The PTEN–PI3K Axis in Cancer


{kind=link}
{kind=link}
Abstract
:1. The PI3K Pathway: Molecular Hubs and Biological Functions
2. The Tumors Suppressor PTEN
2.1. Mutations, Lipid Function and In Vivo Studies
2.2. PIP3 is More than AKT
3. The Proto-Oncogene PI3K and PTEN
3.1. PIK3CA Mutations and Functions Vis a Vis PTEN Regulation
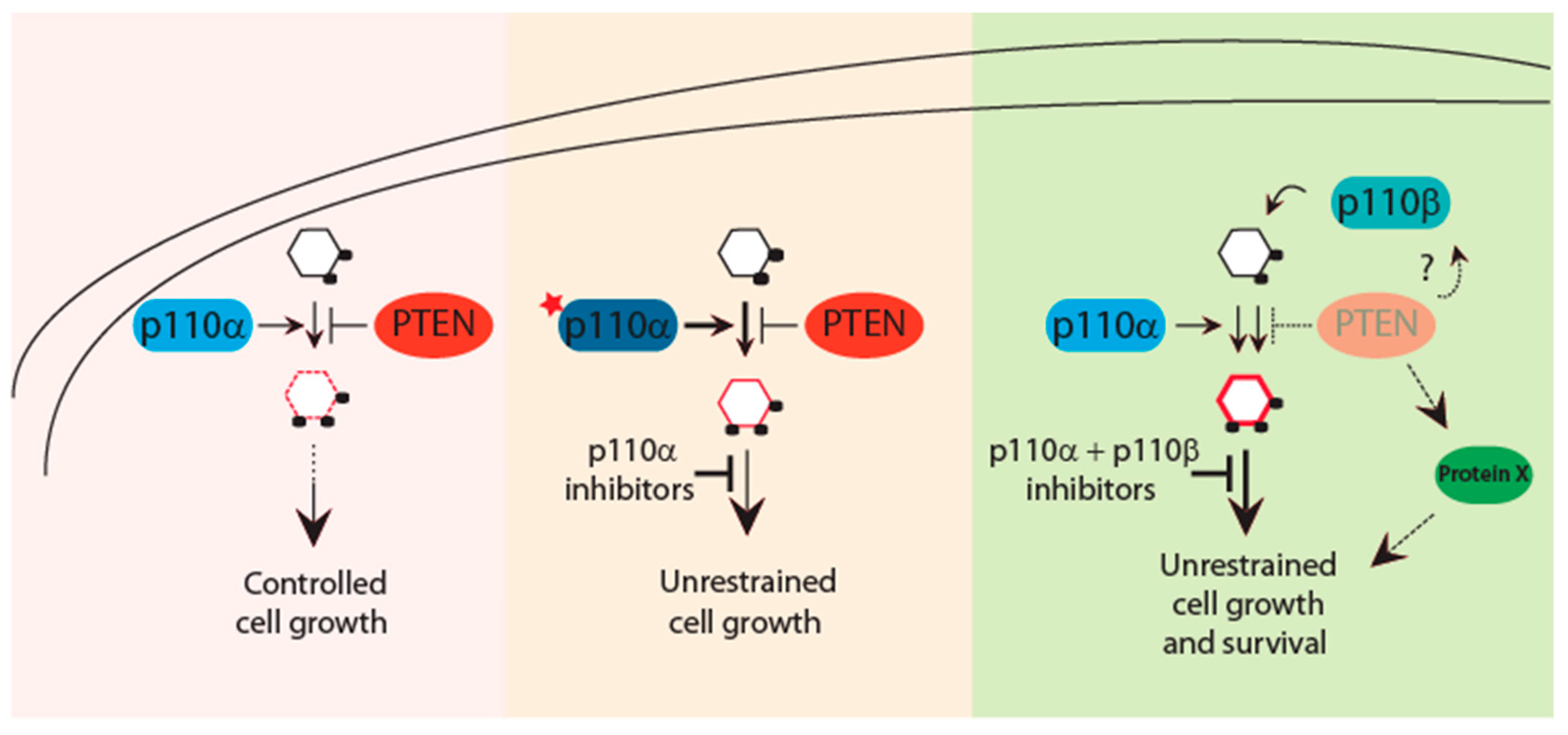
3.2. p110 Isoforms, Targeted Therapies and PTEN
4. PTEN Beyond PI3K
4.1. PIP3-Independent PTEN Functions
4.2. PTEN Lipid and Protein Phosphatase Activity Side-by-Side
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Vanhaesebroeck, B.; Guillermet-Guibert, J.; Graupera, M.; Bilanges, B. The emerging mechanisms of isoform-specific PI3K signalling. Nat. Rev. Mol. Cell Boil. 2010, 11, 329–341. [Google Scholar] [CrossRef]
- Samuels, Y. High Frequency of Mutations of the PIK3CA Gene in Human Cancers. Science 2004, 304, 554. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, M.S.; Stojanov, P.; Mermel, C.H.; Robinson, J.T.; Garraway, L.A.; Golub, T.R.; Meyerson, M.; Gabriel, S.B.; Lander, E.S.; Getz, G. Discovery and saturation analysis of cancer genes across 21 tumor types. Nature 2014, 505, 495–501. [Google Scholar] [CrossRef] [PubMed]
- Fruman, D.A.; Chiu, H.; Bagrodia, S.; Cantley, L.C., Abraham; Abraham, R.T. The PI3K Pathway in Human Disease. Cell 2017, 170, 605–635. [Google Scholar] [CrossRef]
- Manning, B.D.; Toker, A. AKT/PKB Signaling: Navigating the Network. Cell 2017, 169, 381–405. [Google Scholar] [CrossRef]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef]
- Liu, P.; Gan, W.; Chin, Y.R.; Ogura, K.; Guo, J.; Zhang, J.; Wang, B.; Blenis, J.; Cantley, L.C.; Toker, A.; et al. PtdIns(3,4,5)P3-dependent Activation of the mTORC2 Kinase Complex. Cancer Discov. 2015, 5, 1194–1209. [Google Scholar] [CrossRef]
- Menon, S.; Dibble, C.C.; Talbott, G.; Hoxhaj, G.; Valvezan, A.J.; Takahashi, H.; Cantley, L.C.; Manning, B.D. Spatial Control of the TSC Complex Integrates Insulin and Nutrient Regulation of mTORC1 at the Lysosome. Cell 2014, 156, 771–785. [Google Scholar] [CrossRef]
- Lee, Y.R.; Chen, M.; Pandolfi, P.P. The functions and regulation of the PTEN tumor suppressor: New modes and prospects. Nat. Rev. Mol. Cell Biol. 2018, 19, 547–562. [Google Scholar] [CrossRef]
- Hollander, M.C.; Blumenthal, G.M.; Dennis, P.A. PTEN loss in the continuum of common cancers, rare syndromes and mouse models. Nat. Rev. 2011, 11, 289–301. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, A.; De La Pompa, J.L.; Stambolic, V.; Elia, A.J.; Sasaki, T.; Barrantes, I.D.B.; Ho, A.; Wakeham, A.; Ltie, A.; Khoo, W.; et al. High cancer susceptibility and embryonic lethality associated with mutation of the PTEN tumor suppressor gene in mice. Curr. Boil. 1998, 8, 1169–1178. [Google Scholar] [CrossRef]
- Podsypanina, K.; Ellenson, L.H.; Nemes, A.; Gu, J.; Tamura, M.; Yamada, K.M.; Cordon-Cardo, C.; Cattoretti, G.; Fisher, P.E.; Parsons, R. Mutation of Pten/Mmac1 in mice causes neoplasia in multiple organ systems. PNAS 1999, 96, 1563–1568. [Google Scholar] [CrossRef]
- Di Cristofano, A.; Pesce, B.; Cordon-Cardo, C.; Pandolfi, P.P. Pten is essential for embryonic development and tumor suppression. Nat. Genet. 1998, 19, 348–355. [Google Scholar] [CrossRef]
- Bonneau, D.; Longy, M. Mutations of the human PTEN gene. Hum. Mutat. 2000, 16, 109–122. [Google Scholar] [CrossRef]
- Yehia, L.; Ngeow, J.; Eng, C. PTEN-opathies: From biological insights to evidence-based precision medicine. J. Clin. Investig. 2019, 129, 452–464. [Google Scholar] [CrossRef]
- Maehama, T.; Dixon, J.E. The Tumor Suppressor, PTEN/MMAC1, Dephosphorylates the Lipid Second Messenger, Phosphatidylinositol 3,4,5-Trisphosphate. J. Boil. Chem. 1998, 273, 13375–13378. [Google Scholar] [CrossRef]
- Li, J. PTEN, a Putative Protein Tyrosine Phosphatase Gene Mutated in Human Brain, Breast, and Prostate Cancer. Science 1997, 275, 1943–1947. [Google Scholar] [CrossRef] [PubMed]
- Steck, P.A.; Pershouse, M.A.; Jasser, S.A.; Yung, W.K.; Lin, H.; Ligon, A.H.; Langford, L.A.; Baumgard, M.L.; Hattier, T.; Davis, T.; et al. Identification of a candidate tumor suppressor gene, MMAC1, at chromosome 10q23.3 that is mutated in multiple advanced cancers. Nat. Genet. 1997, 15, 356–362. [Google Scholar]
- Stambolic, V.; Suzuki, A.; De La Pompa, J.L.; Brothers, G.M.; Mirtsos, C.; Sasaki, T.; Ruland, J.; Penninger, J.M.; Siderovski, D.P.; Mak, T.W. Negative Regulation of PKB/Akt-Dependent Cell Survival by the Tumor Suppressor PTEN. Cell 1998, 95, 29–39. [Google Scholar] [CrossRef]
- Papa, A.; Wan, L.; Bonora, M.; Salmena, L.; Song, M.S.; Hobbs, R.M.; Lunardi, A.; Webster, K.; Ng, C.; Newton, R.H.; et al. Cancer-associated PTEN mutants act in a dominant negative manner to suppress PTEN protein function. Cell 2014, 157, 595–610. [Google Scholar] [CrossRef]
- Wang, H.; Karikomi, M.; Naidu, S.; Rajmohan, R.; Caserta, E.; Chen, H.-Z.; Rawahneh, M.; Moffitt, J.; Stephens, J.A.; Fernandez, S.A.; et al. Allele-specific tumor spectrum in Pten knockin mice. PNAS 2010, 107, 5142–5147. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Robinson, G.W.; Lesche, R.; Martinez-Diaz, H.; Jiang, Z.; Rozengurt, N.; Wagner, K.-U.; Wu, D.-C.; Lane, T.F.; Liu, X.; et al. Conditional loss of PTEN leads to precocious development and neoplasia in the mammary gland. Development 2002, 129, 4159–4170. [Google Scholar]
- Chen, Z.; Trotman, L.C.; Shaffer, D.; Lin, H.-K.; Dotan, Z.A.; Niki, M.; Koutcher, J.A.; Scher, H.I.; Ludwig, T.; Gerald, W.; et al. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature 2005, 436, 725–730. [Google Scholar] [CrossRef] [PubMed]
- Alimonti, A.; Carracedo, A.; Clohessy, J.G.; Trotman, L.C.; Nardella, C.; Egia, A.; Salmena, L.; Sampieri, K.; Haveman, W.J.; Brogi, E.; et al. Subtle variations in Pten dose determine cancer susceptibility. Nat. Genet. 2010, 42, 454–458. [Google Scholar] [CrossRef]
- Newton, R.H.; Lu, Y.; Papa, A.; Whitcher, G.H.; Kang, Y.J.; Yan, C.; Pandolfi, P.P.; Turka, L.A. Suppression of T-cell lymphomagenesis in mice requires PTEN phosphatase activity. Blood 2015, 125, 852–855. [Google Scholar] [CrossRef]
- Garcia-Cao, I.; Song, M.S.; Hobbs, R.M.; Laurent, G.; Giorgi, C.; De Boer, V.C.; Anastasiou, D.; Ito, K.; Sasaki, A.T.; Rameh, L.; et al. Systemic Elevation of PTEN Induces a Tumor-Suppressive Metabolic State. Cell 2012, 149, 49–62. [Google Scholar] [CrossRef]
- Chin, Y.R.; Yuan, X.; Balk, S.P.; Toker, A. PTEN-deficient tumors depend on AKT2 for maintenance and survival. Cancer Discov. 2014, 4, 942–955. [Google Scholar] [CrossRef]
- Chen, M.L.; Xu, P.Z.; Peng, X.D.; Chen, W.S.; Guzman, G.; Yang, X.; Di Cristofano, A.; Pandolfi, P.P.; Hay, N. The deficiency of Akt1 is sufficient to suppress tumor development in Pten+/- mice. Genes Dev. 2006, 20, 1569–1574. [Google Scholar] [CrossRef]
- Sanidas, I.; Polytarchou, C.; Hatziapostolou, M.; Ezell, S.A.; Kottakis, F.; Hu, L.; Guo, A.; Xie, J.; Comb, M.J.; Iliopoulos, D.; et al. Phosphoproteomics screen reveals Akt isoform-specific signals linking RNA processing to lung cancer. Mol. Cell 2014, 53, 577–590. [Google Scholar] [CrossRef]
- Gonzalez, E.; McGraw, T.E. The Akt kinases: Isoform specificity in metabolism and cancer. Cell Cycle 2009, 8, 2502–2508. [Google Scholar] [CrossRef]
- Costa, C.; Ebi, H.; Martini, M.; Beausoleil, S.A.; Faber, A.C.; Jakubik, C.T.; Huang, A.; Wang, Y.; Nishtala, M.; Hall, B. Measurement of PIP3 levels reveals an unexpected role for p110β in early adaptive responses to p110α-specific inhibitors in luminal breast cancer. Cancer Cell 2015, 27, 97–108. [Google Scholar] [CrossRef]
- Vasudevan, K.M.; Barbie, D.A.; Davies, M.A.; Rabinovsky, R.; McNear, C.J.; Kim, J.J.; Hennessy, B.T.; Tseng, H.; Pochanard, P.; Kim, S.Y.; et al. AKT-independent signaling downstream of oncogenic PIK3CA mutations in human cancer. Cancer Cell 2009, 16, 21–32. [Google Scholar] [CrossRef]
- Ebi, H.; Costa, C.; Faber, A.C.; Nishtala, M.; Kotani, H.; Juric, D.; Della Pelle, P.; Song, Y.; Yano, S.; Mino-Kenudson, M.; et al. PI3K regulates MEK/ERK signaling in breast cancer via the Rac-GEF, P-Rex1. PNAS 2013, 110, 21124–21129. [Google Scholar] [CrossRef]
- Hu, H.; Juvekar, A.; Lyssiotis, C.A.; Lien, E.C.; Albeck, J.G.; Oh, D.; Varma, G.; Hung, Y.P.; Ullas, S.; Lauring, J.; et al. Phosphoinositide 3-Kinase Regulates Glycolysis through Mobilization of Aldolase from the Actin cytoskeleton. Cell 2016, 164, 433–446. [Google Scholar] [CrossRef]
- Zhao, L.; Vogt, P.K. Class I PI3K in oncogenic cellular transformation. Oncogene 2008, 27, 5486–5496. [Google Scholar] [CrossRef]
- Huang, C.H.; Mandelker, D.; Schmidt-Kittler, O.; Samuels, Y.; Velculescu, V.E.; Kinzler, K.W.; Vogelstein, B.; Gabelli, S.B.; Amzel, L.M. The structure of a human p110α/p85α complex elucidates the effects of oncogenic PI3Kα mutations. Science 2007, 318, 1744–8174. [Google Scholar] [CrossRef]
- Miled, N.; Yan, Y.; Hon, W.-C.; Perisic, O.; Zvelebil, M.; Inbar, Y.; Schneidman-Duhovny, D.; Wolfson, H.J.; Backer, J.M.; Williams, R.L. Mechanism of Two Classes of Cancer Mutations in the Phosphoinositide 3-Kinase Catalytic Subunit. Science 2007, 317, 239–242. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.H.; Mandelker, D.; Gabelli, S.B.; Amzel, L.M. Insights into the oncogenic effects of PIK3CA mutations from the structure of p110α/p85α. Cell Cycle 2008, 7, 1151–1156. [Google Scholar] [CrossRef] [PubMed]
- Van Keymeulen, A.; Lee, M.Y.; Ousset, M.; Brohée, S.; Rorive, S.; Giraddi, R.R.; Wuidart, A.; Bouvencourt, G.; Dubois, C.; Salmon, I.; et al. Reactivation of multipotency by oncogenic PIK3CA induces breast tumor heterogeneity. Nature 2015, 525, 119–123. [Google Scholar] [CrossRef]
- Koren, S.; Reavie, L.; Couto, J.P.; De Silva, D.; Stadler, M.B.; Roloff, T.; Britschgi, A.; Eichlisberger, T.; Kohler, H.; Aina, O.; et al. PIK3CA(H1047R) induces multipotency and multi-lineage mammary tumors. Nature 2015, 525, 114–118. [Google Scholar] [CrossRef] [PubMed]
- Koren, S.; Bentires-Alj, M. Mouse models of PIK3CA mutations: One mutation initiates heterogeneous mammary tumors. FEBS J. 2013, 280, 2758–2765. [Google Scholar] [CrossRef]
- Chagpar, R.B.; Links, P.H.; Pastor, M.C.; Furber, L.A.; Hawrysh, A.D.; Chamberlain, M.D.; Anderson, D.H.; Chamberlain, M. Direct positive regulation of PTEN by the p85 subunit of phosphatidylinositol 3-kinase. PNAS 2010, 107, 5471–5476. [Google Scholar] [CrossRef]
- Rabinovsky, R.; Pochanard, P.; McNear, C.; Brachmann, S.M.; Duke-Cohan, J.S.; Garraway, L.A.; Sellers, W.R. p85 Associates with Unphosphorylated PTEN and the PTEN-Associated Complex. Mol. Cell. Boil. 2009, 29, 5377–5388. [Google Scholar] [CrossRef] [PubMed]
- Cheung, L.W.T.; Walkiewicz, K.W.; Besong, T.M.D.; Guo, H.; Hawke, D.H.; Arold, S.T.; Mills, G.B. Regulation of the PI3K pathway through a p85α monomer-homodimer equilibrium. Elife 2015, 4, e06866. [Google Scholar] [CrossRef]
- Heinrich, F.; Chakravarthy, S.; Nanda, H.; Papa, A.; Pandolfi, P.P.; Ross, A.H.; Harishchandra, R.K.; Gericke, A.; Lösche, M. The PTEN tumor suppressor forms homodimers in solution. Structure 2015, 23, 1952–1957. [Google Scholar] [CrossRef] [PubMed]
- Isakoff, S.J.; Engelman, J.A.; Irie, H.Y.; Luo, J.; Brachmann, S.M.; Pearline, R.V.; Cantley, L.C.; Brugge, J.S. Breast cancer-associated PIK3CA mutations are oncogenic in mammary epithelial cells. Cancer Res. 2005, 65, 10992–11000. [Google Scholar] [CrossRef] [PubMed]
- Tikoo, A.; Roh, V.; Montgomery, K.G.; Ivetac, I.; Waring, P.; Pelzer, R.; Hare, L.; Shackleton, M.; Humbert, P.; Phillips, W.A. Physiological levels of Pik3ca(H1047R) mutation in the mouse mammary gland results in ductal hyperplasia and formation of ERα-positive tumors. PLoS ONE 2012, 7, e36924. [Google Scholar] [CrossRef] [PubMed]
- Rodon, J.; Dienstmann, R.; Serra, V.; Tabernero, J. Development of PI3K inhibitors: Lessons learned from early clinical trials. Nat. Rev. Clin. Oncol. 2013, 10, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Toska, E.; Baselga, J. Pharmacology in the Era of Targeted Therapies: The Case of PI3K Inhibitors. Clin. Res. 2016, 22, 2099–2101. [Google Scholar] [CrossRef] [PubMed]
- Pons-Tostivint, E.; Thibault, B.; Guillermet-Guibert, J. Targeting PI3K Signaling in Combination Cancer Therapy. Trends Cancer 2017, 3, 454–469. [Google Scholar] [CrossRef] [PubMed]
- Weigelt, B.; Downward, J. Genomic Determinants of PI3K Pathway Inhibitor Response in Cancer. Front. Oncol. 2012, 2, 109. [Google Scholar] [CrossRef]
- Gonzalez-Angulo, A.M.; Blumenschein, G.R., Jr. Defining biomarkers to predict sensitivity to PI3K/Akt/mTOR pathway inhibitors in breast cancer. Cancer Treat. Rev. 2013, 39, 313–320. [Google Scholar] [CrossRef]
- Fruman, D.A.; Rommel, C. PI3K and Cancer: Lessons, Challenges and Opportunities. Nat. Rev. Drug Discov. 2014, 13, 140–156. [Google Scholar] [CrossRef]
- Juric, D.; Castel, P.; Griffith, M.; Griffith, O.L.; Won, H.H.; Ellis, H.; Ebbesen, S.H.; Ainscough, B.J.; Ramu, A.; Iyer, G.; et al. Convergent loss of PTEN leads to clinical resistance to a PI(3)Kα inhibitor. Nature 2015, 18, 240–244. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Chen, S.; Asara, J.M.; Balk, S.P. Phosphoinositide 3-kinase pathway activation in phosphate and tensin homolog (PTEN)-deficient prostate cancer cells is independent of receptor tyrosine kinases and mediated by the p110β and p110δ catalytic subunits. J. Biol. Chem. 2010, 285, 14980–14989. [Google Scholar] [CrossRef] [PubMed]
- Wee, S.; Wiederschain, D.; Maira, S.-M.; Loo, A.; Miller, C.; Debeaumont, R.; Stegmeier, F.; Yao, Y.-M.; Lengauer, C. PTEN-deficient cancers depend on PIK3CB. PNAS 2008, 105, 13057–13062. [Google Scholar] [CrossRef]
- Wang, Q.; Liu, P.; Spangle, J.M.; Von, T.; Roberts, T.M.; Lin, N.U.; Krop, I.E.; Winer, E.P.; Zhao, J.J. PI3K-p110α mediates resistance to HER2-targeted therapy in HER2+, PTEN-deficient breast cancers. Oncogene 2016, 35, 3607–3612. [Google Scholar] [CrossRef]
- Schwartz, S.; Wongvipat, J.; Trigwell, C.B.; Hancox, U.; Carver, B.S.; Rodrik-Outmezguine, V.; Will, M.; Yellen, P.; de Stanchina, E.; Baselga, J.; et al. Feedback suppression of PI3Kα signaling in PTEN-mutated tumors is relieved by selective inhibition of PI3Kβ. Cancer Cell 2015, 27, 109–122. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Tenorio, G.; Alkhori, L.; Olsson, B.; Waltersson, M.A.; Nordenskjöld, B.; Rutqvist, L.E.; Skoog, L.; Stål, O. PIK3CA Mutations and PTEN Loss Correlate with Similar Prognostic Factors and Are Not Mutually Exclusive in Breast Cancer. Clin. Cancer Res. 2007, 13, 3577–3584. [Google Scholar]
- Stemke-Hale, K.; Gonzalez-Angulo, A.M.; Lluch, A.; Neve, R.M.; Kuo, W.-L.; Davies, M.; Carey, M.; Hu, Z.; Guan, Y.; Sahin, A.; et al. An Integrative Genomic and Proteomic Analysis of PIK3CA, PTEN, and AKT Mutations in Breast Cancer. Cancer Res. 2008, 68, 6084–6091. [Google Scholar] [CrossRef]
- Pearson, H.B.; Li, J.; Méniel, V.S.; Fennell, C.M.; Waring, P.; Montgomery, K.G.; Rebello, R.J.; MacPherson, A.A.; Koushyar, S.; Furic, L.; et al. Identification of Pik3ca Mutation as a Genetic Driver of Prostate Cancer That Cooperates with Pten Loss to Accelerate Progression and Castration-Resistant Growth. Cancer Discov. 2018, 8, 764–779. [Google Scholar] [CrossRef]
- Papa, A.; Pandolfi, P.P. Phosphatase-Independent Functions of the Tumor Suppressor PTEN. In Protein Tyrosine Phosphatases in Cancer; Neel, B.G., Tonks, N.K., Eds.; Springer: New York, NY, USA, 2016; pp. 247–260. [Google Scholar]
- Tanaka, H.; Yoshida, M.; Tanimura, H.; Fujii, T.; Sakata, K.; Tachibana, Y.; Ohwada, J.; Ebiike, H.; Kuramoto, S.; Morita, K.; et al. The Selective Class I PI3K Inhibitor CH5132799 Targets Human Cancers Harboring Oncogenic PIK3CA Mutations. Clin. Res. 2011, 17, 3272–3281. [Google Scholar] [CrossRef]
- Brachmann, S.M.; Hofmann, I.; Schnell, C.; Fritsch, C.; Wee, S.; Lane, H.; Wang, S.; Garcia-Echeverria, C.; Maira, S.-M. Specific apoptosis induction by the dual PI3K/mTor inhibitor NVP-BEZ235 in HER2 amplified and PIK3CA mutant breast cancer cells. PNAS 2009, 106, 22299–22304. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Zhang, J.; Sampieri, K.; Clohessy, J.G.; Mendez, L.; González-Billalabeitia, E.; Liu, X.-S.; Lee, Y.-R.; Fung, J.; Katon, J.M.; et al. An aberrant SREBP-dependent lipogenic program promotes metastatic prostate cancer. Nat. Genet. 2018, 50, 206–218. [Google Scholar] [CrossRef] [PubMed]
- Myers, M.P.; Stolarov, J.P.; Eng, C.; Li, J.; Wang, S.I.; Wigler, M.H.; Parsons, R.; Tonks, N.K. P-TEN, the tumor suppressor from human chromosome 10q23, is a dual-specificity phosphatase. PNAS 1997, 94, 9052–9057. [Google Scholar] [CrossRef]
- Zhang, S.; Huang, W.-C.; Li, P.; Guo, H.; Poh, S.-B.; Brady, S.W.; Xiong, Y.; Tseng, L.-M.; Li, S.-H.; Ding, Z.; et al. Combating trastuzumab resistance by targeting SRC, a common node downstream of multiple resistance pathways. Nat. Med. 2011, 17, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Davidson, L.; Maccario, H.; Perera, N.M.; Yang, X.; Spinelli, L.; Tibarewal, P.; Glancy, B.; Gray, A.; Weijer, C.J.; Downes, C.P.; et al. Suppression of cellular proliferation and invasion by the concerted lipid and protein phosphatase activities of PTEN. Oncogene 2010, 29, 687–797. [Google Scholar] [CrossRef] [PubMed]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Papa, A.; Pandolfi, P.P. The PTEN–PI3K Axis in Cancer. Biomolecules 2019, 9, 153. https://doi.org/10.3390/biom9040153
Papa A, Pandolfi PP. The PTEN–PI3K Axis in Cancer. Biomolecules. 2019; 9(4):153. https://doi.org/10.3390/biom9040153
Chicago/Turabian StylePapa, Antonella, and Pier Paolo Pandolfi. 2019. "The PTEN–PI3K Axis in Cancer" Biomolecules 9, no. 4: 153. https://doi.org/10.3390/biom9040153
APA StylePapa, A., & Pandolfi, P. P. (2019). The PTEN–PI3K Axis in Cancer. Biomolecules, 9(4), 153. https://doi.org/10.3390/biom9040153