The Main Metabolites of Fluorouracil + Adriamycin + Cyclophosphamide (FAC) Are Not Major Contributors to FAC Toxicity in H9c2 Cardiac Differentiated Cells

 ,
,  ,
,  ,
,  ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
2.2.1. Cell Culture Experimental Protocols
2.2.2. Experimental Protocol Paradigm
2.2.3. Cytotoxicity Tests
MTT Reduction Assay
Lysosomal Neutral Red Uptake Assay
2.2.4. Microscopic Observation of the Cells
Contrast Phase Microscopy
Hoechst Nuclear Staining
2.2.5. Mitochondrial Membrane Potential
2.2.6. Statistical Analysis
3. Results
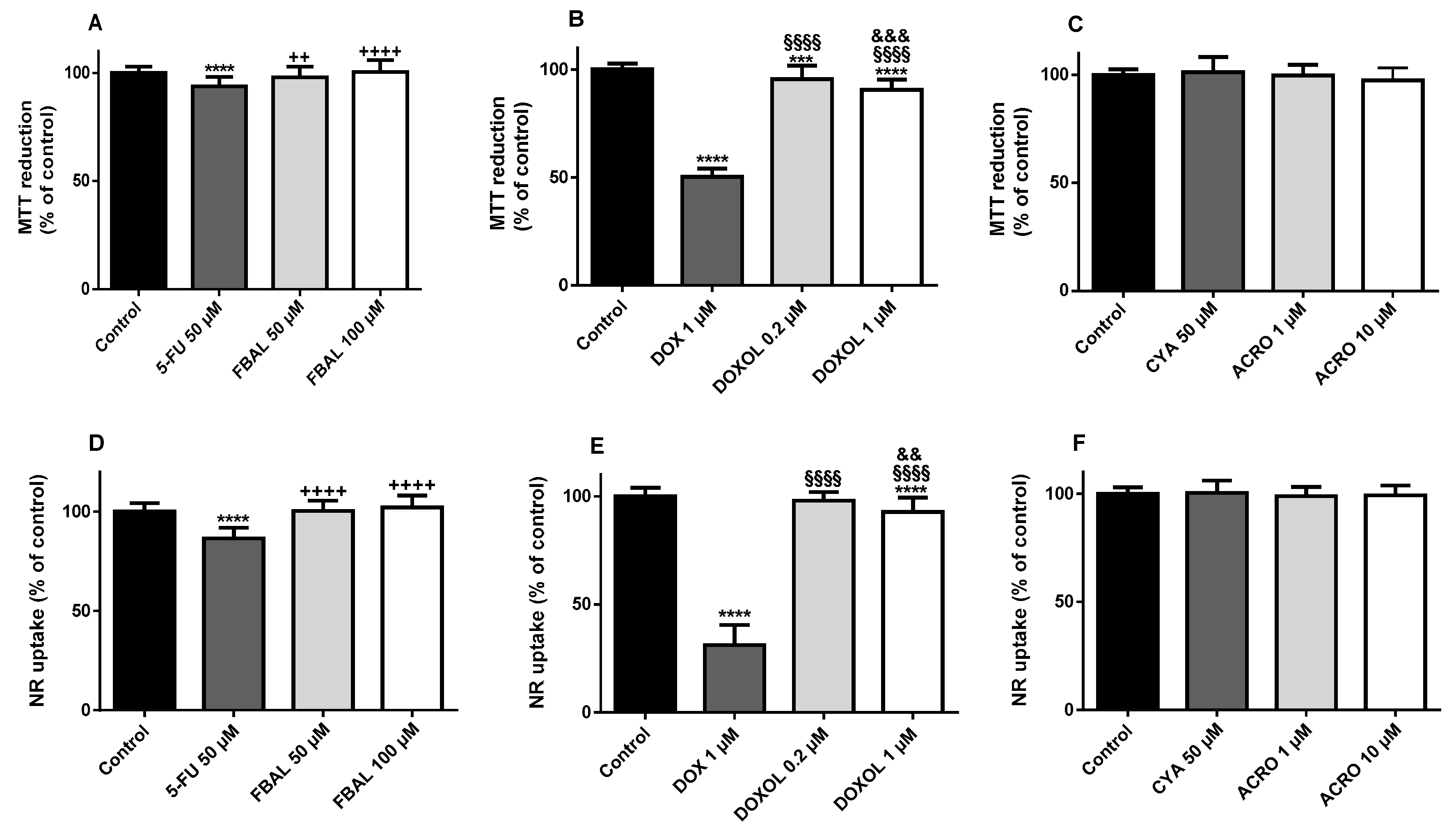
3.1. Both α-Fluoro-β-Alanine and Doxorubicinol Were Less Cytotoxic to Differentiated H9c2 Cells Than Their Parent Drugs
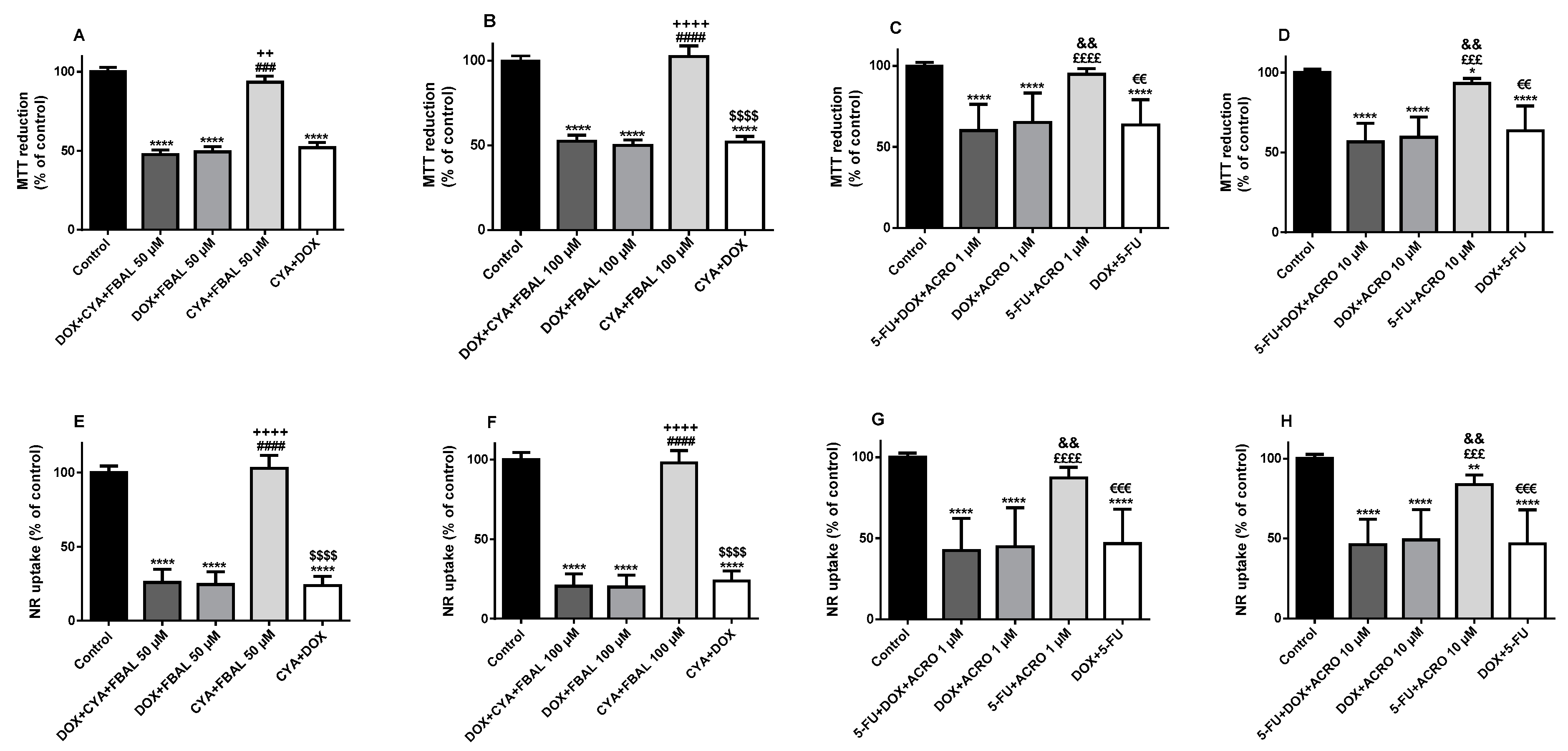
3.2. In Combination with 5-Fluorouracil, Cyclophosphamide or Their Metabolites, Doxorubicin Remained Key to the Cytotoxicity Observed in Differentiated H9c2 Cells
3.3. In the Combination 5-Fluorouracil + Cyclophosphamide + Doxorubicinol, 5-Fluorouracil Was Determinant to the Cytotoxicity Observed in Differentiated H9c2 Cells and Doxorubicinol Did Not Increase Its Cytotoxicity
3.4. All Combinations of α-Fluoro-β-Alanine, Doxorubicinol and Acrolein and All Mixtures Containing Doxorubicinol at the Highest Concentration Caused Cytotoxicity in Differentiated H9c2 Cells
3.5. The Mixture of the Metabolites of 5-Fluorouracil, Doxorubicin and Cyclophosphamide (α-Fluoro-β-Alanine 100 µM + Doxorubicinol 1 µM + Acrolein 10 µM) Caused a Significant Decrease in Mitochondria Potential in Differentiated H9c2 Cells
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Shih, Y.C.; Smieliauskas, F.; Geynisman, D.M.; Kelly, R.J.; Smith, T.J. Trends in the cost and use of targeted cancer therapies for the privately insured nonelderly: 2001 to 2011. J. Clin. Oncol. 2015, 33, 2190–2196. [Google Scholar] [CrossRef] [PubMed]
- Anampa, J.; Makower, D.; Sparano, J.A. Progress in adjuvant chemotherapy for breast cancer: An overview. BMC Med. 2015, 13, 195. [Google Scholar] [CrossRef] [PubMed]
- Chao, T.C.; Wang, W.S.; Yen, C.C.; Chiou, T.J.; Liu, J.H.; Chen, P.M. A dose-escalating pilot study of sterically stabilized, pegylated liposomal doxorubicin (Lipo-Dox) in patients with metastatic breast cancer. Cancer Investig. 2003, 21, 837–847. [Google Scholar] [CrossRef]
- Tampaki, E.C.; Tampakis, A.; Alifieris, C.E.; Krikelis, D.; Pazaiti, A.; Kontos, M.; Trafalis, D.T. Efficacy and safety of neoadjuvant treatment with bevacizumab, liposomal doxorubicin, cyclophosphamide and paclitaxel combination in locally/regionally advanced, HER2-negative, grade III at premenopausal status breast cancer: A phase II study. Clin. Drug Investig. 2018, 38, 639–648. [Google Scholar] [CrossRef] [PubMed]
- Veronese, P.; Hachul, D.T.; Scanavacca, M.I.; Hajjar, L.A.; Wu, T.C.; Sacilotto, L.; Veronese, C.; Darrieux, F. Effects of anthracycline, cyclophosphamide and taxane chemotherapy on QTc measurements in patients with breast cancer. PLoS ONE 2018, 13, e0196763. [Google Scholar] [CrossRef]
- Martin, M.; Villar, A.; Sole-Calvo, A.; Gonzalez, R.; Massuti, B.; Lizon, J.; Camps, C.; Carrato, A.; Casado, A.; Candel, M.T.; et al. Doxorubicin in combination with fluorouracil and cyclophosphamide (i.v. FAC regimen, day 1, 21) versus methotrexate in combination with fluorouracil and cyclophosphamide (i.v. CMF regimen, day 1, 21) as adjuvant chemotherapy for operable breast cancer: A study by the GEICAM group. Ann. Oncol. 2003, 14, 833–842. [Google Scholar]
- Liutkauskiene, S.; Grizas, S.; Jureniene, K.; Suipyte, J.; Statnickaite, A.; Juozaityte, E. Retrospective analysis of the impact of anthracycline dose reduction and chemotherapy delays on the outcomes of early breast cancer molecular subtypes. BMC Cancer 2018, 18, 453. [Google Scholar] [CrossRef]
- Hrynchak, I.; Sousa, E.; Pinto, M.; Costa, V.M. The importance of drug metabolites synthesis: The case-study of cardiotoxic anticancer drugs. Drug Metab. Rev. 2017, 49, 158–196. [Google Scholar] [CrossRef]
- Reis-Mendes, A.F.; Sousa, E.; de Lourdes Bastos, M.; Costa, V.M. The role of the metabolism of anticancer drugs in their induced-cardiotoxicity. Curr. Drug Metab. 2015, 17, 75–90. [Google Scholar] [CrossRef]
- Costa, V.M.; Carvalho, F.; Duarte, J.A.; Bastos, M.L.; Remiao, F. The heart as a target for xenobiotic toxicity: The cardiac susceptibility to oxidative stress. Chem. Res. Toxicol. 2013, 26, 1285–1311. [Google Scholar] [CrossRef]
- Sorrentino, M.F.; Kim, J.; Foderaro, A.E.; Truesdell, A.G. 5-Fluorouracil induced cardiotoxicity: Review of the literature. Cardiol. J. 2012, 19, 453–458. [Google Scholar] [CrossRef]
- Tahover, E.; Patil, Y.P.; Gabizon, A.A. Emerging delivery systems to reduce doxorubicin cardiotoxicity and improve therapeutic index: Focus on liposomes. Anticancer Drugs 2015, 26, 241–258. [Google Scholar] [CrossRef]
- Minotti, G.; Menna, P.; Salvatorelli, E.; Cairo, G.; Gianni, L. Anthracyclines: Molecular advances and pharmacologic developments in antitumor activity and cardiotoxicity. Pharmacol. Rev. 2004, 56, 185–229. [Google Scholar] [CrossRef]
- Wadia, S. Acute cyclophosphamide hemorrhagic myopericarditis: Dilemma case report, literature review and proposed diagnostic criteria. J. Clin. Diagn. Res. 2015, 9, OE01–OE3. [Google Scholar] [CrossRef]
- Dalley, D.N.; Levi, J.A.; Aroney, R.S. Combination chemotherapy with cyclophosphamide, adriamycin, and 5-fluorouracil (CAF) in advanced breast carcinoma. Med. J. Aust. 1980, 1, 216–218. [Google Scholar]
- Bontenbal, M.; Creemers, G.J.; Braun, H.J.; de Boer, A.C.; Janssen, J.T.; Leys, R.B.; Ruit, J.B.; Goey, S.H.; van der Velden, P.C.; Kerkhofs, L.G.; et al. Phase II to III study comparing doxorubicin and docetaxel with fluorouracil, doxorubicin, and cyclophosphamide as first-line chemotherapy in patients with metastatic breast cancer: Results of a Dutch community setting trial for the clinical trial group of the comprehensive cancer centre. J. Clin. Oncol. 2005, 23, 7081–7088. [Google Scholar]
- Martin, M.; Ruiz, A.; Ruiz Borrego, M.; Barnadas, A.; Gonzalez, S.; Calvo, L.; Margeli Vila, M.; Anton, A.; Rodriguez-Lescure, A.; Segui-Palmer, M.A.; et al. Fluorouracil, doxorubicin, and cyclophosphamide (FAC) versus FAC followed by weekly paclitaxel as adjuvant therapy for high-risk, node-negative breast cancer: Results from the GEICAM/2003-02 study. J. Clin. Oncol. 2013, 31, 2593–2599. [Google Scholar] [CrossRef]
- Kaithwas, G.; Dubey, K.; Pillai, K.K. Effect of aloe vera (Aloe barbadensis Miller) gel on doxorubicin-induced myocardial oxidative stress and calcium overload in albino rats. Indian J. Exp. Biol. 2011, 49, 260–268. [Google Scholar]
- Amin, K.A.; Mohamed, B.M.; El-Wakil, M.A.; Ibrahem, S.O. Impact of breast cancer and combination chemotherapy on oxidative stress, hepatic and cardiac markers. J. Breast Cancer 2012, 15, 306–312. [Google Scholar] [CrossRef]
- Koti, B.C.; Vishwanathswamy, A.H.; Wagawade, J.; Thippeswamy, A.H. Cardioprotective effect of lipistat against doxorubicin induced myocardial toxicity in albino rats. Indian J. Exp. Biol. 2009, 47, 41–46. [Google Scholar]
- Kolaric, K.; Bradamante, V.; Cervek, J.; Cieslinska, A.; Cisarz-Filipcak, E.; Denisov, L.E.; Donat, D.; Drosik, K.; Gershanovic, M.; Hudziec, P.; et al. A phase II trial of cardioprotection with cardioxane (ICRF-187) in patients with advanced breast cancer receiving 5-fluorouracil, doxorubicin and cyclophosphamide. Oncology 1995, 52, 251–255. [Google Scholar] [CrossRef]
- Buzdar, A.U.; Kau, S.W.; Smith, T.L.; Hortobagyi, G.N. Ten-year results of FAC adjuvant chemotherapy trial in breast cancer. Am. J. Clin. Oncol. 1989, 12, 123–128. [Google Scholar] [CrossRef]
- Bustova, I. Risk of cardiotoxicity of combination treatment radiotherapy and chemotherapy of locally advanced breast carcinoma stage III. Klin. Onkol. 2009, 22, 17–21. [Google Scholar]
- Mackey, J.R.; Martin, M.; Pienkowski, T.; Rolski, J.; Guastalla, J.P.; Sami, A.; Glaspy, J.; Juhos, E.; Wardley, A.; Fornander, T.; et al. Adjuvant docetaxel, doxorubicin, and cyclophosphamide in node-positive breast cancer: 10-Year follow-up of the phase 3 randomised BCIRG 001 trial. Lancet Oncol. 2013, 14, 72–80. [Google Scholar] [CrossRef]
- Lamberti, M.; Porto, S.; Marra, M.; Zappavigna, S.; Grimaldi, A.; Feola, D.; Pesce, D.; Naviglio, S.; Spina, A.; Sannolo, N.; et al. 5-Fluorouracil induces apoptosis in rat cardiocytes through intracellular oxidative stress. J. Exp. Clin. Cancer Res. 2012, 31, 60. [Google Scholar] [CrossRef]
- Durak, I.; Karaayvaz, M.; Kavutcu, M.; Cimen, M.Y.; Kacmaz, M.; Buyukkocak, S.; Ozturk, H.S. Reduced antioxidant defense capacity in myocardial tissue from guinea pigs treated with 5-fluorouracil. J. Toxicol. Environ. Health A 2000, 59, 585–589. [Google Scholar]
- Logan, R.M.; Stringer, A.M.; Bowen, J.M.; Gibson, R.J.; Sonis, S.T.; Keefe, D.M. Serum levels of NFκB and pro-inflammatory cytokines following administration of mucotoxic drugs. Cancer Biol. Ther. 2008, 7, 1139–1145. [Google Scholar] [CrossRef]
- Reers, S.; Pfannerstill, A.C.; Rades, D.; Maushagen, R.; Andratschke, M.; Pries, R.; Wollenberg, B. Cytokine changes in response to radio-/chemotherapeutic treatment in head and neck cancer. Anticancer Res. 2013, 33, 2481–2489. [Google Scholar]
- Raghu Nadhanan, R.; Abimosleh, S.M.; Su, Y.W.; Scherer, M.A.; Howarth, G.S.; Xian, C.J. Dietary emu oil supplementation suppresses 5-fluorouracil chemotherapy-induced inflammation, osteoclast formation, and bone loss. Am. J. Physiol. Endocrinol. Metab. 2012, 302, E1440–E1449. [Google Scholar] [CrossRef]
- Malet-Martino, M.; Martino, R. Clinical studies of three oral prodrugs of 5-fluorouracil (capecitabine, UFT, S-1): A review. Oncologist 2002, 7, 288–323. [Google Scholar] [CrossRef]
- Miura, K.; Kinouchi, M.; Ishida, K.; Fujibuchi, W.; Naitoh, T.; Ogawa, H.; Ando, T.; Yazaki, N.; Watanabe, K.; Haneda, S.; et al. 5-FU metabolism in cancer and orally-administrable 5-FU drugs. Cancers 2010, 2, 1717–1730. [Google Scholar] [CrossRef]
- Nies, A.T.; Magdy, T.; Schwab, M.; Zanger, U.M. Role of ABC transporters in fluoropyrimidine-based chemotherapy response. Adv. Cancer Res. 2015, 125, 217–243. [Google Scholar]
- Jamieson, D.; Lee, J.; Cresti, N.; Jackson, R.; Griffin, M.; Sludden, J.; Verrill, M.; Boddy, A.V. Pharmacogenetics of adjuvant breast cancer treatment with cyclophosphamide, epirubicin and 5-fluorouracil. Cancer Chemother. Pharmacol. 2014, 74, 667–674. [Google Scholar] [CrossRef]
- Muneoka, K.; Shirai, Y.; Yokoyama, N.; Wakai, T.; Hatakeyama, K. 5-Fluorouracil cardiotoxicity induced by α-fluoro-β-alanine. Int. J. Clin. Oncol. 2005, 10, 441–443. [Google Scholar] [CrossRef] [PubMed]
- Hull, W.E.; Port, R.E.; Herrmann, R.; Britsch, B.; Kunz, W. Metabolites of 5-fluorouracil in plasma and urine, as monitored by 19F nuclear magnetic resonance spectroscopy, for patients receiving chemotherapy with or without methotrexate pretreatment. Cancer Res. 1988, 48, 1680–1688. [Google Scholar]
- Yamada, Y.; Hamaguchi, T.; Goto, M.; Muro, K.; Matsumura, Y.; Shimada, Y.; Shirao, K.; Nagayama, S. Plasma concentrations of 5-fluorouracil and F-β-alanine following oral administration of S-1, a dihydropyrimidine dehydrogenase inhibitory fluoropyrimidine, as compared with protracted venous infusion of 5-fluorouracil. Br. J. Cancer 2003, 89, 816–820. [Google Scholar] [CrossRef]
- Joerger, M.; Huitema, A.D.; Meenhorst, P.L.; Schellens, J.H.; Beijnen, J.H. Pharmacokinetics of low-dose doxorubicin and metabolites in patients with AIDS-related Kaposi sarcoma. Cancer Chemother. Pharmacol. 2005, 55, 488–496. [Google Scholar] [CrossRef]
- Joerger, M.; Huitema, A.D.; Richel, D.J.; Dittrich, C.; Pavlidis, N.; Briasoulis, E.; Vermorken, J.B.; Strocchi, E.; Martoni, A.; Sorio, R.; et al. Population pharmacokinetics and pharmacodynamics of doxorubicin and cyclophosphamide in breast cancer patients: A study by the EORTC-PAMM-NDDG. Clin. Pharmacokinet. 2007, 46, 1051–1068. [Google Scholar] [CrossRef]
- Ren, S.; Kalhorn, T.F.; Slattery, J.T. Inhibition of human aldehyde dehydrogenase 1 by the 4-hydroxycyclophosphamide degradation product acrolein. Drug Metab. Dispos. 1999, 27, 133–137. [Google Scholar]
- Takanashi, S.; Bachur, N.R. Adriamycin metabolism in man. Evidence from urinary metabolites. Drug Metab. Dispos. 1976, 4, 79–87. [Google Scholar]
- Stewart, D.J.; Grewaal, D.; Green, R.M.; Mikhael, N.; Goel, R.; Montpetit, V.A.; Redmond, M.D. Concentrations of doxorubicin and its metabolites in human autopsy heart and other tissues. Anticancer Res. 1993, 13, 1945–1952. [Google Scholar] [PubMed]
- Murayama, Y.; Nagashima, M. [Systemic chemotherapy of mammary carcinoma: Plasma and tissue concentrations of 5-fluorouracil and adriamycin, and result of CAF and CMF therapy of breast cancer. Gan To Kagaku Ryoho 1984, 11, 415–419, (Abstract). [Google Scholar] [PubMed]
- Danesi, R.; Fogli, S.; Gennari, A.; Conte, P.; Del Tacca, M. Pharmacokinetic-pharmacodynamic relationships of the anthracycline anticancer drugs. Clin. Pharmacokinet. 2002, 41, 431–444. [Google Scholar] [CrossRef] [PubMed]
- Blum, R.H.; Carter, S.K. Adriamycin. A new anticancer drug with significant clinical activity. Ann. Intern. Med. 1974, 80, 249–259. [Google Scholar] [CrossRef]
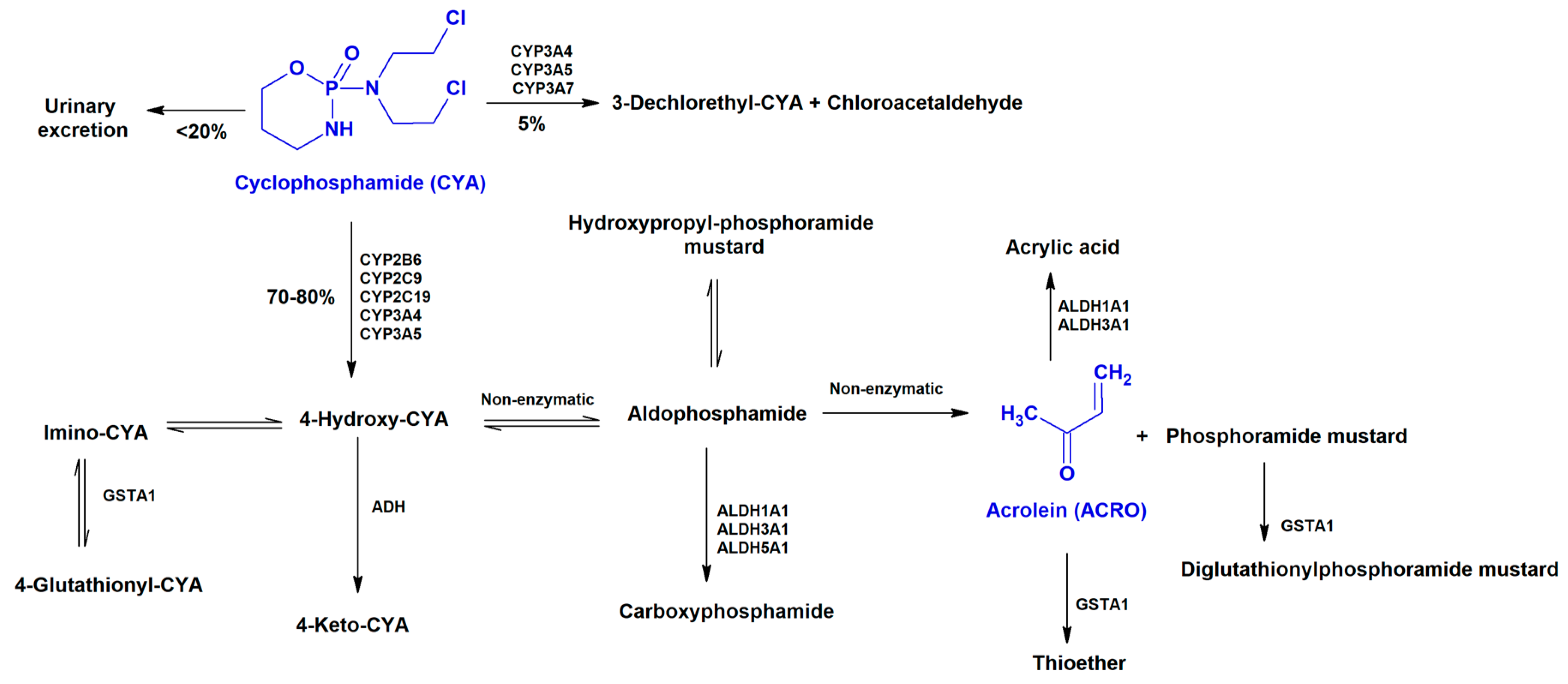
- Sladek, N.E. Metabolism of oxazaphosphorines. Pharmacol. Ther. 1988, 37, 301–355. [Google Scholar] [CrossRef]
- de Jonge, M.E.; Huitema, A.D.; Rodenhuis, S.; Beijnen, J.H. Clinical pharmacokinetics of cyclophosphamide. Clin. Pharmacokinet. 2005, 44, 1135–1164. [Google Scholar] [CrossRef] [PubMed]
- Afsar, N.A.; Ufer, M.; Haenisch, S.; Remmler, C.; Mateen, A.; Usman, A.; Ahmed, K.Z.; Ahmad, H.R.; Cascorbi, I. Relationship of drug metabolizing enzyme genotype to plasma levels as well as myelotoxicity of cyclophosphamide in breast cancer patients. Eur. J. Clin. Pharmacol. 2012, 68, 389–395. [Google Scholar] [CrossRef] [PubMed]
- Dorr, R.T.; Lagel, K. Effect of sulfhydryl compounds and glutathione depletion on rat heart myocyte toxicity induced by 4-hydroperoxycyclophosphamide and acrolein in vitro. Chem. Biol. Interact. 1994, 93, 117–128. [Google Scholar] [CrossRef]
- Kurauchi, K.; Nishikawa, T.; Miyahara, E.; Okamoto, Y.; Kawano, Y. Role of metabolites of cyclophosphamide in cardiotoxicity. BMC Res. Notes 2017, 10, 406. [Google Scholar] [CrossRef] [PubMed]
- Boyd, V.L.; Robbins, J.D.; Egan, W.; Ludeman, S.M. 31P nuclear magnetic resonance spectroscopic observation of the intracellular transformations of oncostatic cyclophosphamide metabolites. J. Med. Chem. 1986, 29, 1206–1210. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Sun, Y.; Asahi, M.; Otsu, K. Acrolein, an environmental toxin, induces cardiomyocyte apoptosis via elevated intracellular calcium and free radicals. Cell Biochem. Biophys. 2011, 61, 131–136. [Google Scholar] [CrossRef]
- Ismahil, M.A.; Hamid, T.; Haberzettl, P.; Gu, Y.; Chandrasekar, B.; Srivastava, S.; Bhatnagar, A.; Prabhu, S.D. Chronic oral exposure to the aldehyde pollutant acrolein induces dilated cardiomyopathy. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H2050–H2060. [Google Scholar] [CrossRef] [PubMed]
- McDonald, G.B.; Slattery, J.T.; Bouvier, M.E.; Ren, S.; Batchelder, A.L.; Kalhorn, T.F.; Schoch, H.G.; Anasetti, C.; Gooley, T. Cyclophosphamide metabolism, liver toxicity, and mortality following hematopoietic stem cell transplantation. Blood 2003, 101, 2043–2048. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Wang, H. Oxazaphosphorine bioactivation and detoxification the role of xenobiotic receptors. Acta Pharm. Sin. B 2012, 2, 107–117. [Google Scholar] [CrossRef] [PubMed]
- Pereira-Oliveira, M.; Reis-Mendes, A.; Carvalho, F.; Remiao, F.; Pinto, M.; Bastos, M.L.; Costa, V.M. Doxorubicin is key for the cardiotoxicity of fac (5-fluorouracil + adriamycin + cyclophosphamide) combination in H9c2 differentiated cells. Biomolecules 2019, 9, 21. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, T.; Miyahara, E.; Kurauchi, K.; Watanabe, E.; Ikawa, K.; Asaba, K.; Tanabe, T.; Okamoto, Y.; Kawano, Y. Mechanisms of fatal cardiotoxicity following high-dose cyclophosphamide therapy and a method for its prevention. PLoS ONE 2015, 10, e0131394. [Google Scholar] [CrossRef] [PubMed]
- Kimes, B.W.; Brandt, B.L. Properties of a clonal muscle cell line from rat heart. Exp. Cell Res. 1976, 98, 367–381. [Google Scholar] [CrossRef]
- Ruiz, M.; Courilleau, D.; Jullian, J.C.; Fortin, D.; Ventura-Clapier, R.; Blondeau, J.P.; Garnier, A. A cardiac-specific robotized cellular assay identified families of human ligands as inducers of PGC-1α expression and mitochondrial biogenesis. PLoS ONE 2012, 7, e46753. [Google Scholar] [CrossRef]
- Reis-Mendes, A.; Gomes, A.S.; Carvalho, R.A.; Carvalho, F.; Remiao, F.; Pinto, M.; Bastos, M.L.; Sousa, E.; Costa, V.M. Naphthoquinoxaline metabolite of mitoxantrone is less cardiotoxic than the parent compound and it can be a more cardiosafe drug in anticancer therapy. Arch. Toxicol. 2017, 91, 1871–1890. [Google Scholar] [CrossRef] [PubMed]
- Menard, C.; Pupier, S.; Mornet, D.; Kitzmann, M.; Nargeot, J.; Lory, P. Modulation of l-type calcium channel expression during retinoic acid-induced differentiation of H9c2 cardiac cells. J. Biol. Chem. 1999, 274, 29063–29070. [Google Scholar] [CrossRef]
- Pereira, S.L.; Ramalho-Santos, J.; Branco, A.F.; Sardao, V.A.; Oliveira, P.J.; Carvalho, R.A. Metabolic remodeling during H9c2 myoblast differentiation: Relevance for in vitro toxicity studies. Cardiovasc. Toxicol. 2011, 11, 180–190. [Google Scholar] [CrossRef]
- Branco, A.F.; Sampaio, S.F.; Moreira, A.C.; Holy, J.; Wallace, K.B.; Baldeiras, I.; Oliveira, P.J.; Sardao, V.A. Differentiation-dependent doxorubicin toxicity on H9c2 cardiomyoblasts. Cardiovasc. Toxicol. 2012, 12, 326–340. [Google Scholar] [CrossRef]
- Sardao, V.A.; Oliveira, P.J.; Holy, J.; Oliveira, C.R.; Wallace, K.B. Doxorubicin-induced mitochondrial dysfunction is secondary to nuclear p53 activation in H9c2 cardiomyoblasts. Cancer Chemother. Pharmacol. 2009, 64, 811–827. [Google Scholar] [CrossRef]
- Sardao, V.A.; Oliveira, P.J.; Holy, J.; Oliveira, C.R.; Wallace, K.B. Morphological alterations induced by doxorubicin on H9c2 myoblasts: Nuclear, mitochondrial, and cytoskeletal targets. Cell Biol. Toxicol. 2009, 25, 227–243. [Google Scholar] [CrossRef]
- Branco, A.F.; Pereira, S.P.; Gonzalez, S.; Gusev, O.; Rizvanov, A.A.; Oliveira, P.J. Gene expression profiling of H9c2 myoblast differentiation towards a cardiac-like phenotype. PLoS ONE 2015, 10, e0129303. [Google Scholar] [CrossRef]
- Repetto, G.; del Peso, A.; Zurita, J.L. Neutral red uptake assay for the estimation of cell viability/cytotoxicity. Nat. Protoc. 2008, 3, 1125–1131. [Google Scholar] [CrossRef]
- Almeida, D.; Pinho, R.; Correia, V.; Soares, J.; Bastos, M.L.; Carvalho, F.; Capela, J.P.; Costa, V.M. Mitoxantrone is more toxic than doxorubicin in SH-SY5Y human cells: A ‘chemobrain’ in vitro study. Pharmaceuticals 2018, 11, 41. [Google Scholar] [CrossRef]
- Reers, M.; Smith, T.W.; Chen, L.B. J-aggregate formation of a carbocyanine as a quantitative fluorescent indicator of membrane potential. Biochemistry 1991, 30, 4480–4486. [Google Scholar] [CrossRef]
- Cossarizza, A.; Baccarani-Contri, M.; Kalashnikova, G.; Franceschi, C. A new method for the cytofluorimetric analysis of mitochondrial membrane potential using the J-aggregate forming lipophilic cation 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolcarbocyanine iodide (JC-1). Biochem. Biophys. Res. Commun. 1993, 197, 40–45. [Google Scholar] [CrossRef]
- Franck, C.; Malfertheiner, P.; Venerito, M. Safe administration of S-1 after 5-fluorouracil-induced cardiotoxicity in a patient with colorectal cancer. BMJ Case Rep. 2017, 2017, bcr-2016-219162. [Google Scholar] [CrossRef]
- Matsubara, I.; Kamiya, J.; Imai, S. Cardiotoxic effects of 5-fluorouracil in the guinea pig. Jpn. J. Pharmacol. 1980, 30, 871–879. [Google Scholar] [CrossRef]
- Fischel, J.L.; Formento, P.; Ciccolini, J.; Etienne-Grimaldi, M.C.; Milano, G. Lack of contribution of dihydrofluorouracil and α-fluoro-β-alanine to the cytotoxicity of 5′-deoxy-5-fluorouridine on human keratinocytes. Anticancer Drugs 2004, 15, 969–974. [Google Scholar] [CrossRef]
- Bains, O.S.; Szeitz, A.; Lubieniecka, J.M.; Cragg, G.E.; Grigliatti, T.A.; Riggs, K.W.; Reid, R.E. A correlation between cytotoxicity and reductase-mediated metabolism in cell lines treated with doxorubicin and daunorubicin. J. Pharmacol. Exp. Ther. 2013, 347, 375–387. [Google Scholar] [CrossRef]
- Licata, S.; Saponiero, A.; Mordente, A.; Minotti, G. Doxorubicin metabolism and toxicity in human myocardium: Role of cytoplasmic deglycosidation and carbonyl reduction. Chem. Res. Toxicol. 2000, 13, 414–420. [Google Scholar] [CrossRef]
- Minotti, G.; Cavaliere, A.F.; Mordente, A.; Rossi, M.; Schiavello, R.; Zamparelli, R.; Possati, G. Secondary alcohol metabolites mediate iron delocalization in cytosolic fractions of myocardial biopsies exposed to anticancer anthracyclines. Novel linkage between anthracycline metabolism and iron-induced cardiotoxicity. J. Clin. Investig. 1995, 95, 1595–1605. [Google Scholar] [CrossRef]
- Minotti, G.; Recalcati, S.; Mordente, A.; Liberi, G.; Calafiore, A.M.; Mancuso, C.; Preziosi, P.; Cairo, G. The secondary alcohol metabolite of doxorubicin irreversibly inactivates aconitase/iron regulatory protein-1 in cytosolic fractions from human myocardium. FASEB J. 1998, 12, 541–552. [Google Scholar] [CrossRef]
- Olson, L.E.; Bedja, D.; Alvey, S.J.; Cardounel, A.J.; Gabrielson, K.L.; Reeves, R.H. Protection from doxorubicin-induced cardiac toxicity in mice with a null allele of carbonyl reductase 1. Cancer Res. 2003, 63, 6602–6606. [Google Scholar]
- Forrest, G.L.; Gonzalez, B.; Tseng, W.; Li, X.; Mann, J. Human carbonyl reductase overexpression in the heart advances the development of doxorubicin-induced cardiotoxicity in transgenic mice. Cancer Res. 2000, 60, 5158–5164. [Google Scholar]
- Minotti, G.; Cairo, G.; Monti, E. Role of iron in anthracycline cardiotoxicity: New tunes for an old song? FASEB J. 1999, 13, 199–212. [Google Scholar] [CrossRef]
- Xie, Y.; Hou, W.; Song, X.; Yu, Y.; Huang, J.; Sun, X.; Kang, R.; Tang, D. Ferroptosis: Process and function. Cell Death Differ. 2016, 23, 369–379. [Google Scholar] [CrossRef]
- Zhou, F.; Hao, G.; Zhang, J.; Zheng, Y.; Wu, X.; Hao, K.; Niu, F.; Luo, D.; Sun, Y.; Wu, L.; et al. Protective effect of 23-hydroxybetulinic acid on doxorubicin-induced cardiotoxicity: A correlation with the inhibition of carbonyl reductase-mediated metabolism. Br. J. Pharmacol. 2015, 172, 5690–5703. [Google Scholar] [CrossRef]
- Sun, Y.; Ito, S.; Nishio, N.; Tanaka, Y.; Chen, N.; Isobe, K. Acrolein induced both pulmonary inflammation and the death of lung epithelial cells. Toxicol. Lett. 2014, 229, 384–392. [Google Scholar] [CrossRef]
- Horton, N.D.; Biswal, S.S.; Corrigan, L.L.; Bratta, J.; Kehrer, J.P. Acrolein causes inhibitor κB-independent decreases in nuclear factor κB activation in human lung adenocarcinoma (A549) cells. J. Biol. Chem. 1999, 274, 9200–9206. [Google Scholar] [CrossRef]
- Biswal, S.; Acquaah-Mensah, G.; Datta, K.; Wu, X.; Kehrer, J.P. Inhibition of cell proliferation and AP-1 activity by acrolein in human A549 lung adenocarcinoma cells due to thiol imbalance and covalent modifications. Chem. Res. Toxicol. 2002, 15, 180–186. [Google Scholar] [CrossRef]
- LoPachin, R.M.; Gavin, T.; Petersen, D.R.; Barber, D.S. Molecular mechanisms of 4-hydroxy-2-nonenal and acrolein toxicity: Nucleophilic targets and adduct formation. Chem. Res. Toxicol. 2009, 22, 1499–1508. [Google Scholar] [CrossRef]
- Damiani, R.M.; Moura, D.J.; Viau, C.M.; Caceres, R.A.; Henriques, J.A.; Saffi, J. Pathways of cardiac toxicity: Comparison between chemotherapeutic drugs doxorubicin and mitoxantrone. Arch. Toxicol. 2016, 90, 2063–2076. [Google Scholar] [CrossRef]
- Hansen, J.M.; Go, Y.M.; Jones, D.P. Nuclear and mitochondrial compartmentation of oxidative stress and redox signaling. Annu. Rev. Pharmacol. Toxicol. 2006, 46, 215–234. [Google Scholar] [CrossRef]
- Gervasi, P.G.; Agrillo, M.R.; Citti, L.; Danesi, R.; Del Tacca, M. Superoxide anion production by adriamycinol from cardiac sarcosomes and by mitochondrial nadh dehydrogenase. Anticancer Res. 1986, 6, 1231–1235. [Google Scholar]
- Olson, R.D.; Mushlin, P.S.; Brenner, D.E.; Fleischer, S.; Cusack, B.J.; Chang, B.K.; Boucek, R.J., Jr. Doxorubicin cardiotoxicity may be caused by its metabolite, doxorubicinol. Proc. Natl. Acad. Sci. USA 1988, 85, 3585–3589. [Google Scholar] [CrossRef]
- Boucek, R.J., Jr.; Olson, R.D.; Brenner, D.E.; Ogunbunmi, E.M.; Inui, M.; Fleischer, S. The major metabolite of doxorubicin is a potent inhibitor of membrane-associated ion pumps. A correlative study of cardiac muscle with isolated membrane fractions. J. Biol. Chem. 1987, 262, 15851–15856. [Google Scholar]
- Zhang, D.; Ma, J. Mitochondrial dynamics in rat heart induced by 5-fluorouracil. Med. Sci. Monit. 2018, 24, 6666–6672. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | Patients and Dose | Plasma Concentrations | Reference |
|---|---|---|---|
| FBAL |
| 3.29–18.26 μM | [34] |
| 19–170 μM | [35] | |
| 8.01 ± 0.12 μM–10.81 ± 3.27 μM | [36] | |
| DOXOL |
| 0.056 ± 0.005 μM | [37] |
| 0.1 μM | [38] | |
| ACRO |
| 6.2–10.2 μM | [39] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reis-Mendes, A.; Carvalho, F.; Remião, F.; Sousa, E.; Bastos, M.d.L.; Costa, V.M. The Main Metabolites of Fluorouracil + Adriamycin + Cyclophosphamide (FAC) Are Not Major Contributors to FAC Toxicity in H9c2 Cardiac Differentiated Cells. Biomolecules 2019, 9, 98. https://doi.org/10.3390/biom9030098
Reis-Mendes A, Carvalho F, Remião F, Sousa E, Bastos MdL, Costa VM. The Main Metabolites of Fluorouracil + Adriamycin + Cyclophosphamide (FAC) Are Not Major Contributors to FAC Toxicity in H9c2 Cardiac Differentiated Cells. Biomolecules. 2019; 9(3):98. https://doi.org/10.3390/biom9030098
Chicago/Turabian StyleReis-Mendes, Ana, Félix Carvalho, Fernando Remião, Emília Sousa, Maria de Lourdes Bastos, and Vera Marisa Costa. 2019. "The Main Metabolites of Fluorouracil + Adriamycin + Cyclophosphamide (FAC) Are Not Major Contributors to FAC Toxicity in H9c2 Cardiac Differentiated Cells" Biomolecules 9, no. 3: 98. https://doi.org/10.3390/biom9030098
APA StyleReis-Mendes, A., Carvalho, F., Remião, F., Sousa, E., Bastos, M. d. L., & Costa, V. M. (2019). The Main Metabolites of Fluorouracil + Adriamycin + Cyclophosphamide (FAC) Are Not Major Contributors to FAC Toxicity in H9c2 Cardiac Differentiated Cells. Biomolecules, 9(3), 98. https://doi.org/10.3390/biom9030098