The α-Tubulin gene TUBA1A in Brain Development: A Key Ingredient in the Neuronal Isotype Blend

Abstract
:1. Introduction
2. Overview of Microtubules and Tubulin Isotypes
2.1. Microtubule Basics
2.2. Tubulin Isotypes
2.3. Tubulin Post-Translational Modifications
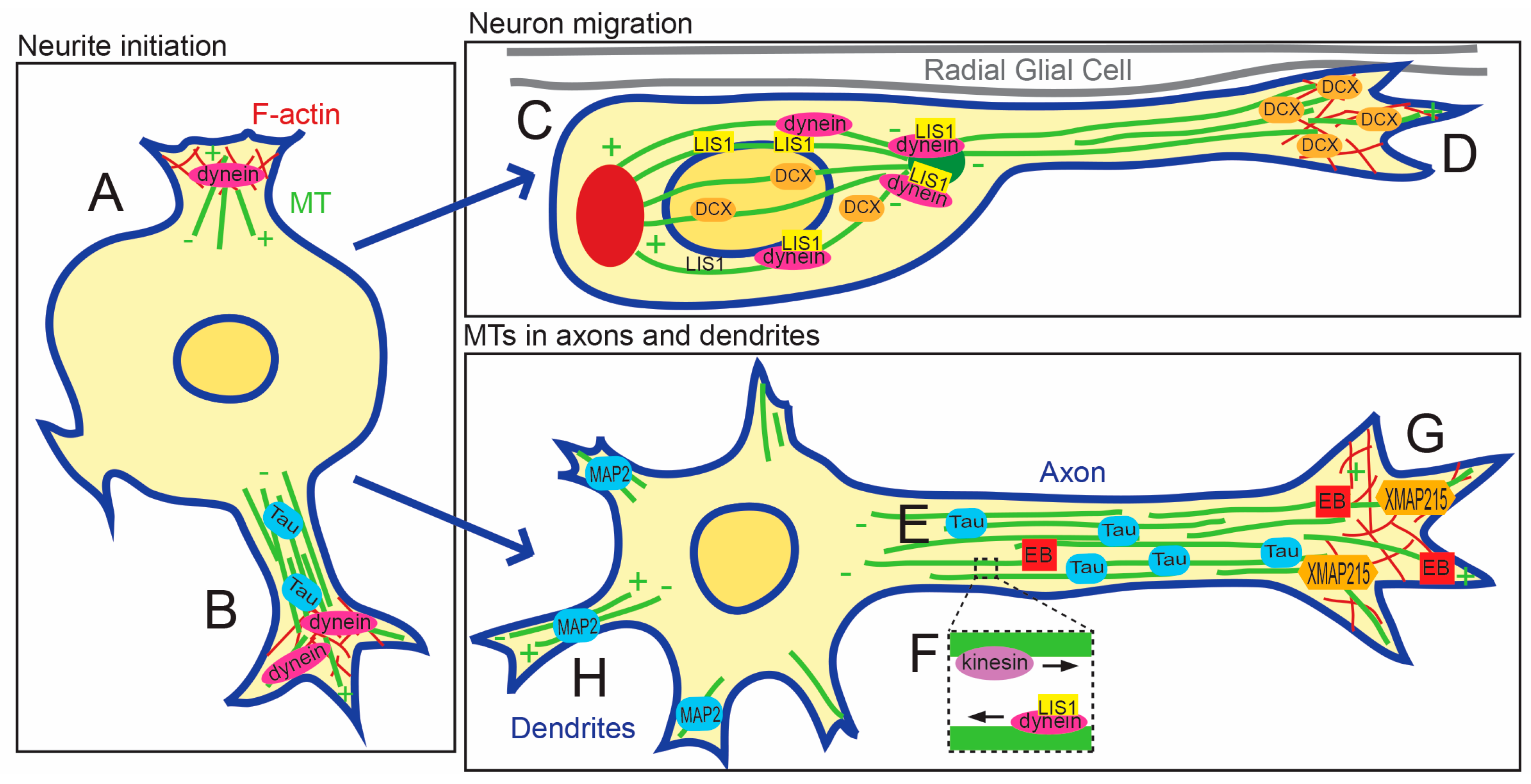
3. Roles of Microtubules during Neuronal Development and Adulthood
4. Tubulinopathies Reveal Essential Role of TUBA1A in Brain Formation and Function
4.1. TUBA1A Mutations Linked to Lissencephaly
4.2. TUBA1A Mutations Linked to Polymicrogyria
4.3. TUBA1A Mutations Linked to Microcephaly
4.4. TUBA1A Mutations Linked to Cerebellar Dysplasia
4.5. Summary of TUBA1A Mutations Associated with Brain Malformations
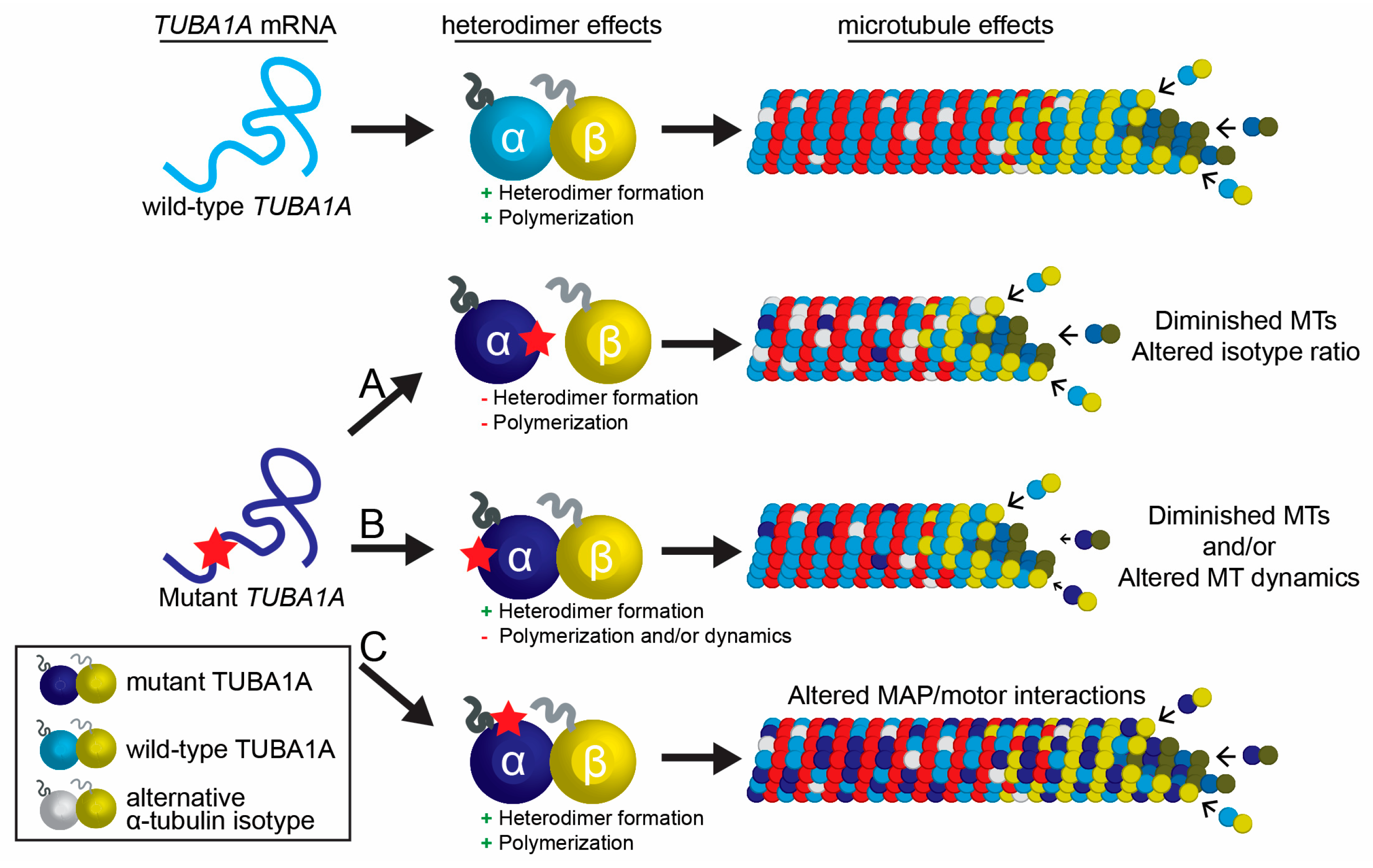
4.6. Cellular Impact of TUBA1A Mutations
5. TUBA1A Expression: A Burst of Tubulin to Fuel Morphogenesis?
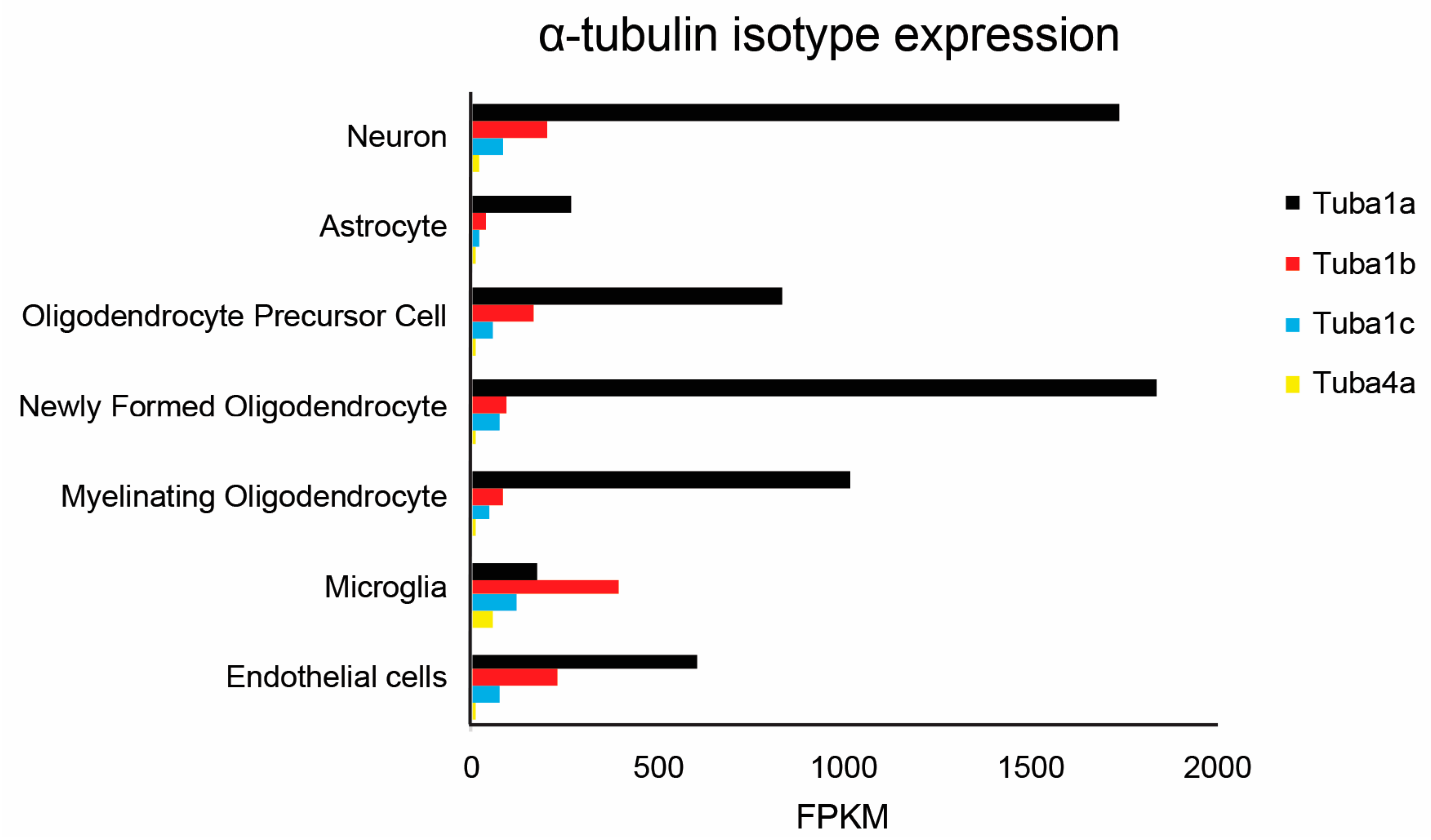
5.1. TUBA1A Expression Pattern
5.2. Mechanisms Regulating TUBA1A Expression
5.3. TUBA1A in Regeneration
6. Concluding Remarks
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Dent, E.W.; Kalil, K. Axon branching requires interactions between dynamic microtubules and actin filaments. J. Neurosci. 2001, 21, 9757–9769. [Google Scholar] [PubMed]
- Hu, J.; Bai, X.; Bowen, J.R.; Dolat, L.; Korobova, F.; Yu, W.; Baas, P.W.; Svitkina, T.; Gallo, G.; Spiliotis, E.T. Septin-driven coordination of actin and microtubule remodeling regulates the collateral branching of axons. Curr. Biol. 2012, 22, 1109–1115. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Liu, M.; Mozgova, O.I.; Yu, W.Q.; Baas, P.W. Mitotic Motors Coregulate Microtubule Patterns in Axons and Dendrites. J. Neurosci. 2012, 32, 14033–14049. [Google Scholar] [CrossRef] [PubMed]
- Fanara, P.; Husted, K.H.; Selle, K.; Wong, P.Y.A.; Banerjee, J.; Brandt, R.; Hellerstein, M.K. Changes in microtubule turnover accompany synaptic plasticity and memory formation in response to contextual fear conditioning in mice. Neuroscience 2010, 168, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Hoogenraad, C.C.; Bradke, F. Control of neuronal polarity and plasticity—A renaissance for microtubules? Trends Cell Biol. 2009, 19, 669–676. [Google Scholar] [CrossRef] [PubMed]
- Jaworski, J.; Kapitein, L.C.; Gouveia, S.M.; Dortland, B.R.; Wulf, P.S.; Grigoriev, I.; Camera, P.; Spangler, S.A.; Di Stefano, P.; Demmers, J.; et al. Dynamic Microtubules Regulate Dendritic Spine Morphology and Synaptic Plasticity. Neuron 2009, 61, 85–100. [Google Scholar] [CrossRef] [PubMed]
- Khodiyar, V.K.; Maltais, L.J.; Ruef, B.J.; Sneddon, K.M.B.; Smith, J.R.; Shimoyama, M.; Cabral, F.; Dumontet, C.; Dutcher, S.K.; Harvey, R.J.; et al. A revised nomenclature for the human and rodent alpha-tubulin gene family. Genomics 2007, 90, 285–289. [Google Scholar] [CrossRef] [PubMed]
- Findeisen, P.; Mühlhausen, S.; Dempewolf, S.; Hertzog, J.; Zietlow, A.; Carlomagno, T.; Kollmar, M. Six subgroups and extensive recent duplications characterize the evolution of the eukaryotic tubulin protein family. Genome Biol. Evol. 2014, 6, 2274–2288. [Google Scholar] [CrossRef] [PubMed]
- Hall, J.L.; Cowan, N.J. Structural features and restricted expression of a human alpha-tubulin gene. Nucleic Acids Res. 1985, 13, 207–223. [Google Scholar] [CrossRef] [PubMed]
- Lewis, S.A.; Lee, M.G.S.; Cowan, N.J. Five mouse tubulin isotypes and their regulated expression during development. J. Cell Biol. 1985, 101, 852–861. [Google Scholar] [CrossRef] [PubMed]
- Keays, D.A.; Tian, G.; Poirier, K.; Huang, G.-J.; Siebold, C.; Cleak, J.; Oliver, P.L.; Fray, M.; Harvey, R.J.; Molnár, Z.; et al. Mutations in alpha-tubulin cause abnormal neuronal migration in mice and lissencephaly in humans. Cell 2007, 128, 45–57. [Google Scholar] [CrossRef] [PubMed]
- Poirier, K.; Keays, D.A.; Francis, F.; Saillour, Y.; Bahi, N.; Manouvrier, S.; Fallet-Bianco, C.; Pasquier, L.; Toutain, A.; Tuy, F.P.D.; et al. Large spectrum of lissencephaly and pachygyria phenotypes resulting from de novo missense mutations in tubulin alpha 1A (TUBA1A). Hum. Mutat. 2007, 28, 1055–1064. [Google Scholar] [CrossRef] [PubMed]
- Bahi-Buisson, N.; Poirier, K.; Boddaert, N.; Saillour, Y.; Castelnau, L.; Philip, N.; Buyse, G.; Villard, L.; Joriot, S.; Marret, S.; et al. Refinement of cortical dysgeneses spectrum associated with TUBA1A mutations. J. Med. Genet. 2008, 45, 647–653. [Google Scholar] [CrossRef] [PubMed]
- Fallet-Bianco, C.; Loeuillet, L.; Poirier, K.; Loget, P.; Chapon, F.; Pasquier, L.; Saillour, Y.; Beldjord, C.; Chelly, J.; Francis, F. Neuropathological phenotype of a distinct form of lissencephaly associated with mutations in TUBA1A. Brain 2008, 131, 2304–2320. [Google Scholar] [CrossRef] [PubMed]
- Morris-Rosendahl, D.J.; Najm, J.; Lachmeijer, A.M.A.; Sztriha, L.; Martins, M.; Kuechler, A.; Haug, V.; Zeschnigk, C.; Martin, P.; Santos, M.; et al. Refining the phenotype of alpha-1a Tubulin (TUBA1A) mutation in patients with classical lissencephaly. Clin. Genet. 2008, 74, 425–433. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.A.; Pilz, D.T.; Babatz, T.D.; Cushion, T.D.; Harvey, K.; Topf, M.; Yates, L.; Robb, S.; Uyanik, G.; Mancini, G.M.S.; et al. TUBA1A mutations cause wide spectrum lissencephaly (smooth brain) and suggest that multiple neuronal migration pathways converge on alpha tubulins. Hum. Mol. Genet. 2010, 19, 2817–2827. [Google Scholar] [CrossRef] [PubMed]
- Lecourtois, M.; Poirier, K.; Friocourt, G.; Jaglin, X.; Goldenberg, A.; Saugier-Veber, P.; Chelly, J.; Laquerrière, A. Human lissencephaly with cerebellar hypoplasia due to mutations in TUBA1A: Expansion of the foetal neuropathological phenotype. Acta Neuropathol. 2010, 119, 779–789. [Google Scholar] [CrossRef] [PubMed]
- Poirier, K.; Saillour, Y.; Fourniol, F.; Francis, F.; Souville, I.; Valence, S.; Desguerre, I.; Marie Lepage, J.; Boddaert, N.; Jacquemont, M.L.; et al. Expanding the spectrum of TUBA1A-related cortical dysgenesis to Polymicrogyria. Eur. J. Hum. Genet. 2013, 21, 381–385. [Google Scholar] [CrossRef] [PubMed]
- Cushion, T.D.; Dobyns, W.B.; Mullins, J.G.L.; Stoodley, N.; Chung, S.K.; Fry, A.E.; Hehr, U.; Gunny, R.; Aylsworth, A.S.; Prabhakar, P.; et al. Overlapping cortical malformations and mutations in TUBB2B and TUBA1A. Brain 2013, 136, 536–548. [Google Scholar] [CrossRef] [PubMed]
- Bahi-Buisson, N.; Poirier, K.; Fourniol, F.; Saillour, Y.; Valence, S.; Lebrun, N.; Hully, M.; Bianco, C.F.; Boddaert, N.; Elie, C.; et al. The wide spectrum of tubulinopathies: What are the key features for the diagnosis? Brain 2014, 137, 1676–1770. [Google Scholar] [CrossRef] [PubMed]
- Fallet-Bianco, C.; Laquerrière, A.; Poirier, K.; Razavi, F.; Guimiot, F.; Dias, P.; Loeuillet, L.; Lascelles, K.; Beldjord, C.; Carion, N.; et al. Mutations in tubulin genes are frequent causes of various foetal malformations of cortical development including microlissencephaly. Acta Neuropathol. Commun. 2014, 2, 69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kraemer, J.A.; Erb, M.L.; Waddling, C.A.; Montabana, E.A.; Zehr, E.A.; Wang, H.; Nguyen, K.; Pham, D.S.L.; Agard, D.A.; Pogliano, J. A phage tubulin assembles dynamic filaments by an atypical mechanism to center viral DNA within the host cell. Cell 2012, 149, 1488–1499. [Google Scholar] [CrossRef] [PubMed]
- Amos, L.A.; van den Ent, F.; Löwe, J. Structural/functional homology between the bacterial and eukaryotic cytoskeletons. Curr. Opin. Cell Biol. 2004, 16, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Nogales, E.; Wolf, S.G.; Downing, K.H. Structure of the alpha beta tubulin dimer by electron crystallography. Nature 1998, 391, 199–203. [Google Scholar] [CrossRef] [PubMed]
- Spiegelman, B.M.; Penningroth, S.M.; Kirschner, M.W. Turnover of tubulin and the N site GTP in Chinese hamster ovary cells. Cell 1977, 12, 587–600. [Google Scholar] [CrossRef]
- Carlier, M.F.; Pantaloni, D. Kinetic analysis of guanosine 5′-triphosphate hydrolysis associated with tubulin polymerization. Biochemistry 1981, 20, 1918–1924. [Google Scholar] [CrossRef] [PubMed]
- Alushin, G.M.; Lander, G.C.; Kellogg, E.H.; Zhang, R.; Baker, D.; Nogales, E. High-resolution microtubule structures reveal the structural transitions in αβ-tubulin upon GTP hydrolysis. Cell 2014, 157, 1117–1129. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Alushin, G.M.; Brown, A.; Nogales, E. Mechanistic Origin of Microtubule Dynamic Instability and Its Modulation by EB Proteins. Cell 2015, 162, 849–859. [Google Scholar] [CrossRef] [PubMed]
- Geyer, E.A.; Burns, A.; Lalonde, B.A.; Ye, X.; Piedra, F.-A.; Huffaker, T.C.; Rice, L.M. A mutation uncouples the tubulin conformational and GTPase cycles, revealing allosteric control of microtubule dynamics. eLife 2015, 4, e10113. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.-W.; Nogales, E. Nucleotide-dependent bending flexibility of tubulin regulates microtubule assembly. Nature 2005, 435, 911–915. [Google Scholar] [CrossRef] [PubMed]
- Rice, L.M.; Montabana, E.A.; Agard, D.A. The lattice as allosteric effector: Structural studies of alphabeta- and gamma-tubulin clarify the role of GTP in microtubule assembly. Proc. Natl. Acad. Sci. USA 2008, 105, 5378–5383. [Google Scholar] [CrossRef] [PubMed]
- Nogales, E.; Whittaker, M.; Milligan, R.A.; Downing, K.H. High-resolution model of the microtubule. Cell 1999, 96, 79–88. [Google Scholar] [CrossRef]
- Sept, D.; Baker, N.A.; McCammon, J.A. The physical basis of microtubule structure and stability. Protein Sci. 2003, 12, 2257–2261. [Google Scholar] [CrossRef] [PubMed]
- Kononova, O.; Kholodov, Y.; Theisen, K.E.; Marx, K.A.; Dima, R.I.; Ataullakhanov, F.I.; Grishchuk, E.L.; Barsegov, V. 52 Tubulin bond energies and microtubule biomechanics determined from nanoindentation in silico. J. Biomol. Struct. Dyn. 2015, 33 (Suppl. 1), 35–36. [Google Scholar] [CrossRef] [PubMed]
- Mandelkow, E.M.; Mandelkow, E.; Milligan, R.A. Microtubule dynamics and microtubule caps: A time-resolved cryo-electron microscopy study. J. Cell Biol. 1991, 114, 977–991. [Google Scholar] [CrossRef] [PubMed]
- Hirokawa, N.; Takemura, R. Molecular motors and mechanisms of directional transport in neurons. Nat. Rev. Neurosci. 2005, 6, 201–214. [Google Scholar] [CrossRef] [PubMed]
- Conde, C.; Cáceres, A. Microtubule assembly, organization and dynamics in axons and dendrites. Nat. Rev. Neurosci. 2009, 10, 319–332. [Google Scholar] [CrossRef] [PubMed]
- Moon, H.M.; Wynshaw-Boris, A. Cytoskeleton in action: Lissencephaly, a neuronal migration disorder. Wiley Interdiscip. Rev. Dev. Biol. 2014, 2, 229–245. [Google Scholar] [CrossRef] [PubMed]
- Dehmelt, L.; Halpain, S. Actin and Microtubules in Neurite Initiation: Are MAPs the Missing Link? J. Neurobiol. 2004, 58, 18–33. [Google Scholar] [CrossRef] [PubMed]
- Hancock, W.O. The Kinesin-1 Chemomechanical Cycle: Stepping Toward a Consensus. Biophys. J. 2016, 110, 1216–1225. [Google Scholar] [CrossRef] [PubMed]
- Hirokawa, N.; Tanaka, Y. Kinesin superfamily proteins (KIFs): Various functions and their relevance for important phenomena in life and diseases. Exp. Cell Res. 2015, 334, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Tsai, J.-W.; Lian, W.-N.; Kemal, S.; Kriegstein, A.R.; Vallee, R.B. Kinesin 3 and cytoplasmic dynein mediate interkinetic nuclear migration in neural stem cells. Nat. Neurosci. 2010, 13, 1463–1471. [Google Scholar] [CrossRef] [PubMed]
- Homma, N.; Takei, Y.; Tanaka, Y.; Nakata, T.; Terada, S.; Kikkawa, M.; Noda, Y.; Hirokawa, N. Kinesin superfamily protein 2A (KIF2A) functions in suppression of collateral branch extension. Cell 2003, 114, 229–239. [Google Scholar] [CrossRef]
- Schmidt, H.; Carter, A.P. Review: Structure and mechanism of the dynein motor ATPase. Biopolymers 2016, 105, 557–567. [Google Scholar] [CrossRef] [PubMed]
- Schlager, M.A.; Hoang, H.T.; Urnavicius, L.; Bullock, S.L.; Carter, A.P. In vitro reconstitution of a highly processive recombinant human dynein complex. EMBO J. 2014, 33, 1855–1868. [Google Scholar] [CrossRef] [PubMed]
- McKenney, R.J.; Huynh, W.; Tanenbaum, M.E.; Bhabha, G.; Vale, R.D. Activation of cytoplasmic dynein motility by dynactin-cargo adapter complexes. Science 2014, 345, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Ayloo, S.; Lazarus, J.E.; Dodda, A.; Tokito, M.; Ostap, E.M.; Holzbaur, E.L.F. Dynactin functions as both a dynamic tether and brake during dynein-driven motility. Nat. Commun. 2014, 5, 4807. [Google Scholar] [CrossRef] [PubMed]
- McKenney, R.J.; Vershinin, M.; Kunwar, A.; Vallee, R.B.; Gross, S.P. LIS1 and NudE induce a persistent dynein force-producing state. Cell 2010, 141, 304–314. [Google Scholar] [CrossRef] [PubMed]
- Toropova, K.; Zou, S.; Roberts, A.J.; Redwine, W.B.; Goodman, B.S.; Reck-Peterson, S.L.; Leschziner, A.E. Lis1 regulates dynein by sterically blocking its mechanochemical cycle. eLife 2014, 3. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, P.A.; Ackermann, B.E.; Vershinin, M.; McKenney, R.J. Differential effects of the dynein-regulatory factor Lissencephaly-1 on processive dynein-dynactin motility. J. Biol. Chem. 2017, 292, 12245–12255. [Google Scholar] [CrossRef] [PubMed]
- Yao, X.; Arst, H.N.; Wang, X.; Xiang, X. Discovery of a vezatin-like protein for dynein-mediated early endosome transport. Mol. Biol. Cell 2015, 26, 3816–3827. [Google Scholar] [CrossRef] [PubMed]
- Olenick, M.A.; Tokito, M.; Boczkowska, M.; Dominguez, R.; Holzbaur, E.L.F. Hook Adaptors Induce Unidirectional Processive Motility by Enhancing the Dynein-Dynactin Interaction. J. Biol. Chem. 2016, 291, 18239–18251. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, C.M.; Vale, R.D. Assembly and activation of dynein-dynactin by the cargo adaptor protein Hook3. J. Cell Biol. 2016, 214, 309–318. [Google Scholar] [CrossRef] [PubMed]
- Redwine, W.B.; DeSantis, M.E.; Hollyer, I.; Htet, Z.M.; Tran, P.T.; Swanson, S.K.; Florens, L.; Washburn, M.P.; Reck-Peterson, S.L. The human cytoplasmic dynein interactome reveals novel activators of motility. eLife 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Yu, I.; Garnham, C.P.; Roll-Mecak, A. Writing and Reading the Tubulin Code. J. Biol. Chem. 2015, 290, 17163–17172. [Google Scholar] [CrossRef] [PubMed]
- Janke, C. The tubulin code: Molecular components, readout mechanisms, and functions. J. Cell Biol. 2014, 206, 461–472. [Google Scholar] [CrossRef] [PubMed]
- Duan, J.; Gorovsky, M.A. Both carboxy-terminal tails of alpha- and beta-tubulin are essential, but either one will suffice. Curr. Biol. 2002, 12, 313–316. [Google Scholar] [CrossRef]
- Peris, L.; Thery, M.; Fauré, J.; Saoudi, Y.; Lafanechère, L.; Chilton, J.K.; Gordon-Weeks, P.; Galjart, N.; Bornens, M.; Wordeman, L.; et al. Tubulin tyrosination is a major factor affecting the recruitment of CAP-Gly proteins at microtubule plus ends. J. Cell Biol. 2006, 174, 839–849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caudron, F.; Andrieux, A.; Job, D.; Boscheron, C. A new role for kinesin-directed transport of Bik1p (CLIP-170) in Saccharomyces cerevisiae. J. Cell Sci. 2008, 121, 1506–1513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aiken, J.; Sept, D.; Costanzo, M.; Boone, C.; Cooper, J.A.; Moore, J.K. Genome-wide analysis reveals novel and discrete functions for tubulin carboxy-terminal tails. Curr. Biol. 2014, 24, 1295–1303. [Google Scholar] [CrossRef] [PubMed]
- Fees, C.P.; Aiken, J.; O’Toole, E.T.; Giddings, T.H.; Moore, J.K. The negatively charged carboxy-terminal tail of β-tubulin promotes proper chromosome segregation. Mol. Biol. Cell 2016, 27, 1786–1796. [Google Scholar] [CrossRef] [PubMed]
- Löwe, J.; Li, H.; Downing, K.H.; Nogales, E. Refined structure of alpha beta-tubulin at 3.5 A resolution. J. Mol. Biol. 2001, 313, 1045–1057. [Google Scholar] [CrossRef] [PubMed]
- Eddé, B.; Rossier, J.; Le Caer, J.P.; Desbruyères, E.; Gros, F.; Denoulet, P. Posttranslational glutamylation of alpha-tubulin. Science 1990, 247, 83–85. [Google Scholar] [CrossRef] [PubMed]
- Cowan, N.J.; Dobner, P.R.; Fuchs, E.V.; Cleveland, D.W. Expression of human alpha-tubulin genes: Interspecies conservation of 3′ untranslated regions. Mol. Cell. Biol. 1983, 3, 1738–1745. [Google Scholar] [CrossRef] [PubMed]
- Cleveland, D.W.; Lopata, M.A.; MacDonald, R.J.; Cowan, N.J.; Rutter, W.J.; Kirschner, M.W. Number and evolutionary conservation of alpha- and beta-tubulin and cytoplasmic beta- and gamma-actin genes using specific cloned cDNA probes. Cell 1980, 20, 95–105. [Google Scholar] [CrossRef]
- Baraban, M.; Anselme, I.; Schneider-Maunoury, S.; Giudicelli, F. Zebrafish Embryonic Neurons Transport Messenger RNA to Axons and Growth Cones In Vivo. J. Neurosci. 2013, 33, 15726–15734. [Google Scholar] [CrossRef] [PubMed]
- Denoulet, P.; Eddé, B.; Gros, F. Differential expression of several neurospecific beta-tubulin mRNAs in the mouse brain during development. Gene 1986, 50, 289–297. [Google Scholar] [CrossRef]
- Leandro-García, L.J.; Leskelä, S.; Landa, I.; Montero-Conde, C.; López-Jiménez, E.; Letón, R.; Cascón, A.; Robledo, M.; Rodríguez-Antona, C. Tumoral and tissue-specific expression of the major human beta-tubulin isotypes. Cytoskeleton (Hoboken) 2010, 67, 214–223. [Google Scholar] [CrossRef] [PubMed]
- Joshi, H.C.; Cleveland, D.W. Differential utilization of beta-tubulin isotypes in differentiating neurites. J. Cell Biol. 1989, 109, 663–673. [Google Scholar] [CrossRef] [PubMed]
- Perry, B.; Jensen-Smith, H.C.; Ludueña, R.F.; Hallworth, R. Selective expression of beta tubulin isotypes in gerbil vestibular sensory epithelia and neurons. J. Assoc. Res. Otolaryngol. 2003, 4, 329–338. [Google Scholar] [CrossRef] [PubMed]
- Hao, L.; Thein, M.; Brust-Mascher, I.; Civelekoglu-Scholey, G.; Lu, Y.; Acar, S.; Prevo, B.; Shaham, S.; Scholey, J.M. Intraflagellar transport delivers tubulin isotypes to sensory cilium middle and distal segments. Nat. Cell Biol. 2011, 13, 790–798. [Google Scholar] [CrossRef] [PubMed]
- Miller, F.D.; Naus, C.C.; Durand, M.; Bloom, F.E.; Milner, R.J. Isotypes of alpha-tubulin are differentially regulated during neuronal maturation. J. Cell Biol. 1987, 105, 3065–3073. [Google Scholar] [CrossRef] [PubMed]
- Hutchens, J.A.; Hoyle, H.D.; Turner, F.R.; Raff, E.C. Structurally similar Drosophila alpha-tubulins are functionally distinct in vivo. Mol. Biol. Cell 1997, 8, 481–500. [Google Scholar] [CrossRef] [PubMed]
- Hurd, D.D.; Miller, R.M.; Núñez, L.; Portman, D.S. Specific alpha- and beta-tubulin isotypes optimize the functions of sensory Cilia in Caenorhabditis elegans. Genetics 2010, 185, 883–896. [Google Scholar] [CrossRef] [PubMed]
- Silva, M.; Morsci, N.; Nguyen, K.C.Q.; Rizvi, A.; Rongo, C.; Hall, D.H.; Barr, M.M. Cell-Specific α-Tubulin Isotype Regulates Ciliary Microtubule Ultrastructure, Intraflagellar Transport, and Extracellular Vesicle Biology. Curr. Biol. 2017, 27, 968–980. [Google Scholar] [CrossRef] [PubMed]
- Burke, D.; Gasdaska, P.; Hartwell, L. Dominant effects of tubulin overexpression in Saccharomyces cerevisiae. Mol. Cell. Biol. 1989, 9, 1049–1059. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, B.; Solomon, F. Phenotypic consequences of tubulin overproduction in Saccharomyces cerevisiae: Differences between alpha-tubulin and beta-tubulin. Mol. Cell. Biol. 1990, 10, 5295–5304. [Google Scholar] [CrossRef] [PubMed]
- Katz, W.; Weinstein, B.; Solomon, F. Regulation of tubulin levels and microtubule assembly in Saccharomyces cerevisiae: Consequences of altered tubulin gene copy number. Mol. Cell. Biol. 1990, 10, 5286–5294. [Google Scholar] [CrossRef] [PubMed]
- Janke, C.; Kneussel, M. Tubulin post-translational modifications: Encoding functions on the neuronal microtubule cytoskeleton. Trends Neurosci. 2010, 33, 362–372. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Brady, S.T. Post-translational modifications of tubulin: Pathways to functional diversity of microtubules. Trends Cell Biol. 2015, 25, 125–136. [Google Scholar] [CrossRef] [PubMed]
- Prota, A.E.; Magiera, M.M.; Kuijpers, M.; Bargsten, K.; Frey, D.; Wieser, M.; Jaussi, R.; Hoogenraad, C.C.; Kammerer, R.A.; Janke, C.; et al. Structural basis of tubulin tyrosination by tubulin tyrosine ligase. J. Cell Biol. 2013, 200, 259–270. [Google Scholar] [CrossRef] [PubMed]
- Szyk, A.; Deaconescu, A.M.; Piszczek, G.; Roll-Mecak, A. Tubulin tyrosine ligase structure reveals adaptation of an ancient fold to bind and modify tubulin. Nat. Struct. Mol. Biol. 2011, 18, 1250–1258. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.; Li, Y.; Slaughter, T.; Black, M.M. Composite microtubules of the axon: Quantitative analysis of tyrosinated and acetylated tubulin along individual axonal microtubules. J. Cell Sci. 1993, 352, 339–352. [Google Scholar]
- Baas, P.W.; Black, M.M. Individual microtubules in the axon consist of domains that differ in both composition and stability. J. Cell Biol. 1990, 111, 495–509. [Google Scholar] [CrossRef] [PubMed]
- Gundersen, G.G.; Khawaja, S.; Bulinski, J.C. Postpolymerization detyrosination of alpha-tubulin: A mechanism for subcellular differentiation of microtubules. J. Cell Biol. 1987, 105, 251–264. [Google Scholar] [CrossRef] [PubMed]
- Robson, S.J.; Burgoyne, R.D. Differential localisation of tyrosinated, detyrosinated, and acetylated alpha-tubulins in neurites and growth cones of dorsal root ganglion neurons. Cell Motil. Cytoskeleton 1989, 12, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.; Slaughter, T.; Black, M.M. Newly assembled microtubules are concentrated in the proximal and distal regions of growing axons. J. Cell Biol. 1992, 119, 867–882. [Google Scholar] [CrossRef] [PubMed]
- Baas, P.W.; Ahmad, F.J.; Pienkowski, T.P.; Brown, A.; Black, M.M. Sites of microtubule stabilization for the axon. J. Neurosci. 1993, 13, 2177–2185. [Google Scholar] [PubMed]
- Erck, C.; Peris, L.; Andrieux, A.; Meissirel, C.; Gruber, A.D.; Vernet, M.; Schweitzer, A.; Saoudi, Y.; Pointu, H.; Bosc, C.; et al. A vital role of tubulin-tyrosine-ligase for neuronal organization. Proc. Natl. Acad. Sci. USA 2005, 102, 7853–7858. [Google Scholar] [CrossRef] [PubMed]
- Honnappa, S.; Okhrimenko, O.; Jaussi, R.; Jawhari, H.; Jelesarov, I.; Winkler, F.K.; Steinmetz, M.O. Key interaction modes of dynamic +TIP networks. Mol. Cell 2006, 23, 663–671. [Google Scholar] [CrossRef] [PubMed]
- Weisbrich, A.; Honnappa, S.; Jaussi, R.; Okhrimenko, O.; Frey, D.; Jelesarov, I.; Akhmanova, A.; Steinmetz, M.O. Structure-function relationship of CAP-Gly domains. Nat. Struct. Mol. Biol. 2007, 14, 959–967. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Crevenna, A.H.; Kunze, I.; Mizuno, N. Structural basis for the extended CAP-Gly domains of p150(glued) binding to microtubules and the implication for tubulin dynamics. Proc. Natl. Acad. Sci. USA 2014, 111, 11347–11352. [Google Scholar] [CrossRef] [PubMed]
- Nirschl, J.J.; Magiera, M.M.; Lazarus, J.E.; Janke, C.; Holzbaur, E.L.F. α-Tubulin Tyrosination and CLIP-170 Phosphorylation Regulate the Initiation of Dynein-Driven Transport in Neurons. Cell Rep. 2016, 14, 2637–2652. [Google Scholar] [CrossRef] [PubMed]
- McKenney, R.J.; Huynh, W.; Vale, R.D.; Sirajuddin, M. Tyrosination of α-tubulin controls the initiation of processive dynein-dynactin motility. EMBO J. 2016, 35, 1175–1185. [Google Scholar] [CrossRef] [PubMed]
- Janke, C.; Rogowski, K.; Wloga, D.; Regnard, C.; Kajava, A.V.; Strub, J.-M.; Temurak, N.; van Dijk, J.; Boucher, D.; van Dorsselaer, A.; et al. Tubulin polyglutamylase enzymes are members of the TTL domain protein family. Science 2005, 308, 1758–1762. [Google Scholar] [CrossRef] [PubMed]
- Van Dijk, J.; Rogowski, K.; Miro, J.; Lacroix, B.; Eddé, B.; Janke, C. A targeted multienzyme mechanism for selective microtubule polyglutamylation. Mol. Cell 2007, 26, 437–448. [Google Scholar] [CrossRef] [PubMed]
- Natarajan, K.; Gadadhar, S.; Souphron, J.; Magiera, M.M.; Janke, C. Molecular interactions between tubulin tails and glutamylases reveal determinants of glutamylation patterns. EMBO Rep. 2017, 18, 1013–1026. [Google Scholar] [CrossRef] [PubMed]
- Rogowski, K.; van Dijk, J.; Magiera, M.M.; Bosc, C.; Deloulme, J.-C.; Bosson, A.; Peris, L.; Gold, N.D.; Lacroix, B.; Grau, M.B.; et al. A family of protein-deglutamylating enzymes associated with neurodegeneration. Cell 2010, 143, 564–578. [Google Scholar] [CrossRef] [PubMed]
- Valenstein, M.L.; Roll-Mecak, A. Graded Control of Microtubule Severing by Tubulin Glutamylation. Cell 2016, 164, 911–921. [Google Scholar] [CrossRef] [PubMed]
- Lacroix, B.; Van Dijk, J.; Gold, N.D.; Guizetti, J.; Aldrian-Herrada, G.; Rogowski, K.; Gerlich, D.W.; Janke, C. Tubulin polyglutamylation stimulates spastin-mediated microtubule severing. J. Cell Biol. 2010, 189, 945–954. [Google Scholar] [CrossRef] [PubMed]
- Ikegami, K.; Heier, R.L.; Taruishi, M.; Takagi, H.; Mukai, M.; Shimma, S.; Taira, S.; Hatanaka, K.; Morone, N.; Yao, I.; et al. Loss of alpha-tubulin polyglutamylation in ROSA22 mice is associated with abnormal targeting of KIF1A and modulated synaptic function. Proc. Natl. Acad. Sci. USA 2007, 104, 3213–3218. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, L.; Eng, J.; Martinez, R.A.; Jackson, S.; Huang, J.; Possin, D.E.; Sopher, B.L.; La Spada, A.R. The zinc-binding domain of Nna1 is required to prevent retinal photoreceptor loss and cerebellar ataxia in Purkinje cell degeneration (pcd) mice. Vision Res. 2008, 48, 1999–2005. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Parris, J.; Li, L.; Morgan, J.I. The carboxypeptidase-like substrate-binding site in Nna1 is essential for the rescue of the Purkinje cell degeneration (pcd) phenotype. Mol. Cell. Neurosci. 2006, 33, 200–213. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Gonzalez, A.; La Spada, A.R.; Treadaway, J.; Higdon, J.C.; Harris, B.S.; Sidman, R.L.; Morgan, J.I.; Zuo, J. Purkinje cell degeneration (pcd) phenotypes caused by mutations in the axotomy-induced gene, Nna1. Science 2002, 295, 1904–1906. [Google Scholar] [CrossRef] [PubMed]
- Brady, S.T.; Tytell, M.; Lasek, R.J. Axonal tubulin and axonal microtubules: Biochemical evidence for cold stability. J. Cell Biol. 1984, 99, 1716–1724. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Kirkpatrick, L.L.; Schilling, A.B.; Helseth, D.L.; Chabot, N.; Keillor, J.W.; Johnson, G.V.W.; Brady, S.T. Transglutaminase and polyamination of tubulin: Posttranslational modification for stabilizing axonal microtubules. Neuron 2013, 78, 109–123. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Schaedel, L.; Portran, D.; Aguilar, A.; Gaillard, J.; Marinkovich, M.P.; Théry, M.; Nachury, M.V. Microtubules acquire resistance from mechanical breakage through intralumenal acetylation. Science 2017, 356, 328–332. [Google Scholar] [CrossRef] [PubMed]
- Portran, D.; Schaedel, L.; Xu, Z.; Théry, M.; Nachury, M.V. Tubulin acetylation protects long-lived microtubules against mechanical ageing. Nat. Cell Biol. 2017, 19, 391–398. [Google Scholar] [CrossRef] [PubMed]
- Topalidou, I.; Keller, C.; Kalebic, N.; Nguyen, K.C.Q.; Somhegyi, H.; Politi, K.A.; Heppenstall, P.; Hall, D.H.; Chalfie, M. Genetically separable functions of the MEC-17 tubulin acetyltransferase affect microtubule organization. Curr. Biol. 2012, 22, 1057–1065. [Google Scholar] [CrossRef] [PubMed]
- Cueva, J.G.; Hsin, J.; Huang, K.C.; Goodman, M.B. Posttranslational acetylation of α-tubulin constrains protofilament number in native microtubules. Curr. Biol. 2012, 22, 1066–1074. [Google Scholar] [CrossRef] [PubMed]
- Reed, N.A.; Cai, D.; Blasius, T.L.; Jih, G.T.; Meyhofer, E.; Gaertig, J.; Verhey, K.J. Microtubule Acetylation Promotes Kinesin-1 Binding and Transport. Curr. Biol. 2006, 16, 2166–2172. [Google Scholar] [CrossRef] [PubMed]
- Alper, J.D.; Decker, F.; Agana, B.; Howard, J. The motility of axonemal dynein is regulated by the tubulin code. Biophys. J. 2014, 107, 2872–2880. [Google Scholar] [CrossRef] [PubMed]
- Dehmelt, L.; Smart, F.M.; Ozer, R.S.; Halpain, S. The role of microtubule-associated protein 2c in the reorganization of microtubules and lamellipodia during neurite initiation. J. Neurosci. 2003, 23, 9479–9490. [Google Scholar] [PubMed]
- Witte, H.; Neukirchen, D.; Bradke, F. Microtubule stabilization specifies initial neuronal polarization. J. Cell Biol. 2008, 180, 619–632. [Google Scholar] [CrossRef] [PubMed]
- Knops, J.; Kosik, K.S.; Lee, G.; Pardee, J.D.; Cohen-Gould, L.; McConlogue, L. Overexpression of tau in a nonneuronal cell induces long cellular processes. J. Cell Biol. 1991, 114, 725–733. [Google Scholar] [CrossRef] [PubMed]
- Del Castillo, U.; Winding, M.; Lu, W.; Gelfand, V.I. Interplay between kinesin-1 and cortical dynein during axonal outgrowth and microtubule organization in Drosophila neurons. eLife 2015, 4, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Baas, P.W.; Black, M.M.; Banker, G.A. Changes in microtubule polarity orientation during the development of hippocampal neurons in culture. J. Cell Biol. 1989, 109, 3085–3094. [Google Scholar] [CrossRef] [PubMed]
- Baas, P.W.; Deitch, J.S.; Black, M.M.; Banker, G.A. Polarity orientation of microtubules in hippocampal neurons: Uniformity in the axon and nonuniformity in the dendrite. Proc. Natl. Acad. Sci. USA 1988, 85, 8335–8339. [Google Scholar] [CrossRef] [PubMed]
- Lowery, L.A.; Stout, A.; Faris, A.E.; Ding, L.; Baird, M.A.; Davidson, M.W.; Danuser, G.; Van Vactor, D. Growth cone-specific functions of XMAP215 in restricting microtubule dynamics and promoting axonal outgrowth. Neural Dev. 2013, 8, 22. [Google Scholar] [CrossRef] [PubMed]
- Hirokawa, N.; Noda, Y.; Tanaka, Y.; Niwa, S. Kinesin superfamily motor proteins and intracellular transport. Nat. Rev. Mol. Cell Biol. 2009, 10, 682–696. [Google Scholar] [CrossRef] [PubMed]
- Vale, R.D.; Schnapp, B.J.; Mitchison, T.; Steuer, E.; Reese, T.S.; Sheetz, M.P. Different axoplasmic proteins generate movement in opposite directions along microtubules in vitro. Cell 1985, 43, 623–632. [Google Scholar] [CrossRef]
- Buck, K.B.; Zheng, J.Q. Growth cone turning induced by direct local modification of microtubule dynamics. J. Neurosci. 2002, 22, 9358–9367. [Google Scholar] [PubMed]
- Rivas, R.J.; Hatten, M.E. Motility and cytoskeletal organization of migrating cerebellar granule neurons. J. Neurosci. 1995, 15, 981–989. [Google Scholar] [PubMed]
- Tsai, J.-W.; Bremner, K.H.; Vallee, R.B. Dual subcellular roles for LIS1 and dynein in radial neuronal migration in live brain tissue. Nat. Neurosci. 2007, 10, 970–979. [Google Scholar] [CrossRef] [PubMed]
- Fry, A.E.; Cushion, T.D.; Pilz, D.T. The genetics of lissencephaly. Am. J. Med. Genet. Part C Semin. Med. Genet. 2014, 166, 198–210. [Google Scholar] [CrossRef] [PubMed]
- Stratton, R.F.; Dobyns, W.B.; Airhart, S.D.; Ledbetter, D.H. New chromosomal syndrome: Miller-Dieker syndrome and monosomy 17p13. Hum. Genet. 1984, 67, 193–200. [Google Scholar] [CrossRef] [PubMed]
- Valiente, M.; Marín, O. Neuronal migration mechanisms in development and disease. Curr. Opin. Neurobiol. 2010, 20, 68–78. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Roberts, A.J.; Leschziner, A.E.; Reck-Peterson, S.L. Lis1 acts as a “clutch” between the ATPase and microtubule-binding domains of the dynein motor. Cell 2012, 150, 975–986. [Google Scholar] [CrossRef] [PubMed]
- De Rouvroit, C.L.; Goffinet, A.M. Neuronal migration. Mech. Dev. 2001, 105, 47–56. [Google Scholar] [CrossRef]
- Horesh, D.; Sapir, T.; Francis, F.; Wolf, S.G.; Caspi, M.; Elbaum, M.; Chelly, J.; Reiner, O. Doublecortin, a stabilizer of microtubules. Hum. Mol. Genet. 1999, 8, 1599–1610. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Serneo, F.F.; Higgins, C.; Gambello, M.J.; Wynshaw-Boris, A.; Gleeson, J.G. Lis1 and doublecortin function with dynein to mediate coupling of the nucleus to the centrosome in neuronal migration. J. Cell Biol. 2004, 165, 709–721. [Google Scholar] [CrossRef] [PubMed]
- Jansen, A.C.; Oostra, A.; Desprechins, B.; De Vlaeminck, Y.; Verhelst, H.; Régal, L.; Verloo, P.; Bockaert, N.; Keymolen, K.; Seneca, S.; et al. TUBA1A mutations: From isolated lissencephaly to familial polymicrogyria. Neurology 2011, 76, 988–992. [Google Scholar] [CrossRef] [PubMed]
- Yokoi, S.; Ishihara, N.; Miya, F.; Tsutsumi, M.; Yanagihara, I.; Fujita, N.; Yamamoto, H.; Kato, M.; Okamoto, N.; Tsunoda, T.; et al. TUBA1A mutation can cause a hydranencephaly-like severe form of cortical dysgenesis. Sci. Rep. 2015, 5, 15165. [Google Scholar] [CrossRef] [PubMed]
- Shimojima, K.; Narita, A.; Maegaki, Y.; Saito, A.; Furukawa, T.; Yamamoto, T. Whole-exome sequencing identifies a de novo TUBA1A mutation in a patient with sporadic malformations of cortical development: A case report. BMC Res. Notes 2014, 7, 465. [Google Scholar] [CrossRef] [PubMed]
- McMichael, G.; Bainbridge, M.N.; Haan, E.; Corbett, M.; Gardner, A.; Thompson, S.; van Bon, B.W.M.; van Eyk, C.L.; Broadbent, J.; Reynolds, C.; et al. Whole-exome sequencing points to considerable genetic heterogeneity of cerebral palsy. Mol. Psychiatry 2015, 20, 176–182. [Google Scholar] [CrossRef] [PubMed]
- Hikita, N.; Hattori, H.; Kato, M.; Sakuma, S.; Morotomi, Y.; Ishida, H.; Seto, T.; Tanaka, K.; Shimono, T.; Shintaku, H.; et al. A case of TUBA1A mutation presenting with lissencephaly and Hirschsprung disease. Brain Dev. 2014, 36, 159–162. [Google Scholar] [CrossRef] [PubMed]
- Oegema, R.; Cushion, T.D.; Phelps, I.G.; Chung, S.-K.; Dempsey, J.C.; Collins, S.; Mullins, J.G.L.; Dudding, T.; Gill, H.; Green, A.J.; et al. Recognizable cerebellar dysplasia associated with mutations in multiple tubulin genes. Hum. Mol. Genet. 2015, 24, 5313–5325. [Google Scholar] [CrossRef] [PubMed]
- Bamba, Y.; Shofuda, T.; Kato, M.; Pooh, R.K.; Tateishi, Y.; Takanashi, J.-I.; Utsunomiya, H.; Sumida, M.; Kanematsu, D.; Suemizu, H.; et al. In vitro characterization of neurite extension using induced pluripotent stem cells derived from lissencephaly patients with TUBA1A missense mutations. Mol. Brain 2016, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Myers, K.A.; Bello-Espinosa, L.E.; Kherani, A.; Wei, X.-C.; Innes, A.M. TUBA1A Mutation Associated With Eye Abnormalities in Addition to Brain Malformation. Pediatr. Neurol. 2015, 53, 442–444. [Google Scholar] [CrossRef] [PubMed]
- Romaniello, R.; Arrigoni, F.; Cavallini, A.; Tenderini, E.; Baschirotto, C.; Triulzi, F.; Bassi, M.-T.; Borgatti, R. Brain malformations and mutations in α- and β-tubulin genes: A review of the literature and description of two new cases. Dev. Med. Child Neurol. 2014, 56, 354–360. [Google Scholar] [CrossRef] [PubMed]
- Kamiya, K.; Tanaka, F.; Ikeno, M.; Okumura, A.; Aoki, S. DTI tractography of lissencephaly caused by TUBA1A mutation. Neurol. Sci. 2014, 35, 801–803. [Google Scholar] [CrossRef] [PubMed]
- Sohal, A.P.S.; Montgomery, T.; Mitra, D.; Ramesh, V. TUBA1A mutation-associated lissencephaly: Case report and review of the literature. Pediatr. Neurol. 2012, 46, 127–131. [Google Scholar] [CrossRef] [PubMed]
- Judkins, A.R.; Martinez, D.; Ferreira, P.; Dobyns, W.B.; Golden, J.A. Polymicrogyria includes fusion of the molecular layer and decreased neuronal populations but normal cortical laminar organization. J. Neuropathol. Exp. Neurol. 2011, 70, 438–443. [Google Scholar] [CrossRef] [PubMed]
- Stutterd, C.A.; Leventer, R.J. Polymicrogyria: A common and heterogeneous malformation of cortical development. Am. J. Med. Genet. Part C Semin. Med. Genet. 2014, 166, 227–239. [Google Scholar] [CrossRef] [PubMed]
- Gressens, P.; Baes, M.; Leroux, P.; Lombet, A.; Van Veldhoven, P.; Janssen, A.; Vamecq, J.; Marret, S.; Evrard, P. Neuronal migration disorder in Zellweger mice is secondary to glutamate receptor dysfunction. Ann. Neurol. 2000, 48, 336–343. [Google Scholar] [CrossRef]
- Barkovich, A.J.; Guerrini, R.; Kuzniecky, R.I.; Jackson, G.D.; Dobyns, W.B. A developmental and genetic classification for malformations of cortical development: Update 2012. Brain 2012, 135, 1348–1369. [Google Scholar] [CrossRef] [PubMed]
- Zanni, G.; Colafati, G.S.; Barresi, S.; Randisi, F.; Talamanca, L.F.; Genovese, E.; Bellacchio, E.; Bartuli, A.; Bernardi, B.; Bertini, E. Description of a novel TUBA1A mutation in Arg-390 associated with asymmetrical polymicrogyria and mid-hindbrain dysgenesis. Eur. J. Paediatr. Neurol. 2013, 17, 361–365. [Google Scholar] [CrossRef] [PubMed]
- Faheem, M.; Naseer, M.I.; Rasool, M.; Chaudhary, A.G.; Kumosani, T.A.; Ilyas, A.M.; Pushparaj, P.; Ahmed, F.; Algahtani, H.A.; Al-Qahtani, M.H.; et al. Molecular genetics of human primary microcephaly: An overview. BMC Med. Genom. 2015, 8 (Suppl. 1), S4. [Google Scholar] [CrossRef] [PubMed]
- Harris, S.R. Measuring head circumference: Update on infant microcephaly. Can. Fam. Physician 2015, 61, 680–684. [Google Scholar] [PubMed]
- Woods, C.G.; Parker, A. Investigating microcephaly. Arch. Dis. Child. 2013, 98, 707–713. [Google Scholar] [CrossRef] [PubMed]
- Tian, G.; Jaglin, X.H.; Keays, D.A.; Francis, F.; Chelly, J.; Cowan, N.J. Disease-associated mutations in TUBA1A result in a spectrum of defects in the tubulin folding and heterodimer assembly pathway. Hum. Mol. Genet. 2010, 19, 3599–3613. [Google Scholar] [CrossRef] [PubMed]
- Tian, G.; Kong, X.-P.; Jaglin, X.H.; Chelly, J.; Keays, D.; Cowan, N.J. A pachygyria-causing alpha-tubulin mutation results in inefficient cycling with CCT and a deficient interaction with TBCB. Mol. Biol. Cell 2008, 19, 1152–1161. [Google Scholar] [CrossRef] [PubMed]
- Belvindrah, R.; Natarajan, K.; Shabajee, P.; Bruel-Jungerman, E.; Bernard, J.; Goutierre, M.; Moutkine, I.; Jaglin, X.H.; Savariradjane, M.; Irinopoulou, T.; et al. Mutation of the α-tubulin Tuba1a leads to straighter microtubules and perturbs neuronal migration. J. Cell Biol. 2017, 216, 2443–2461. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chen, K.; Sloan, S.A.; Bennett, M.L.; Scholze, A.R.; O’Keeffe, S.; Phatnani, H.P.; Guarnieri, P.; Caneda, C.; Ruderisch, N.; et al. An RNA-Sequencing Transcriptome and Splicing Database of Glia, Neurons, and Vascular Cells of the Cerebral Cortex. J. Neurosci. 2014, 34, 11929–11947. [Google Scholar] [CrossRef] [PubMed]
- Gartz Hanson, M.; Aiken, J.; Sietsema, D.V.; Sept, D.; Bates, E.A.; Niswander, L.; Moore, J.K. Novel α-tubulin mutation disrupts neural development and tubulin proteostasis. Dev. Biol. 2016, 409, 406–419. [Google Scholar] [CrossRef] [PubMed]
- Gloster, A.; Wu, W.; Speelman, A.; Weiss, S.; Causing, C.; Pozniak, C.; Reynolds, B.; Chang, E.; Toma, J.G.; Miller, F.D. The T alpha 1 alpha-tubulin promoter specifies gene expression as a function of neuronal growth and regeneration in transgenic mice. J. Neurosci. 1994, 14, 7319–7330. [Google Scholar] [PubMed]
- Bamji, S.X.; Miller, F.D. Comparison of the expression of a Tα1:nlacZ transgene and Tα1 α-tubulin mRNA in the mature central nervous system. J. Comp. Neurol. 1996, 374, 52–69. [Google Scholar] [CrossRef]
- Gloster, A.; El-Bizri, H.; Bamji, S.X.; Rogers, D.; Miller, F.D. Early induction of Talpha1 alpha-tubulin transcription in neurons of the developing nervous system. J. Comp. Neurol. 1999, 405, 45–60. [Google Scholar] [CrossRef]
- Coksaygan, T.; Magnus, T.; Cai, J.; Mughal, M.; Lepore, A.; Xue, H.; Fischer, I.; Rao, M.S. Neurogenesis in Talpha-1 tubulin transgenic mice during development and after injury. Exp. Neurol. 2006, 197, 475–485. [Google Scholar] [CrossRef] [PubMed]
- Keays, D.A.; Cleak, J.; Huang, G.J.; Edwards, A.; Braun, A.; Treiber, C.D.; Pidsley, R.; Flint, J. The role of Tuba1a in adult hippocampal neurogenesis and the formation of the dentate gyrus. Dev. Neurosci. 2010, 32, 268–277. [Google Scholar] [CrossRef] [PubMed]
- Goldman, D.; Hankin, M.; Li, Z.; Dai, X.; Ding, J. Transgenic zebrafish for studying nervous system development and regeneration. Transgenic Res. 2001, 10, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Veldman, M.B.; Bemben, M.A.; Goldman, D. Tuba1a gene expression is regulated by KLF6/7 and is necessary for CNS development and regeneration in zebrafish. Mol. Cell. Neurosci. 2010, 43, 370–383. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, R.; Reifler, A.; Parent, J.M.; Goldman, D. Conditional gene expression and lineage tracing of tuba1a expressing cells during zebrafish development and retina regeneration. J. Comp. Neurol. 2010, 518, 4196–4212. [Google Scholar] [CrossRef] [PubMed]
- Mathew, T.C.; Miller, F.D. Increased expression of Tα1 α-tubulin mRNA during collateral and NGF-induced sprouting of sympathetic neurons. Dev. Biol. 1990, 141, 84–92. [Google Scholar] [CrossRef]
- Mohiuddin, L.; Fernyhough, P.; Tomlinson, D.R. Acidic fibroblast growth factor enhances neurite outgrowth and stimulates expression of GAP-43 and T alpha 1 alpha-tubulin in cultured neurones from adult rat dorsal root ganglia. Neurosci. Lett. 1996, 215, 111–114. [Google Scholar] [CrossRef]
- Ménard, C.; Hein, P.; Paquin, A.; Savelson, A.; Yang, X.M.; Lederfein, D.; Barnabé-Heider, F.; Mir, A.A.; Sterneck, E.; Peterson, A.C.; et al. An essential role for a MEK-C/EBP pathway during growth factor-regulated cortical neurogenesis. Neuron 2002, 36, 597–610. [Google Scholar] [CrossRef]
- Miller, F.D.; Tetzlaff, W.; Bisby, M.A.; Fawcett, J.W.; Milner, R.J. Rapid induction of the major embryonic alpha-tubulin mRNA, T alpha 1, during nerve regeneration in adult rats. J. Neurosci. 1989, 9, 1452–1463. [Google Scholar] [PubMed]
- Kost, S.A.; Oblinger, M.M. Immature corticospinal neurons respond to axotomy with changes in tubulin gene expression. Brain Res. Bull. 1993, 30, 469–475. [Google Scholar] [CrossRef]
- Mikucki, S.A.; Oblinger, M.M. Corticospinal neurons exhibit a novel pattern of cytoskeletal gene expression after injury. J. Neurosci. Res. 1991, 30, 213–225. [Google Scholar] [CrossRef] [PubMed]
- Tetzlaff, W.; Alexander, S.W.; Miller, F.D.; Bisby, M.A. Response of facial and rubrospinal neurons to axotomy: Changes in mRNA expression for cytoskeletal proteins and GAP-43. J. Neurosci. 1991, 11, 2528–2544. [Google Scholar] [PubMed]
- Theriault, E.; Tetzlaff, W.; Tator, C.H. Elevated gene expression in the red nucleus after spinal cord compression injury. Neuroreport 1992, 3, 559–562. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Mathew, T.C.; Miller, F.D. Evidence that the loss of homeostatic signals induces regeneration-associated alterations in neuronal gene expression. Dev. Biol. 1993, 158, 456–466. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Toma, J.G.; Chan, H.; Smith, R.; Miller, F.D. Disruption of fast axonal transport in vivo leads to alterations in Schwann cell gene expression. Dev. Biol. 1994, 163, 423–439. [Google Scholar] [CrossRef] [PubMed]
- Mathew, T.C.; Miller, F.D. Induction of T alpha 1 alpha-tubulin mRNA during neuronal regeneration is a function of the amount of axon lost. Dev. Biol. 1993, 158, 467–474. [Google Scholar] [CrossRef] [PubMed]
- Ramón y Cajal, S. Degeneration and Regeneration of the Nervous System; Oxford University Press: New York, NY, USA, 1959. [Google Scholar]
- Richardson, P.M.; Issa, V.M.; Aguayo, A.J. Regeneration of long spinal axons in the rat. J. Neurocytol. 1984, 13, 165–182. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.D.; Gloster, A.; Miller, F.D. Transcriptional repression of the growth-associated T alpha 1 alpha-tubulin gene by target contact. J. Neurosci. Res. 1997, 48, 477–487. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| TUBA1A | TUBA1B | |||||
|---|---|---|---|---|---|---|
| Organism | Human | Mouse | Rat | Human | Mouse | Rat |
| Aliases | TUBA1A [7] b-α-1 [7,12] | Tuba1a [7] Tuba1 [7] M-α-1 [10] | Tuba1a [7] Tuba1 [7] α-T14 [9] T-α-1 [13] | TUBA1B [7] k-α-1 [7,12] | Tuba1b [7] M-α-2 [10] Tuba2 [7] | Tuba1b [7] T26 [13] |
| Human Gene | Gene Accession | Protein Accession | Identity to TUBA1A | CTT Amino Acid Sequence | Mouse Gene | Identity to Human Isotype |
|---|---|---|---|---|---|---|
| TUBA1A | NM_006009 | NP_006000 | - | MAALEKDYEEVGVDSVEGEGEEEGEEY | Tuba1a | 451/451 |
| TUBA1B | NM_006082 | NP_006073 | 449/451 | MAALEKDYEEVGVDSVEGEGEEEGEEY | Tuba1b | 451/451 |
| TUBA1C | NM_032704 | NP_116093 | 442/451 | MAALEKDYEEVGADSADGEDEGEEY | Tuba1c | 446/449 |
| TUBA3C | NM_006001 | NP_005992 | 440/451 | LAALEKDYEEVGVDSVEAEAEEGEEY | ||
| TUBA3D | NM_080386 | NP_525125 | 440/451 | LAALEKDYEEVGVDSVEAEAEEGEEY | ||
| TUBA3E | NM_207312 | NP_997195 | 435/451 | LAALEKDCEEVGVDSVEAEAEEGEEY | ||
| TUBA4A | NM_006000 | NP_005991 | 432/450 | MAALEKDYEEVGIDSYEDEDEGEE | Tuba4a | 448/448 |
| TUBA8 | NM_018943 | NP_061816 | 399/439 | LAALEKDYEEVGTDSFEEENEGEEF | Tuba8 | 446/449 |
| TUBAL3 | NM_024803 | NP_079079 | 329/444 | LAALERDYEEVAQSF | Tubal3 | 369/446 |
| Reference | Mutation | Case Number | Gender/Age | Cortical Phenotype | Corpus Callosum Defect | Other Brain Malformations |
|---|---|---|---|---|---|---|
| [132] | I5L | not reported | F/7 | perisylvian pachygyria | thin | brainstem mildly hypoplastic |
| [132] | I5L | not reported | F/2 | perisylvian pachygyria | splenium hypoplastic | brainstem and cerebellum vermis mildly hypoplastic |
| [133] | C25F | K3373 | M/2 | lissencephaly, thin cortex | agenesis | poorly differentiated dysmorphic basal ganglia, slightly hypoplastic cerebellar vermis, ventricle dilation |
| [134] | E27Q * | not reported | F/0.5 | simplified gyral pattern, diffuse pachygyria | hypoplastic | hypoplastic cerebellar vermis, lateral ventricle dilation |
| [15] | E55K | FR08-D5604 | M/3 | microcephaly, lissencephaly posterior, pachygyria anterior | partial agenesis | severe vermis hypoplasia, flattened isthmus and pons, hypoplastic hippocampus, trigones and occipital horns dilated |
| [20,21] | T56M | LIS_TUB_003_foetus18 | M/24.3 GW | microcephaly, lissencephaly; absent cortical plate, 2 layered | complete agenesis | severe vermis hypoplasia; hypoplastic basal ganglia, severe hypoplasia of cerebellum and pons, optic nerve hypoplasia |
| [133] | R64W * | NCU_F41 | F/3 | extremely thin cerebral parenchyma | agenesis | optic nerve hypoplasia, hypoplastic brainstem, agenesis of cerebellum |
| [19] | L70S | not reported | F/2 weeks | lissencephaly, diffuse polymicrogyria-like | absent | cerebellar hypoplasia, enlarged lateral ventricles |
| [20] | P72S | LIS_TUB_012_foetus22 | F/37.8 GW | severe lissencephaly | dysmorphic/hypoplastic | severe vermis hypoplasia |
| [16] | L92V | CM-66 | fetus | lissencephaly with cerebellar hypoplasia; microcephaly | absent | small brainstem, cerebellum, and corticospinal tract, severe ventricular dilation |
| [20,21] | N101S | LIS_TUB_079_foetus25 | M/25 GW | microcephaly, lissencephaly; 2–3 layered cortex, poorly differentiated | complete agenesis | severe vermis and hemispheric dysplasia; severe hypoplasia and dysplasia of cerebellum, severe hypoplasia of pons |
| [20] | E113K | LIS_TUB_031 | M/11 | central pachygyria | normal | normal cerebellum |
| [135] | R123C * | 106115P | M | pachygyria | not reported | not reported |
| [16] | V137D | CM-107 | not reported | pachygyria with cerebellar hypoplasia | absent | malformed hippocampus, thin brainstem, severe cerebellar hypoplasia |
| [135] | L152Q * | 169451P | F | pachygyria | not reported | not reported |
| [12] | I188L | LIS_TUB_026 | F/2 | laminar heterotopia | thin, partial agenesis | vermis and brainstem hypoplasia, severe ventricular dilation |
| [136] | C200Y | not reported | F/8 | lissencephaly | agenesis | abnormal hippocampus, dysmorphic and hypoplastic basal ganglia and thalamus, hypoplastic cerebellum, enlarged lateral ventricles |
| [132] | Y210C | not reported | M/1.5 | lissencephaly anterior, pachygyria posterior | thin | brainstem and cerebellum vermis mildly hypoplastic |
| [137] | R214H | UW168-3 | F/4 | diffuse irregular gyration and sulcation | partial agenesis | hypoplasia of vermis, asymmetric pons, dysmorphic basal ganglia, cranial nerve hypoplasia, enlarged lateral ventricles |
| [16] | D218Y | LR07-213 | not reported | lissencephaly with cerebellar hypoplasia, microcephaly | absent | thin brainstem, hypoplasia of cerebellar vermis |
| [137] | I219V | UW167-3 | F/10 | diffuse (L > R) irregular gyration and sulcation | partial agenesis | hypoplasia of vermis, asymmetric pons, dysmorphic basal ganglia, cranial nerve hypoplasia, enlarged lateral ventricles |
| [14] | I238V | LIS_TUB_022_foetus05 | M/fetus | two-layered cortex, poorly differentiated | complete agenesis | disorganized hippocampus, internal capsule absent on one side, hypoplastic brainstem and corticospinal tract |
| [12] | P263T | not reported | M/fetus | lissencephaly, microcephaly | agenesis | abnormal hippocampus, cerebellum, vermis and brainstem hypoplasia, severe ventricular dilation |
| [14] | P263T | LIS_TUB_025_foetus06 | M/fetus | two-layered cortex, poorly differentiated | complete agenesis | disorganized hippocampus, hypoplastic internal capsule, hypoplastic brainstem and corticospinal tract |
| [11] | R264C | not reported | not reported | pachygyria | agenesis | abnormal hippocampus, abnormal vermis, brainstem hypoplasia |
| [12] | R264C | LIS_TUB_037 | M/4 | pachygyria | present, abnormal shape | vermis and brainstem hypoplasia, mild ventricular dilation |
| [12] | R264C | LIS_TUB_036 | M/2 | pachygyria | present, abnormal shape | vermis hypoplasia, mild ventricular dilation |
| [13] | R264C | LIS_TUB_041 | M/7 | perisylvian pachygyria | posterior agenesis | severe dysgenesis of internal capsule |
| [13] | R264C | LIS_TUB_040 | M/1.5 | perisylvian pachygyria | mild hypoplasia | moderate dysgenesis of internal capsule |
| [20] | R264C | LIS_TUB_033 | F/1.5 | central pachygyria | normal | mild vermis hypoplasia |
| [20] | R264C | LIS_TUB_034 | F/6.5 | central pachygyria | hypogenetic | normal cerebellum |
| [20] | R264C | LIS_TUB_035 | M/6 | central pachygyria | normal | mild vermis hypoplasia |
| [138] | R264C | Patient B | M/2 | grade 4 agyria, pachygyria (P > A gradient) | present, abnormal shape | hypoplastic basal ganglia |
| [20,21] | R264H | LIS_TUB_002_foetus20 | F/24 GW | microcephaly, lissencephaly; absent cortical plate, 2 layered | complete agenesis | severe vermis hypoplasia; moderate hypoplasia of cerebellum, severe pons hypoplasia |
| [16] | A270T | LR07-244 | not reported | pachygyria with cerebellar hypoplasia | absent | malformed hippocampus, thin brainstem, severe cerebellar hypoplasia |
| [139] | A270S | not reported | M/19 mo | mild posterior simplified cerebral gyral pattern | agenesis | severe hypoplastic cerebellar vermis, mildly dysplastic and hypoplastic cerebellar hemispheres, mildly hypoplastic brainstem, dysplastic basal ganglia, thalami, hypoplastic optic nerves, absent olfactory bulbs, lateral and third ventricle dilated |
| [12] | L286F | not reported | M/fetus | lissencephaly | agenesis | abnormal hippocampus, vermis and brainstem hypoplasia, severe ventricular dilation |
| [14] | L286F | LIS_TUB_007_foetus04 | fetus | two-layered cortex, poorly differentiated | complete agenesis | absent hippocampus, olfactory bulb, and internal capsule, hypoplastic brainstem and corticospinal tract |
| [17] | V303G | LIS_TUB_006_foetus03 | fetus | pachygyria, microcephaly | short and thin | thin brainstem, pons and medulla flattened, hypoplastic cerebellum and corticospinal tracts, severe ventricular dilation |
| [20,21] | R320H | LIS_TUB_005_foetus01 | M/25 GW | microcephaly, lissencephaly; absent cortical plate | partial agenesis | severe vermis and hemispheric dysplasia/severe hypoplasia and dysplasia of cerebellum, severe hypoplasia (neuronal over migration) spinal cord anterior horn hypoplasia |
| [21] | R320H | LIS_TUB_081_foetus26 | M/26 GW | absent cortical plate, 2 layered | complete agenesis | severe hypoplasia and dysplasia of cerebellum, severe hypoplasia of pons |
| [20,21] | K326N | LIS_TUB_004_foetus08 | M/23 GW | microcephaly, lissencephaly; thick 2-layered cortex | partial agenesis; complete agenesis | severe vermis and hemispheric dysplasia/hypoplastic basal ganglia, severe hypoplasia and dysplasia of cerebellum, severe pons hypoplasia |
| [16] | N329S | LR05-388 | not reported | lissencephaly with cerebellar hypoplasia, microcephaly | absent | thin brainstem, hypoplasia of cerebellar vermis |
| [138] | N329S | Patient A | M/0 | grade 1 lissencephaly with cerebellar hypoplasia | agenesis | hypoplastic basal ganglia, cerebellum and brain stem |
| [20,21] | V371E | LIS_TUB_080_foetus24 | F/23.3 GW | microcephaly, lissencephaly; 2–3 layered cortex, poorly differentiated | complete agenesis | severe vermis hypoplasia; hypoplastic basal ganglia, severe hypoplasia of cerebellum, severe pons hypoplasia |
| [140] | A387V | not reported | F/5 | pachygyria with SBH | thin | simplified hippocampus, highly dysmorphic brainstem, flattened pons, mildly hypoplastic cerebellar vermis |
| [20] | A369T | LIS_TUB_030 | M/11 | central pachygyria | dysmorphic/hypoplastic | mild vermis hypoplasia |
| [13] | L397P | LIS_TUB_039 | M/5.5 | perisylvian pachygyria | posterior agenesis | severe vermis dysplasia, severe dysgenesis of internal capsule |
| [12] | R402C | not reported | M/fetus | lissencephaly | abnormally thick | abnormal hippocampus, vermis and brainstem hypoplasia, severe ventricular dilation |
| [15] | R402C | FR04-D4148 | F/11 | microcephaly, lissencephaly | thin, rostrum absent, splenium hypoplastic | mild vermis and pons hypoplasia, trigones and occipital horns dilated |
| [16] | R402C | LP95-073 | not reported | lissencephaly | dysmorphic but intact | classic lissencephaly, round hippocampi with rounded rim |
| [16] | R402C | LR07-008 | not reported | lissencephaly | dysmorphic but intact | classic lissencephaly, round hippocampi with rounded rim |
| [16] | R402C | LR06-210 | not reported | lissencephaly | dysmorphic but intact | classic lissencephaly, round hippocampi with rounded rim |
| [16] | R402C | LR08-035 | not reported | lissencephaly | dysmorphic but intact | classic lissencephaly, round hippocampi with rounded rim |
| [16] | R402C | LR06-064 | not reported | lissencephaly | dysmorphic but intact | classic lissencephaly, round hippocampi with rounded rim |
| [20] | R402C | LIS_TUB_019 | M/10 | moderate lissencephaly | dysmorphic, hypoplastic | mild vermis hypoplasia |
| [14] | R402C | LIS_TUB_021_foetus07 | M/fetus | thick four-layered cortex | abnormally thick and short | disorganized hippocampus, hypoplastic vermis, brainstem, abnormal corticospinal tract |
| [11,12] | R402H | LIS_TUB_023 | M/11 | lissencephaly | agenesis; thin, partial agenesis | abnormal vermis, brainstem hypoplasia/abnormal hippocampus, vermis and brainstem hypoplasia, severe ventricular dilation |
| [16] | R402H | LP97-039 | not reported | lissencephaly with cerebellar hypoplasia | dysmorphic but intact | moderate cerebellar vermis hypoplasia, classic lissencephaly, round hippocampi with rounded rim, |
| [16] | R402H | LP97-041 | not reported | lissencephaly with cerebellar hypoplasia | dysmorphic but intact | moderate cerebellar vermis hypoplasia, classic lissencephaly, round hippocampi with rounded rim |
| [20,21] | R402H | LIS_TUB_017_foetus02 | M/29 GW | severe lissencephaly | complete agenesis | mild vermis and pons hypoplasia |
| [20] | R402H | LIS_TUB_014 | M/4 | severe lissencephaly | hypogenetic | severe hemispheric hypoplasia |
| [20] | R402H | LIS_TUB_015 | M/1 | severe lissencephaly | hypogenetic | severe hemispheric hypoplasia |
| [20] | R402H | LIS_TUB_016 | F/1.5 | severe lissencephaly | hypogenetic | vermis dysplasia |
| [141] | R402H | not reported | M/1 | lissencephaly | not reported | severe dysplasia of brainstem and cerebellum |
| [15] | R402L | RE07-S1605 | M/3 | microcephaly, lissencephaly posterior, pachygyria anterior | thin | mild vermis and pons hypoplasia, retrocerebellar cyst, abnormal hippocampus, dilated lateral ventricles, occipital horns, anterior horns |
| [142] | R402L | not reported | F/1 | microcephaly, lissencephaly posterior, pachygyria anterior | mild hypoplasia | severe hypoplasia of cerebellum, mild hypoplasia of brainstem, moderate ventricular dilation |
| [20,21] | V409A | LIS_TUB_011_foetus23 | M/32 GW | severe lissencephaly with cerebellar hypoplasia | complete agenesis | severe vermis hypoplasia/severe hypoplasia of cerebellum and pons |
| [20] | V409I | LIS_TUB_032 | M/10 | central pachygyria | dysmorphic, hypoplastic | normal cerebellum |
| [12] | S419L | LIS_TUB_024 | M/18 | pachygyria | abnormal shape | abnormal hippocampus, vermis hypoplasia, severe ventricular dilation |
| [13] | R422C | LIS_TUB_042 | F/4.5 | perisylvian pachygyria | mild hypoplasia | mild vermis hypoplasia, severe dysgenesis of internal capsule |
| [13] | R422H | LIS_TUB_020 | F/7 | posterior pachygyria | mild hypoplasia | mild vermis hypoplasia, moderate dysgenesis of internal capsule |
| [15] | R422H | FR05-D4607 | M/9 | pachygyria with SBH | partial agenesis | severe vermis hypoplasia, dandy-walker malformation, hypoplasia of pons, abnormal hippocampus, dilated lateral ventricles, enlarged 4th ventricle |
| [15] | R422H | FR07-D5526 | F/5 | microcephaly, pachygyria with SBH | partial agenesis | moderate vermis hypoplasia, mild pons hypoplasia, abnormal hippocampus, dilated lateral ventricles, enlarged 4th ventricle |
| [16] | R422H | LR05-052 | not reported | pachygyria with cerebellar hypoplasia | absent | malformed hippocampus, thin brainstem, severe cerebellar hypoplasia |
| [16] | R422H | LR08-340 | not reported | pachygyria with cerebellar hypoplasia | absent | malformed hippocampus, thin brainstem, severe cerebellar hypoplasia |
| [20,21] | R422H | LIS_TUB_018_foetus10 | F/28 GW | severe lissencephaly | complete agenesis | mild vermis and pons hypoplasia |
| [16] | M425K | LR08-388 | not reported | lissencephaly with cerebellar hypoplasia, microcephaly | absent | thin brainstem, hypoplasia of cerebellar vermis |
| [20] | E429Q | LIS_TUB_001_foetus09 | F/25 GW | microcephaly, lissencephaly | complete agenesis | severe vermis hypoplasia |
| [21] | E429Q | LIS_TUB_004_foetus09 | F/25 GW | 4 layered cortex | complete agenesis | hypoplastic basal ganglia, severe hypoplasia of cerebellum and pons |
| [13] | G436R | LIS_TUB_038 | M/7 | perisylvian pachygyria | mild hypoplasia | mild vermis hypoplasia, severe dysgenesis of internal capsule |
| References | Mutation | Case Number | Gender/Age | Cortical Phenotype | Corpus Callosum Defect | Other Brain Malformations |
|---|---|---|---|---|---|---|
| [19] | L70S | not reported | F/2 weeks | lissencephaly, diffuse polymicrogyria-like | absent | cerebellar hypoplasia, enlarged lateral ventricles |
| [21] | P72S | LIS_TUB_012_foetus22 | F/37.8 GW | unlayered generalized and asymmetric polymicrogyria | hypoplastic | severe hypoplasia of cerebellum and pons |
| [20] | R123C | LIS_TUB_044 | F/3 | central polymicrogyria-like cortical dysplasia | normal | vermis dysplasia |
| [20,21] | S158L | LIS_TUB_053_foetus21 | F/24.5 GW | unlayered generalized and asymmetric polymicrogyria | complete agenesis | severe vermis and hemispheric dysplasia/hypoplastic basal ganglia, severe hypoplastic and dysmorphic cerebellum, hypoplasia olivary heterotopia |
| [18] | Y161H | LIS_TUB_047 | F/11 | asymmetrical perisylvian polymicrogyria | moderate hypoplasia | dysmorphic basal ganglia, dysplastic vermis and pons |
| [20,21] | R214H | LIS_TUB_043_foetus11 | M/23 GW | central polymicrogyria-like cortical dysplasia; unlayered central and asymmetric polymicrogyria | complete agenesis | normal cerebellum; mild vermian hypoplasia, mild dysplastic olivary nuclei |
| [18] | V235L | LIS_TUB_046 | M/7.5 | asymmetrical perisylvian polymicrogyria | moderate hypoplasia | dysmorphic basal ganglia |
| [19] | A333V | not reported | M/7 | right focal polymicrogyria-like | thin | vermis hypoplasia, mild brainstem hypoplasia, 4th ventricle enlarged |
| [20] | V353I | LIS_TUB_059 | M/4 | simplified gyral pattern with focal polymicrogyria | partial agenesis | normal |
| [18] | R390C | LIS_TUB_045 | M/1 | asymmetrical perisylvian polymicrogyria | severe hypoplasia | dysmorphic basal ganglia, dysplastic vermis, severe hypoplasia of brainstem |
| [147] | R390H | not reported | F/3 | focal polymicrogyria | thin and incomplete | hypoplasia of left internal capsule, severe hypoplastic and asymmetric brainstem, vermis dysplasia, enlarged lateral ventricles |
| [20] | D396Y | LIS_TUB_078 | F/4 | central polymicrogyria-like cortical dysplasia | partial agenesis | severe vermis dysplasia |
| Reference | Mutation | Case Number | Gender/Age | Cortical Phenotype | OFC | Corpus Callosum Defect | Other Brain Malformations |
|---|---|---|---|---|---|---|---|
| [132] | I5L | not reported | F/7 | perisylvian pachygyria | −2 SD | thin | brainstem mildly hypoplastic |
| [134] | E27Q * | not reported | F/5 | simplified gyral pattern, diffuse pachygyria | −1 SD at birth, −3.3 SD at 2 mo. | hypoplastic | hypoplastic cerebellar vermis, lateral ventricle dilation |
| [15] | E55K | FR08-D5604 | M/3 | lissencephaly posterior, pachygyria anterior | −7 SD | partial agenesis | severe vermis hypoplasia, flattened isthmus and pons, hypoplastic hippocampus, trigones and occipital horns dilated |
| [20] | T56M | LIS_TUB_003_foetus18 | M/24.3 GW | lissencephaly | microcephaly | complete agenesis | severe vermis hypoplasia |
| [21] | T56M | LIS_TUB_003_foetus18 | M/24.3 GW | absent cortical plate, 2 layered | <3rd centile | complete agenesis | hypoplastic basal ganglia, severe hypoplasia of cerebellum and pons, optic nerve hypoplasia |
| [133] | R64W * | NCU_F41 | F/3 | extremely thin cerebral parenchyma | −2.4 SD | agenesis | optic nerve hypoplasia, hypoplastic brainstem, agenesis of cerebellum |
| [19] | L70S | not reported | F/2 weeks | lissencephaly, diffuse polymicrogyria-like | 2–9 centile | absent | cerebellar hypoplasia, enlarged lateral ventricles |
| [21] | P72S | LIS_TUB_012_foetus22 | F/37.8 GW | unlayered generalized and asymmetric polymicrogyria | 5th centile | hypoplastic | severe hypoplasia of cerebellum and pons |
| [16] | L92V | CM-66 | fetus | lissencephaly with cerebellar hypoplasia | microcephaly | absent | small brainstem, cerebellum, and corticospinal tract, severe ventricular dilation |
| [20] | N101S | LIS_TUB_079_foetus25 | M/25 GW | lissencephaly | microcephaly | complete agenesis | severe vermis and hemispheric dysplasia |
| [21] | N101S | LIS_TUB_079_foetus25 | M/25 GW | 2–3 layered cortex, poorly differentiated | <3rd centile | complete agenesis | severe hypoplasia and dysplasia of cerebellum, severe hypoplasia of pons |
| [20] | E113K | LIS_TUB_031 | M/11 | central pachygyria | −3 SD | normal | normal cerebellum |
| [20] | R123C | LIS_TUB_044 | F/3 | central polymicrogyria-like cortical dysplasia | −3 SD | normal | vermis dysplasia |
| [18] | Y161H | LIS_TUB_047 | F/11 | asymmetrical perisylvian polymicrogyria | 3rd centile | moderate hypoplasia | dysmorphic basal ganglia, dysplastic vermis and pons |
| [12] | I188L | LIS_TUB_026 | F/2 | laminar heterotopia | −4 SD | thin, partial agenesis | vermis and brainstem hypoplasia, severe ventricular dilation |
| [136] | C200Y * reported as C402Y | not reported | F/8 | lissencephaly | −3.5 SD | agenesis | abnormal hippocampus, dysmorphic and hypoplastic basal ganglia and thalamus, hypoplastic cerebellum, enlarged lateral ventricles |
| [132] | Y210C | not reported | M/1.5 | lissencephaly anterior, pachygyria posterior | −3 SD | thin | brainstem and cerebellum vermis mildly hypoplastic |
| [137] | R214H | UW168-3 | F/4 | diffuse irregular gyration and sulcation | <−2.5 SD | partial agenesis | hypoplasia of vermis, asymmetric pons, dysmorphic basal ganglia, cranial nerve hypoplasia, enlarged lateral ventricles |
| [16] | D218Y | LR07-213 | Not reported | lissencephaly with cerebellar hypoplasia | microcephaly | absent | thin brainstem, hypoplasia of cerebellar vermis |
| [12] | P263T | not reported | M/fetus | lissencephaly | microcephaly | agenesis | abnormal hippocampus, cerebellum, vermis and brainstem hypoplasia, severe ventricular dilation |
| [12] | R264C | LIS_TUB_037 | M/4 | pachygyria | −4.5 SD | present, abnormal shape | vermis and brainstem hypoplasia, mild ventricular dilation |
| [12] | R264C | LIS_TUB_036 | M/2 | pachygyria | −4 SD | present, abnormal shape | vermis hypoplasia, mild ventricular dilation |
| [13] | R264C | LIS_TUB_041 | M/7 | perisylvian pachygyria | −4 SD | posterior agenesis | severe dysgenesis of internal capsule |
| [13] | R264C | LIS_TUB_040 | M/1.5 | perisylvian pachygyria | −4 SD | mild hypoplasia | moderate dysgenesis of internal capsule |
| [20] | R264C | LIS_TUB_033 | F/1.5 | central pachygyria | −5 SD | normal | mild vermis hypoplasia |
| [20] | R264C | LIS_TUB_034 | F/6.5 | central pachygyria | −4 SD | hypogenetic | normal cerebellum |
| [20] | R264C | LIS_TUB_035 | M/6 | central pachygyria | −3 SD | normal | mild vermis hypoplasia |
| [20] | R264H | LIS_TUB_002_foetus20 | F/24 GW | lissencephaly | microcephaly | complete agenesis | severe vermis hypoplasia |
| [21] | R264H | LIS_TUB_002_foetus20 | F/24 GW | absent cortical plate, 2 layered | <3rd centile | complete agenesis | moderate hypoplasia of cerebellum, severe pons hypoplasia |
| [139] | A270S | not reported | M/19 mo | mild posterior simplified cerebral gyral pattern | microcephaly | agenesis | severe hypoplastic cerebellar vermis, mildly dysplastic and hypoplastic cerebellar hemispheres, mildly hypoplastic brainstem, dysplastic basal ganglia, thalami, hypoplastic optic nerves, absent olfactory bulbs, lateral and third ventricle dilated |
| [14] | L286F | LIS_TUB_007_foetus04 | fetus | two-layered cortex, poorly differentiated | <3rd centile | complete agenesis | absent hippocampus, olfactory bulb, and internal capsule, hypoplastic brainstem and corticospinal tract |
| [17] | V303G | LIS_TUB_006_foetus03 | fetus | pachygyria | microcephaly | short and thin | thin brainstem, pons and medulla flattened, hypoplastic cerebellum and corticospinal tracts |
| [20] | R320H | LIS_TUB_005_foetus01 | M/25 GW | lissencephaly | microcephaly | partial agenesis | severe vermis and hemispheric dysplasia |
| [21] | R320H | LIS_TUB_005_foetus01 | M/25 GW | absent cortical plate | <3rd centile | partial agenesis | severe hypoplasia and dysplasia of cerebellum, severe hypoplasia (neuronal over-migration) spinal cord anterior horn hypoplasia |
| [21] | R320H | LIS_TUB_081_foetus26 | M/26 GW | absent cortical plate, 2 layered | <3rd centile | complete agenesis | severe hypoplasia and dysplasia of cerebellum, severe hypoplasia of pons |
| [20] | K326N | LIS_TUB_004_foetus08 | M/23 GW | lissencephaly | microcephaly | partial agenesis | severe vermis and hemispheric dysplasia |
| [21] | K326N | LIS_TUB_004_foetus08 | M/23 GW | thick 2-layered cortex | <3rd centile | complete agenesis | hypoplastic basal ganglia, severe hypoplasia and dysplasia of cerebellum, severe pons hypoplasia |
| [16] | N329S | LR05-388 | Not reported | lissencephaly with cerebellar hypoplasia | microcephaly | absent | thin brainstem, hypoplasia of cerebellar vermis |
| [20] | V353I | LIS_TUB_059 | M/4 | simplified gyral pattern with focal polymicrogyria | −2 SD | partial agenesis | normal |
| [20] | V371E | LIS_TUB_080_foetus24 | F/23.3 GW | lissencephaly | <3rd centile | complete agenesis | severe vermis hypoplasia |
| [21] | V371E | LIS_TUB_080_foetus24 | F/23.3 GW | 2–3 layered cortex, poorly differentiated | <3rd centile | complete agenesis | hypoplastic basal ganglia, severe hypoplasia of cerebellum, severe pons hypoplasia |
| [140] | A387V | Not reported | F/5 | pachygyria with SBH (subcortical band heterotopia) | <3rd centile | thin | simplified hippocampus, highly dysmorphic brainstem, flattened pons, mildly hypoplastic cerebellar vermis |
| [18] | R390C | LIS_TUB_045 | M/1 | asymmetrical perisylvian polymicrogyria | microcephaly | severe hypoplasia | dysmorphic basal ganglia, dysplastic vermis, severe hypoplasia of brainstem |
| [20] | A369T | LIS_TUB_030 | M/11 | central pachygyria | −2 SD | dysmorphic, hypoplastic | mild vermis hypoplasia |
| [20] | D396Y | LIS_TUB_078 | F/4 | central polymicrogyria-like cortical dysplasia | −3 SD | partial agenesis | severe vermis dysplasia |
| [13] | L397P | LIS_TUB_039 | M/5.5 | perisylvian pachygyria | −4 SD | posterior agenesis | severe vermis dysplasia, severe dysgenesis of internal capsule |
| [15] | R402C | FR04-D4148 | F/11 | lissencephaly | −4 SD | thin, rostrum absent, splenium hypoplastic | mild vermis and pons hypoplasia, trigones and occipital horns dilated |
| [20] | R402C | LIS_TUB_019 | M/10 | moderate lissencephaly | −3 SD | dysmorphic, hypoplastic | mild vermis hypoplasia |
| [12] | R402H | LIS_TUB_023 | M/11 | lissencephaly | −3 SD | thin, partial agenesis | abnormal hippocampus, vermis and brainstem hypoplasia, severe ventricular dilation |
| [20] | R402H | LIS_TUB_014 | M/4 | severe lissencephaly | −4 SD | hypogenetic | severe hemispheric hypoplasia |
| [20] | R402H | LIS_TUB_015 | M/1 | severe lissencephaly | −4 SD | hypogenetic | severe hemispheric hypoplasia |
| [20] | R402H | LIS_TUB_016 | F/1.5 | severe lissencephaly | −3 SD | hypogenetic | vermis dysplasia |
| [141] | R402H | not reported | M/1 | lissencephaly | microcephaly | severe dysplasia of brainstem and cerebellum | |
| [15] | R402L | RE07-S1605 | M/3 | lissencephaly posterior, pachygyria anterior | −3.5 SD | thin | mild vermis and pons hypoplasia, retrocerebellar cyst, abnormal hippocampus, dilated lateral ventricles, occipital horns, anterior horns |
| [142] | R402L | Not reported | F/1 | lissencephaly posterior, pachygyria anterior | −4 SD | mild hypoplasia | severe hypoplasia of cerebellum, mild hypoplasia of brainstem |
| [21] | V409A | LIS_TUB_011_foetus23 | M/32 GW | not available | 5th centile | complete agenesis | severe hypoplasia of cerebellum and pons |
| [20] | V409I | LIS_TUB_032 | M/10 | central pachygyria | −2 SD | dysmorphic, hypoplastic | normal cerebellum |
| [13] | R422C | LIS_TUB_042 | F/4.5 | perisylvian pachygyria | −3 SD | mild hypoplasia | mild vermis hypoplasia, severe dysgenesis of internal capsule |
| [13] | R422H | LIS_TUB_020 | F/7 | posterior pachygyria | −4 SD | mild hypoplasia | mild vermis hypoplasia, moderate dysgenesis of internal capsule |
| [15] | R422H | FR05-D4607 | M/9 | pachygyria with SBH | −4 SD | partial agenesis | severe vermis hypoplasia, dandy-walker malformation, hypoplasia of pons, abnormal hippocampus |
| [15] | R422H | FR07-D5526 | F/.5 | pachygyria with SBH | −3.5 SD | partial agenesis | moderate vermis hypoplasia, mild pons hypoplasia, abnormal hippocampus |
| [16] | M425K | LR08-388 | Not reported | lissencephaly with cerebellar hypoplasia | microcephaly | absent | thin brainstem, hypoplasia of cerebellar vermis |
| [20] | E429Q | LIS_TUB_001_foetus09 | F/25 GW | lissencephaly | microcephaly | complete agenesis | severe vermis hypoplasia |
| [21] | E429Q | LIS_TUB_004_foetus09 | F/25 GW | 4 layered cortex | <3rd centile | complete agenesis | hypoplastic basal ganglia, severe hypoplasia of cerebellum and pons |
| [13] | G436R | LIS_TUB_038 | M/7 | perisylvian pachygyria | −3 SD | mild hypoplasia | mild vermis hypoplasia, severe dysgenesis of internal capsule |
| Developmental Time Point | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| E9.5 | E10.5 | E11.5 | E12 | E12.5 | E13.5 | E14.5 | E16.5 | |||
| Developing CNS | brain | brain, unspecified | [159] | [156] | [156] | |||||
| forebrain | [158] | |||||||||
| midbrain | [158] | |||||||||
| hindbrain | [158] | |||||||||
| telencephalon | [12] | |||||||||
| diencephalon | [156] | [156] | [156] | [12] | ||||||
| mesencephalon | [12] | |||||||||
| metencephalon | [156] | [156] | [156] | [12] | ||||||
| myelencephalon | [156] | [156] | [156] | [12] | ||||||
| developing neocortex | [158] | [156] | [156] | [12] | ||||||
| developing striatum | [159] | |||||||||
| developing hippocampus | [159] | |||||||||
| developing thalamus | [159] | |||||||||
| developing amygdala | [159] | |||||||||
| developing hypothalamus | [159] | |||||||||
| developing cerebellum | [159] | [12] | ||||||||
| developing brain stem | [12] | |||||||||
| spinal cord | [158] | [156] | [156] | [159] | [156] | |||||
| retina | [158] | [156] | ||||||||
| Developing PNS | cranial nerves | cranial ganglia | [156] | [156] | ||||||
| trigeminal ganglion | [158] | |||||||||
| fascioacoustic ganglion | [158] | |||||||||
| glossopharyngeal ganglia | [158] | |||||||||
| developing olfactory bulbs | [156] | [159] | [156] | |||||||
| nasal epithelium | [158] | |||||||||
| vomeronasal organ | [158] | |||||||||
| dorsal root ganglia | [158] | [156] | [156] | [12] | ||||||
| sensory ganglia | [156] | [156] | ||||||||
| ANS | sympathetic ganglia | [158] | [156] | [156] | ||||||
| parasympathetic ganglia | [156] | [156] | ||||||||
| heart | [158] | |||||||||
| Postnatal Time Point | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| P0 | P3 | P6 | P10 | P15 | P22 | P32 | Adult | |||
| CNS | brain | brain, unspecified | [10] | [10] | [10] | [10] | [10] | [10] | [10,157] | |
| olfactory system | [159] | [157,159] | ||||||||
| cortex | [159] | [12] | [157] | |||||||
| corpus callosum | [159] | [159] | ||||||||
| striatum | [159] | |||||||||
| rostral migratory stream | [159] | [12] | ||||||||
| basal ganglia | [157] | |||||||||
| basal forebrain | [157] | |||||||||
| hippocampus | [159] | [12] | [157,159] | |||||||
| amygdala | [159] | [157,159] | ||||||||
| hypothalamus | [159] | [157,159] | ||||||||
| thalamus | [159] | [157,159] | ||||||||
| subthalamus | [157] | |||||||||
| midbrain | [159] | [157,159] | ||||||||
| pons | [157] | |||||||||
| medulla | [157] | |||||||||
| cerebellum | [159] | [12] | [157] | |||||||
| brainstem | [12] | [157] | ||||||||
| spinal cord | [159] | [159] | ||||||||
| Other | lung | [10] | [10] | [10] | [10] | [10] | [10] | [10] | ||
| testes | [10] | [10] | [10] | |||||||
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aiken, J.; Buscaglia, G.; Bates, E.A.; Moore, J.K. The α-Tubulin gene TUBA1A in Brain Development: A Key Ingredient in the Neuronal Isotype Blend. J. Dev. Biol. 2017, 5, 8. https://doi.org/10.3390/jdb5030008
Aiken J, Buscaglia G, Bates EA, Moore JK. The α-Tubulin gene TUBA1A in Brain Development: A Key Ingredient in the Neuronal Isotype Blend. Journal of Developmental Biology. 2017; 5(3):8. https://doi.org/10.3390/jdb5030008
Chicago/Turabian StyleAiken, Jayne, Georgia Buscaglia, Emily A. Bates, and Jeffrey K. Moore. 2017. "The α-Tubulin gene TUBA1A in Brain Development: A Key Ingredient in the Neuronal Isotype Blend" Journal of Developmental Biology 5, no. 3: 8. https://doi.org/10.3390/jdb5030008
APA StyleAiken, J., Buscaglia, G., Bates, E. A., & Moore, J. K. (2017). The α-Tubulin gene TUBA1A in Brain Development: A Key Ingredient in the Neuronal Isotype Blend. Journal of Developmental Biology, 5(3), 8. https://doi.org/10.3390/jdb5030008