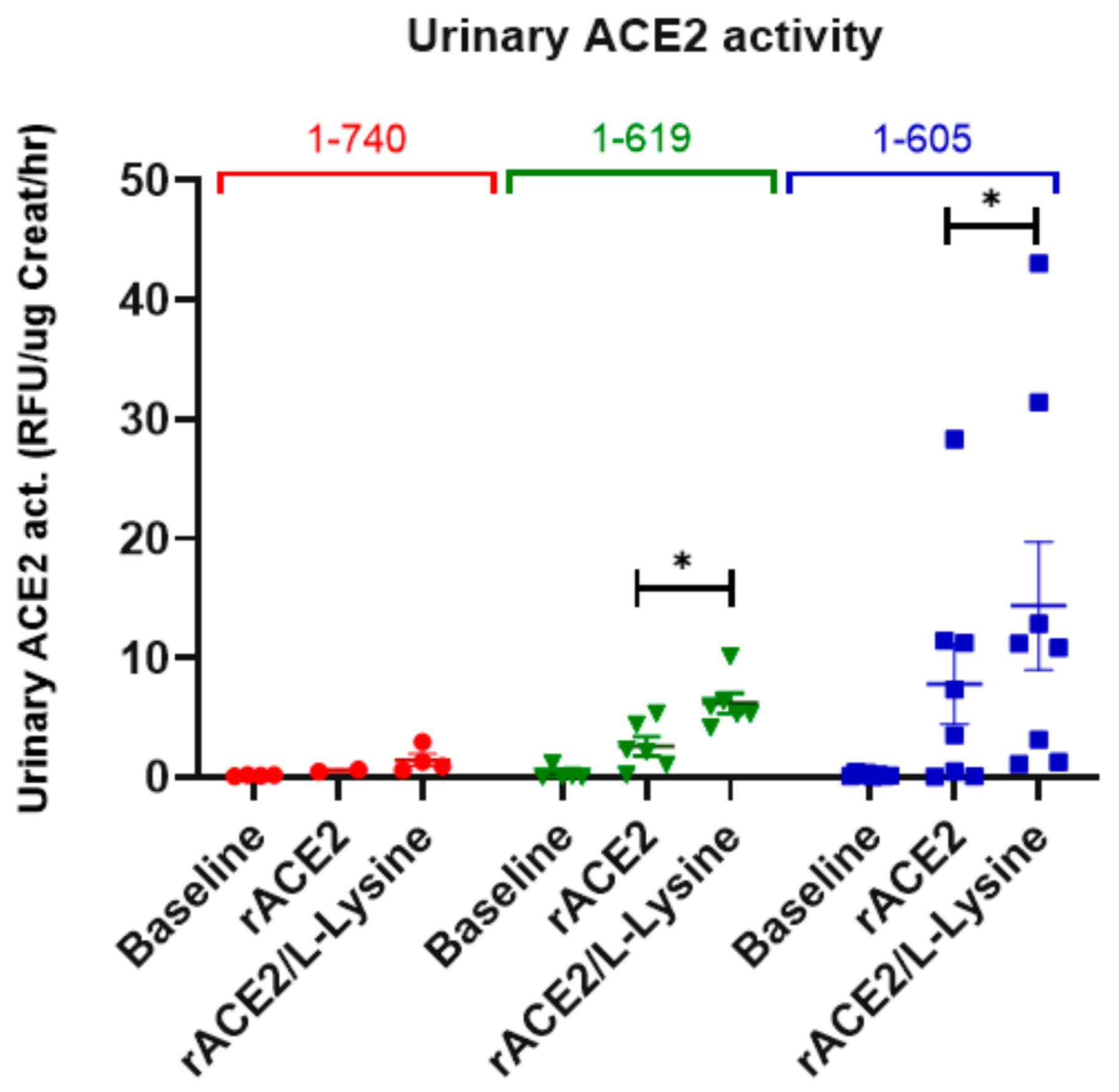
3.6. Recovery of Urinary ACE2 Activity after Administration of Short rACE2 Variants to ACE2-Deficient Mice
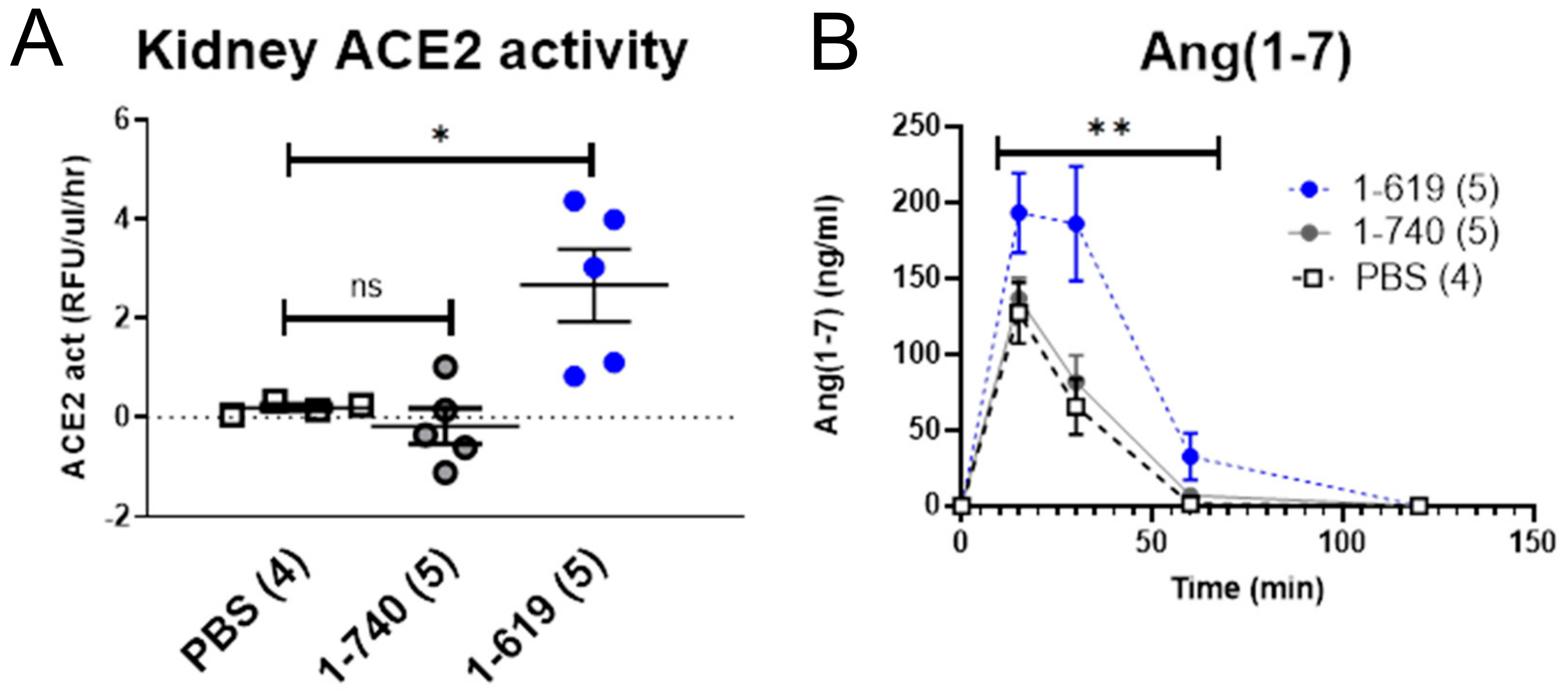
3.7. Glomerular Filtration and Kidney Uptake of Small rACE2 Variants in ACE2-Deficient Mice
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Batlle, D.; Wysocki, J.; Soler, M.J.; Ranganath, K. Angiotensin-converting enzyme 2: Enhancing the degradation of angiotensin II as a potential therapy for diabetic nephropathy. Kidney Int. 2012, 81, 520–528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ortiz-Melo, D.I.; Gurley, S.B. Angiotensin converting enzyme 2 and the kidney. Curr. Opin. Nephrol. Hypertens. 2016, 25, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Santos, R.A.; Simoes e Silva, A.C.; Maric, C.; Silva, D.M.; Machado, R.P.; de Buhr, I.; Heringer-Walther, S.; Pinheiro, S.V.; Lopes, M.T.; Bader, M.; et al. Angiotensin-(1-7) is an endogenous ligand for the G protein-coupled receptor Mas. Proc. Natl. Acad. Sci. USA 2003, 100, 8258–8263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zimmerman, D.; Burns, K.D. Angiotensin-(1-7) in kidney disease: A review of the controversies. Clin. Sci. 2012, 123, 333–346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrario, C.M.; Jessup, J.; Gallagher, P.E.; Averill, D.B.; Brosnihan, K.B.; Ann Tallant, E.; Smith, R.D.; Chappell, M.C. Effects of renin-angiotensin system blockade on renal angiotensin-(1-7) forming enzymes and receptors. Kidney Int. 2005, 68, 2189–2196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wysocki, J.; Ye, M.; Rodriguez, E.; Gonzalez-Pacheco, F.R.; Barrios, C.; Evora, K.; Schuster, M.; Loibner, H.; Brosnihan, K.B.; Ferrario, C.M.; et al. Targeting the degradation of angiotensin II with recombinant angiotensin-converting enzyme 2: Prevention of angiotensin II-dependent hypertension. Hypertension 2010, 55, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Ye, M.; Wysocki, J.; Gonzalez-Pacheco, F.R.; Salem, M.; Evora, K.; Garcia-Halpin, L.; Poglitsch, M.; Schuster, M.; Batlle, D. Murine recombinant angiotensin-converting enzyme 2: Effect on angiotensin II-dependent hypertension and distinctive angiotensin-converting enzyme 2 inhibitor characteristics on rodent and human angiotensin-converting enzyme 2. Hypertension 2012, 60, 730–740. [Google Scholar] [CrossRef] [Green Version]
- Li, N.; Zimpelmann, J.; Cheng, K.; Wilkins, J.A.; Burns, K.D. The role of angiotensin converting enzyme 2 in the generation of angiotensin 1-7 by rat proximal tubules. Am. J. Physiol. Ren. Physiol. 2005, 288, F353–F362. [Google Scholar] [CrossRef] [PubMed]
- Ross, M.J.; Nangaku, M. ACE2 as therapy for glomerular disease: The devil is in the detail. Kidney Int. 2017, 91, 1269–1271. [Google Scholar] [CrossRef] [PubMed]
- Jiang, F.; Yang, J.; Zhang, Y.; Dong, M.; Wang, S.; Zhang, Q.; Liu, F.F.; Zhang, K.; Zhang, C. Angiotensin-converting enzyme 2 and angiotensin 1-7: Novel therapeutic targets. Nat. Rev. Cardiol. 2014, 11, 413–426. [Google Scholar] [CrossRef]
- Towler, P.; Staker, B.; Prasad, S.G.; Menon, S.; Tang, J.; Parsons, T.; Ryan, D.; Fisher, M.; Williams, D.; Dales, N.A.; et al. ACE2 X-ray structures reveal a large hinge-bending motion important for inhibitor binding and catalysis. J. Biol. Chem. 2004, 279, 17996–18007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wysocki, J.; Garcia-Halpin, L.; Ye, M.; Maier, C.; Sowers, K.; Burns, K.D.; Batlle, D. Regulation of urinary ACE2 in diabetic mice. Am. J. Physiol. Ren. Physiol. 2013, 305, F600–F611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wysocki, J.; Ye, M.; Khattab, A.M.; Fogo, A.; Martin, A.; David, N.V.; Kanwar, Y.; Osborn, M.; Batlle, D. Angiotensin-converting enzyme 2 amplification limited to the circulation does not protect mice from development of diabetic nephropathy. Kidney Int. 2017, 91, 1336–1346. [Google Scholar] [CrossRef] [PubMed]
- Haber, P.K.; Ye, M.; Wysocki, J.; Maier, C.; Haque, S.K.; Batlle, D. Angiotensin-converting enzyme 2-independent action of presumed angiotensin-converting enzyme 2 activators: Studies in vivo, ex vivo, and in vitro. Hypertension 2014, 63, 774–782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, P.; Wysocki, J.; Souma, T.; Ye, M.; Ramirez, V.; Zhou, B.; Wilsbacher, L.D.; Quaggin, S.E.; Batlle, D.; Jin, J. Novel ACE2-Fc chimeric fusion provides long-lasting hypertension control and organ protection in mouse models of systemic renin angiotensin system activation. Kidney Int. 2018, 94, 114–125. [Google Scholar] [CrossRef]
- Wysocki, J.; Goodling, A.; Burgaya, M.; Whitlock, K.; Ruzinski, J.; Batlle, D.; Afkarian, M. Urine RAS components in mice and people with type 1 diabetes and chronic kidney disease. Am. J. Physiol. Ren. Physiol. 2017, 313, F487–F494. [Google Scholar] [CrossRef] [Green Version]
- Wysocki, J.; Ye, M.; Batlle, D. Plasma and Kidney Angiotensin Peptides: Importance of the Aminopeptidase A/Angiotensin III Axis. Am. J. Hypertens. 2015, 28, 1418–1426. [Google Scholar] [CrossRef] [Green Version]
- Xiao, F.; Hiremath, S.; Knoll, G.; Zimpelmann, J.; Srivaratharajah, K.; Jadhav, D.; Fergusson, D.; Kennedy, C.R.; Burns, K.D. Increased urinary angiotensin-converting enzyme 2 in renal transplant patients with diabetes. PLoS ONE 2012, 7, e37649. [Google Scholar] [CrossRef] [Green Version]
- Lew, R.A.; Warner, F.J.; Hanchapola, I.; Smith, A.I. Characterization of angiotensin converting enzyme-2 (ACE2) in human urine. Int. J. Pept. Res. 2006, 12, 283–289. [Google Scholar] [CrossRef]
- Xiao, F.; Zimpelmann, J.; Agaybi, S.; Gurley, S.B.; Puente, L.; Burns, K.D. Characterization of angiotensin-converting enzyme 2 ectodomain shedding from mouse proximal tubular cells. PLoS ONE 2014, 9, e85958. [Google Scholar] [CrossRef]
- Mizuiri, S.; Aoki, T.; Hemmi, H.; Arita, M.; Sakai, K.; Aikawa, A. Urinary ACE2 in patients with CKD. Nephrology 2011. [Google Scholar] [CrossRef] [PubMed]
- Ye, M.; Wysocki, J.; William, J.; Soler, M.J.; Cokic, I.; Batlle, D. Glomerular localization and expression of Angiotensin-converting enzyme 2 and Angiotensin-converting enzyme: Implications for albuminuria in diabetes. J. Am. Soc. Nephrol. 2006, 17, 3067–3075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wysocki, J.; Ye, M.; Soler, M.J.; Gurley, S.B.; Xiao, H.D.; Bernstein, K.E.; Coffman, T.M.; Chen, S.; Batlle, D. ACE and ACE2 activity in diabetic mice. Diabetes 2006, 55, 2132–2139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, M.; Wysocki, J.; Naaz, P.; Salabat, M.R.; LaPointe, M.S.; Batlle, D. Increased ACE 2 and decreased ACE protein in renal tubules from diabetic mice: A renoprotective combination? Hypertension 2004, 43, 1120–1125. [Google Scholar] [CrossRef] [Green Version]
- Liu, P.; Wysocki, J.; Serfozo, P.; Ye, M.; Souma, T.; Batlle, D.; Jin, J. A Fluorometric Method of Measuring Carboxypeptidase Activities for Angiotensin II and Apelin 13. Sci. Rep. 2017, 7, 45473. [Google Scholar] [CrossRef] [Green Version]
- Maier, C.; Schadock, I.; Haber, P.K.; Wysocki, J.; Ye, M.; Kanwar, Y.; Flask, C.A.; Yu, X.; Hoit, B.D.; Adams, G.N.; et al. Prolylcarboxypeptidase deficiency is associated with increased blood pressure, glomerular lesions, and cardiac dysfunction independent of altered circulating and cardiac angiotensin II. J. Mol. Med. 2017, 95, 473–486. [Google Scholar] [CrossRef]
- Gurley, S.B.; Allred, A.; Le, T.H.; Griffiths, R.; Mao, L.; Philip, N.; Haystead, T.A.; Donoghue, M.; Breitbart, R.E.; Acton, S.L.; et al. Altered blood pressure responses and normal cardiac phenotype in ACE2-null mice. J. Clin. Investig. 2006, 116, 2218–2225. [Google Scholar] [CrossRef] [Green Version]
- Thelle, K.; Christensen, E.I.; Vorum, H.; Orskov, H.; Birn, H. Characterization of proteinuria and tubular protein uptake in a new model of oral L-lysine administration in rats. Kidney Int. 2006, 69, 1333–1340. [Google Scholar] [CrossRef] [Green Version]
- Tang, J.; Wysocki, J.; Ye, M.; Valles, P.G.; Rein, J.; Shirazi, M.; Bader, M.; Gomez, R.A.; Sequeira-Lopez, M.S.; Afkarian, M.; et al. Urinary Renin in Patients and Mice With Diabetic Kidney Disease. Hypertension 2019, 74, 83–94. [Google Scholar] [CrossRef]
- Lambert, D.W.; Yarski, M.; Warner, F.J.; Thornhill, P.; Parkin, E.T.; Smith, A.I.; Hooper, N.M.; Turner, A.J. Tumor necrosis factor-alpha convertase (ADAM17) mediates regulated ectodomain shedding of the severe-acute respiratory syndrome-coronavirus (SARS-CoV) receptor, angiotensin-converting enzyme-2 (ACE2). J. Biol. Chem. 2005, 280, 30113–30119. [Google Scholar] [CrossRef] [Green Version]
- Salem, E.S.; Grobe, N.; Elased, K.M. Insulin treatment attenuates renal ADAM17 and ACE2 shedding in diabetic Akita mice. Am. J. Physiol. Ren. Physiol. 2014, 306, F629–F639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donoghue, M.; Hsieh, F.; Baronas, E.; Godbout, K.; Gosselin, M.; Stagliano, N.; Donovan, M.; Woolf, B.; Robison, K.; Jeyaseelan, R.; et al. A novel angiotensin-converting enzyme-related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1-9. Circ. Res. 2000, 87, E1–E9. [Google Scholar] [CrossRef] [PubMed]
- Durvasula, R.V.; Petermann, A.T.; Hiromura, K.; Blonski, M.; Pippin, J.; Mundel, P.; Pichler, R.; Griffin, S.; Couser, W.G.; Shankland, S.J. Activation of a local tissue angiotensin system in podocytes by mechanical strain. Kidney Int. 2004, 65, 30–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durvasula, R.V.; Shankland, S.J. Activation of a local renin angiotensin system in podocytes by glucose. Am. J. Physiol. Ren. Physiol. 2008, 294, F830–F839. [Google Scholar] [CrossRef] [Green Version]
- Ingelfinger, J.R.; Zuo, W.M.; Fon, E.A.; Ellison, K.E.; Dzau, V.J. In situ hybridization evidence for angiotensinogen messenger RNA in the rat proximal tubule. An hypothesis for the intrarenal renin angiotensin system. J. Clin. Investig. 1990, 85, 417–423. [Google Scholar] [CrossRef] [Green Version]
- Ba Aqeel, S.; Ye, M.; Wysocki, J.; Sanchez, A.; Khattab, A.; Lores, E.; Rademaker, A.; Gao, X.; Bebu, I.; Nelson, R.G.; et al. Urinary angiotensinogen antedates the development of stage 3 CKD in patients with type 1 diabetes mellitus. Physiol. Rep. 2019, 7, e14242. [Google Scholar] [CrossRef] [Green Version]
- Mills, K.T.; Kobori, H.; Hamm, L.L.; Alper, A.B.; Khan, I.E.; Rahman, M.; Navar, L.G.; Liu, Y.; Browne, G.M.; Batuman, V.; et al. Increased urinary excretion of angiotensinogen is associated with risk of chronic kidney disease. Nephrol. Dial. Transplant. 2012, 27, 3176–3181. [Google Scholar] [CrossRef] [Green Version]
- Urushihara, M.; Kagami, S. Role of the intrarenal renin-angiotensin system in the progression of renal disease. Pediatr. Nephrol. 2017, 32, 1471–1479. [Google Scholar] [CrossRef]
- Kobori, H.; Urushihara, M. Augmented intrarenal and urinary angiotensinogen in hypertension and chronic kidney disease. Pflug. Arch. Eur. J. Physiol. 2013, 465, 3–12. [Google Scholar] [CrossRef] [Green Version]
- Vickers, C.; Hales, P.; Kaushik, V.; Dick, L.; Gavin, J.; Tang, J.; Godbout, K.; Parsons, T.; Baronas, E.; Hsieh, F.; et al. Hydrolysis of biological peptides by human angiotensin-converting enzyme-related carboxypeptidase. J. Biol. Chem. 2002, 277, 14838–14843. [Google Scholar] [CrossRef] [Green Version]
- Mehta, R.L.; Pascual, M.T.; Soroko, S.; Savage, B.R.; Himmelfarb, J.; Ikizler, T.A.; Paganini, E.P.; Chertow, G.M. Program to Improve Care in Acute Renal, D. Spectrum of acute renal failure in the intensive care unit: The PICARD experience. Kidney Int. 2004, 66, 1613–1621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liano, F.; Pascual, J. Outcomes in acute renal failure. Semin. Nephrol. 1998, 18, 541–550. [Google Scholar] [PubMed]
- Basile, D.P.; Anderson, M.D.; Sutton, T.A. Pathophysiology of acute kidney injury. Compr. Physiol. 2012, 2, 1303–1353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Star, R.A. Treatment of acute renal failure. Kidney Int. 1998, 54, 1817–1831. [Google Scholar] [CrossRef] [Green Version]
- Kontogiannis, J.; Burns, K.D. Role of AT1 angiotensin II receptors in renal ischemic injury. Am. J. Physiol. 1998, 274, F79–F90. [Google Scholar] [CrossRef]
- Da Silveira, K.D.; Pompermayer Bosco, K.S.; Diniz, L.R.; Carmona, A.K.; Cassali, G.D.; Bruna-Romero, O.; de Sousa, L.P.; Teixeira, M.M.; Santos, R.A.; Simoes e Silva, A.C.; et al. ACE2-angiotensin-(1-7)-Mas axis in renal ischaemia/reperfusion injury in rats. Clin. Sci. 2010, 119, 385–394. [Google Scholar] [CrossRef] [Green Version]
- Allred, A.J.; Chappell, M.C.; Ferrario, C.M.; Diz, D.I. Differential actions of renal ischemic injury on the intrarenal angiotensin system. Am. J. Physiol. Ren. Physiol. 2000, 279, F636–F645. [Google Scholar] [CrossRef] [Green Version]
- Ba Aqeel, S.H.; Sanchez, A.; Batlle, D. Angiotensinogen as a biomarker of acute kidney injury. Clin. Kidney J. 2017, 10, 759–768. [Google Scholar] [CrossRef] [Green Version]
- Bellomo, R.; Kellum, J.A.; Ronco, C. Acute kidney injury. Lancet 2012, 380, 756–766. [Google Scholar] [CrossRef]
- Mackie, F.E.; Campbell, D.J.; Meyer, T.W. Intrarenal angiotensin and bradykinin peptide levels in the remnant kidney model of renal insufficiency. Kidney Int. 2001, 59, 1458–1465. [Google Scholar] [CrossRef] [Green Version]
- Okui, S.; Yamamoto, H.; Li, W.; Gamachi, N.; Fujita, Y.; Kashiwamura, S.; Miura, D.; Takai, S.; Miyazaki, M.; Urade, M.; et al. Cisplatin-induced acute renal failure in mice is mediated by chymase-activated angiotensin-aldosterone system and interleukin-18. Eur. J. Pharmacol. 2012, 685, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Fang, F.; Liu, G.C.; Zhou, X.; Yang, S.; Reich, H.N.; Williams, V.; Hu, A.; Pan, J.; Konvalinka, A.; Oudit, G.Y.; et al. Loss of ACE2 exacerbates murine renal ischemia-reperfusion injury. PLoS ONE 2013, 8, e71433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saleh, S.; Ain-Shoka, A.A.; El-Demerdash, E.; Khalef, M.M. Protective effects of the angiotensin II receptor blocker losartan on cisplatin-induced kidney injury. Chemotherapy 2009, 55, 399–406. [Google Scholar] [CrossRef]
- El-Sayed el, S.M.; Abd-Ellah, M.F.; Attia, S.M. Protective effect of captopril against cisplatin-induced nephrotoxicity in rats. Pak. J. Pharm. Sci. 2008, 21, 255–261. [Google Scholar]
- Zhang, J.; Rudemiller, N.P.; Patel, M.B.; Wei, Q.; Karlovich, N.S.; Jeffs, A.D.; Wu, M.; Sparks, M.A.; Privratsky, J.R.; Herrera, M.; et al. Competing Actions of Type 1 Angiotensin II Receptors Expressed on T Lymphocytes and Kidney Epithelium during Cisplatin-Induced AKI. J. Am. Soc. Nephrol. 2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deegan, P.M.; Nolan, C.; Ryan, M.P.; Basinger, M.A.; Jones, M.M.; Hande, K.R. The role of the renin-angiotensin system in cisplatin nephrotoxicity. Ren. Fail. 1995, 17, 665–674. [Google Scholar] [CrossRef]
- Efrati, S.; Berman, S.; Hamad, R.A.; Siman-Tov, Y.; Ilgiyaev, E.; Maslyakov, I.; Weissgarten, J. Effect of captopril treatment on recuperation from ischemia/reperfusion-induced acute renal injury. Nephrol. Dial. Transplant. 2012, 27, 136–145. [Google Scholar] [CrossRef] [Green Version]
- Kocak, C.; Kocak, F.E.; Akcilar, R.; Bayat, Z.; Aras, B.; Metineren, M.H.; Yucel, M.; Simsek, H. Effects of captopril, telmisartan and bardoxolone methyl (CDDO-Me) in ischemia-reperfusion-induced acute kidney injury in rats: An experimental comparative study. Clin. Exp. Pharmacol. Physiol. 2016, 43, 230–241. [Google Scholar] [CrossRef]
- Mehrotra, P.; Patel, J.B.; Ivancic, C.M.; Collett, J.A.; Basile, D.P. Th-17 cell activation in response to high salt following acute kidney injury is associated with progressive fibrosis and attenuated by AT-1R antagonism. Kidney Int. 2015, 88, 776–784. [Google Scholar] [CrossRef] [Green Version]
- Cho, J.H.; Choi, S.Y.; Ryu, H.M.; Oh, E.J.; Yook, J.M.; Ahn, J.S.; Jung, H.Y.; Choi, J.Y.; Park, S.H.; Kim, C.D.; et al. Fimasartan attenuates renal ischemia-reperfusion injury by modulating inflammation-related apoptosis. Korean J. Physiol. Pharm. 2018, 22, 661–670. [Google Scholar] [CrossRef] [Green Version]
- Rafiq, K.; Hitomi, H.; Nakano, D.; Nishiyama, A. Pathophysiological roles of aldosterone and mineralocorticoid receptor in the kidney. J. Pharmacol. Sci. 2011, 115, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chrysostomou, A.; Pedagogos, E.; MacGregor, L.; Becker, G.J. Double-blind, placebo-controlled study on the effect of the aldosterone receptor antagonist spironolactone in patients who have persistent proteinuria and are on long-term angiotensin-converting enzyme inhibitor therapy, with or without an angiotensin II receptor blocker. Clin. J. Am. Soc. Nephrol. CJASN 2006, 1, 256–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, X.; Ono, H.; Ono, Y.; Frohlich, E.D. Aldosterone antagonism ameliorates proteinuria and nephrosclerosis independent of glomerular dynamics in L-NAME/SHR model. Am. J. Nephrol. 2004, 24, 242–249. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ACE2 1-740 (n = 6) | ACE2 1-619 (n = 6) | ACE2 1-605 (n = 5) | |
|---|---|---|---|
| Distribution Phase (t1/2α) (min) | 13 ± 2 | 19 ± 5 | 58 ± 24 |
| Elimination Phase (t1/2β) (h) | 1.35 ± 0.21 | 4.2 ± 1.07 * | 4.0 ± 0.5 |
| AUC | 6437 ± 1639 | 27,636 ± 6297 * | 28,257 ± 1901 * |
| MRT (h) | 0.86 ± 0.12 | 2.20 ± 0.8 | 3.30 ± 0.72 * |
| ACE2 1-740 (n = 6) | ACE2 1-619 (n = 6) | ACE2 1-605 (n = 6) | |
|---|---|---|---|
| Distribution Phase (t1/2α) (min) | 123 ± 47 | 70 ± 8 | 62 ± 9 |
| Elimination Phase (t1/2β) (h) | 8.68 ± 0.79 | 5.62 ± 0.32 * | 6.77 ± 0.67 |
| AUC | 5145 ± 683 | 17,792 ± 2288 * | 11,753 ± 1250 * |
| MRT (h) | 15.0 ± 1.71 | 14.9 ± 1.16 | 12.9 ± 0.95 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wysocki, J.; Schulze, A.; Batlle, D. Novel Variants of Angiotensin Converting Enzyme-2 of Shorter Molecular Size to Target the Kidney Renin Angiotensin System. Biomolecules 2019, 9, 886. https://doi.org/10.3390/biom9120886
Wysocki J, Schulze A, Batlle D. Novel Variants of Angiotensin Converting Enzyme-2 of Shorter Molecular Size to Target the Kidney Renin Angiotensin System. Biomolecules. 2019; 9(12):886. https://doi.org/10.3390/biom9120886
Chicago/Turabian StyleWysocki, Jan, Arndt Schulze, and Daniel Batlle. 2019. "Novel Variants of Angiotensin Converting Enzyme-2 of Shorter Molecular Size to Target the Kidney Renin Angiotensin System" Biomolecules 9, no. 12: 886. https://doi.org/10.3390/biom9120886
APA StyleWysocki, J., Schulze, A., & Batlle, D. (2019). Novel Variants of Angiotensin Converting Enzyme-2 of Shorter Molecular Size to Target the Kidney Renin Angiotensin System. Biomolecules, 9(12), 886. https://doi.org/10.3390/biom9120886