
Transcriptome and Metabolite Insights into Domestication Process of Cultivated Barley in China



Abstract
:1. Introduction
2. Results
2.1. Both Cb-C and Wb-T Are the Descendants of Wb-NE
2.2. Wb-T Contributed More to Cb-C Than Wb-NE
2.3. Wb-T Was the Product of Feralization or Hybridization of Cultivated Barley
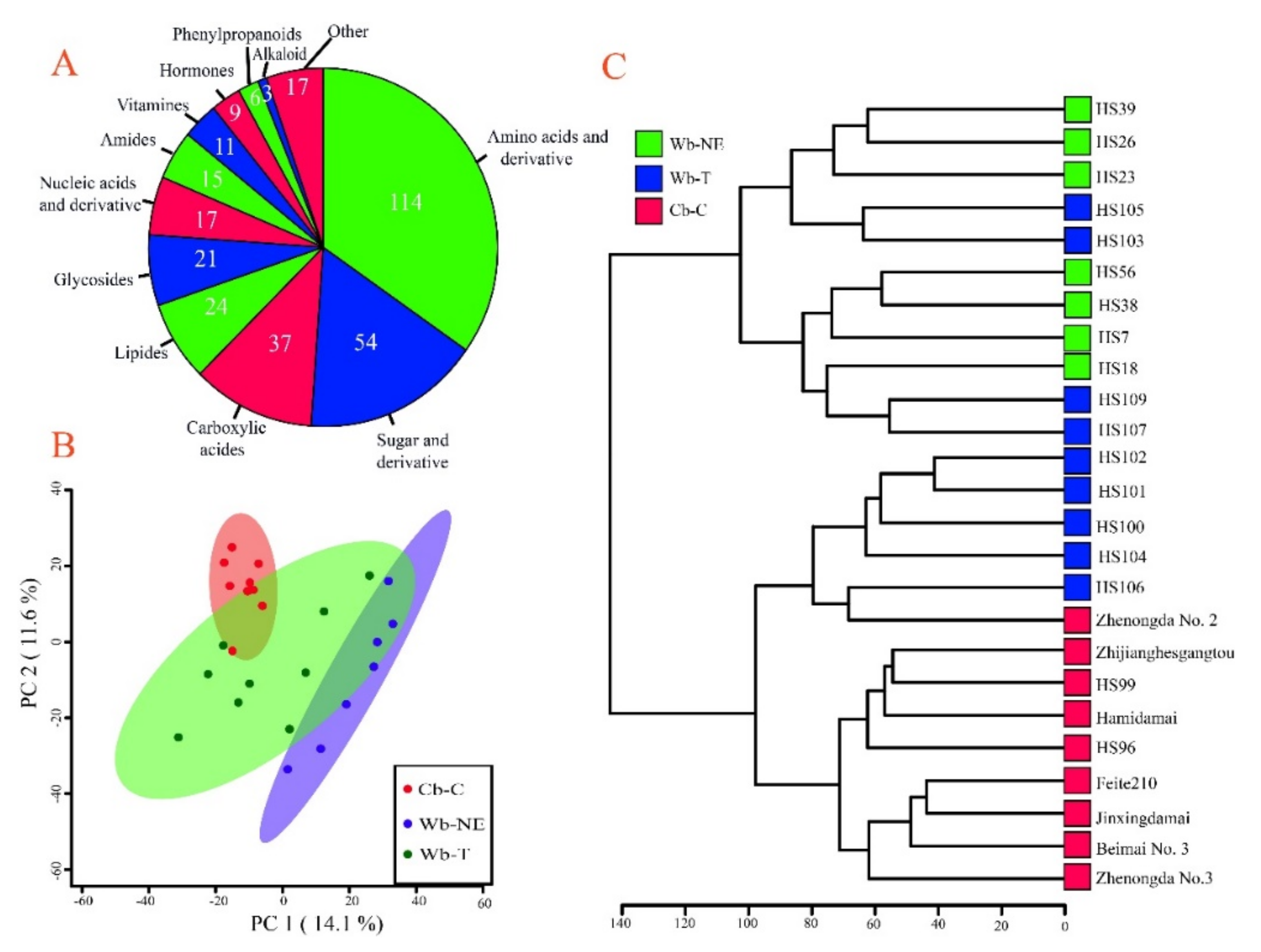
2.4. Different Evolutionary Stages Were Accompanied by Different Sets of Divergent Metabolites
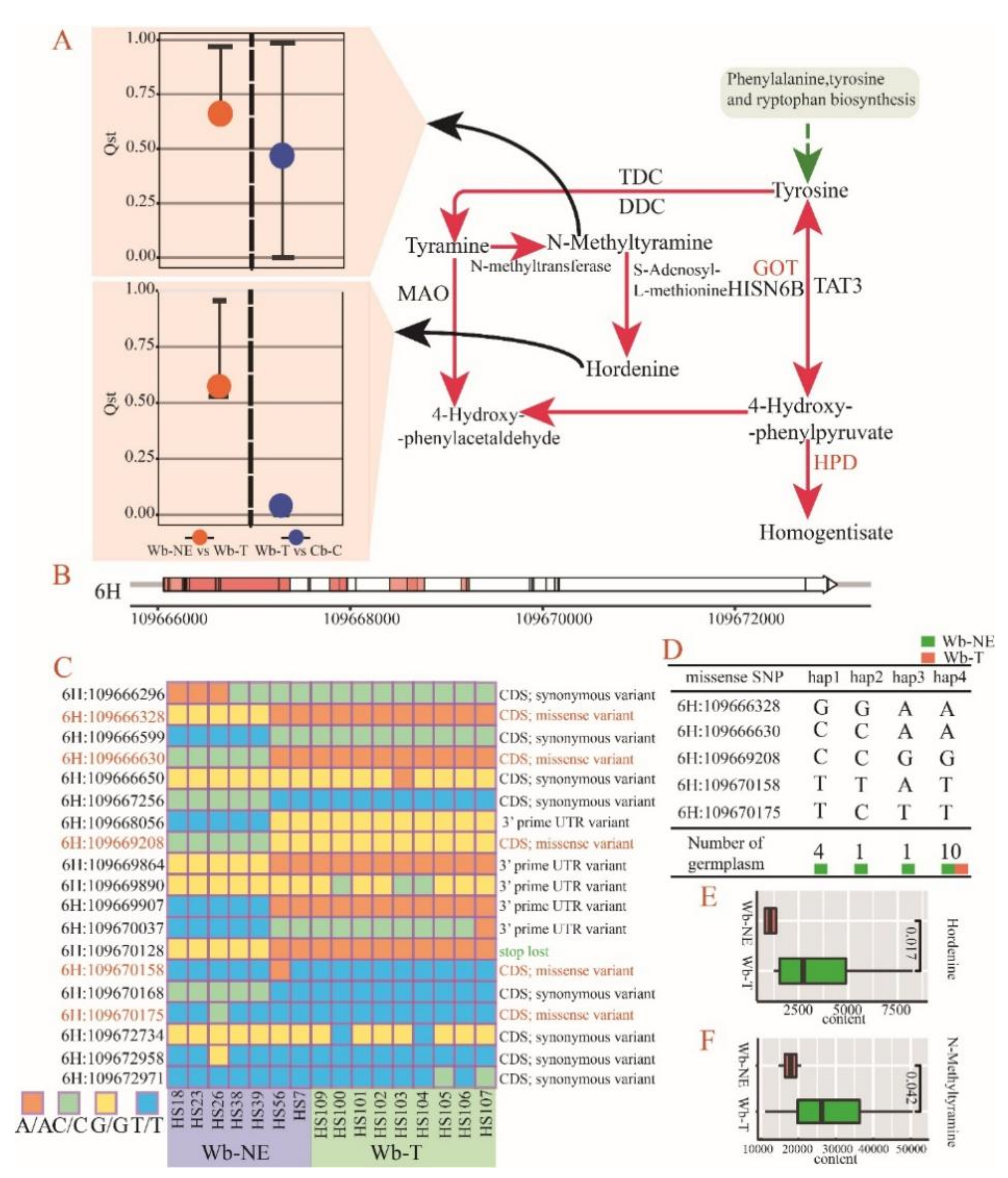
2.5. Positive Selective Genes and SNPs Influencing the Metabolic Divergence
3. Discussion
3.1. Origin and Domestication Process of Cultivated Barley in China
3.2. Metabolic Divergence in the Domestication Process of Cultivated Barley in China
4. Materials and Methods
4.1. Plant Growth Conditions
4.2. RNA Sequencing and Single Nucleotide Polymorphisms (SNPs) Calling
4.3. Genetic Diversity Analysis
4.4. PCA and Population Structure Inference Based on SNPs of RNA-Seq
4.5. Genomic Similarity Analysis
4.6. Demographic History, Migration Event, and Haplotype Detection
4.7. Extraction and Detection of Untargeted Metabolites
4.8. PCA and Hierarchical Clustering Analysis Based on Metabolites
4.9. Qst–Fst Comparison
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Harlan, J.R.; Zohary, D. Distribution of wild wheats and barley. Science 1966, 153, 1074–1080. [Google Scholar] [CrossRef] [PubMed]
- Nevo, E. Evolution of wild barley and barley improvement. In Advance in Barley Sciences, Proceedings of the 11th International Barley Genetics Symposium, Hangzhou, China, 15–20 April 2012; Springer: Berlin/Heidelberg, Germany, 2013; pp. 1–16. [Google Scholar]
- Xu, T.W. On the origin and phylogeny of cultivated barley with preference to the discovery of Ganze wild two-rowed barley Hordeum spontaneum c. Koch. Acta. Genet. Sin. 1975, 2, 129–137. [Google Scholar]
- Xu, T.W. Origin and evolution of cultivated barley in China. Acta. Genet. Sin. 1982, 9, 440–446. [Google Scholar]
- Zohary, D.; Hopf, M.; Weiss, E. Domestication of Plants in the Old World: The Origin and Spread of Domesticated Plants in Southwest Asia, Europe, and the Mediterranean Basin, 4th ed.; Oxford University Press: Oxford, UK, 2012. [Google Scholar]
- Dai, F.; Nevo, E.; Wu, D.Z.; Comadran, J.; Zhou, M.X.; Qiu, L.; Chen, Z.; Beiles, A.; Chen, G.; Zhang, G. Tibet is one of the centers of domestication of cultivated barley. Proc. Natl. Acad. Sci. USA 2012, 109, 16969–16973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, X.F.; Nevo, E.; Sun, D.F.; Sun, G.L. Tibet as a potential domestication center of cultivated barley of China. PLoS ONE 2013, 8, e62700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Åberg, E. Hordeum agriocrithon nova sp., a wild six-rowed barley. Ann. Agric. Coll. Swed. 1938, 6, 159–212. [Google Scholar]
- Ma, D.Q.; Xu, T.W.; Gu, M.Z.; Wu, S.B.; Kang, Y.C. The classification and distribution of wild barley in the Tibet Autonomous Region. Sci. Agric. Sin. 1987, 20, 1–6. [Google Scholar]
- Ma, D.Q.; Xu, T.W. The research on classification and origin of cultivated barley in Tibet Autonomous Region. Sci. Agric. Sin. 1988, 21, 7–14. [Google Scholar] [CrossRef]
- Zeng, X.; Guo, Y.; Xu, Q.; Mascher, M.; Guo, G.; Li, S.; Mao, L.; Liu, Q.; Xia, Z.; Zhou, J.; et al. Origin and evolution of qingke barley in Tibet. Nat. Commun. 2018, 9, 5433–5445. [Google Scholar] [CrossRef]
- Pourkheirandish, M.; Kanamori, H.; Wu, J.; Sakuma, S.; Blattner, F.R.; Komatsuda, T. Elucidation of the origin of “agriocrithon” based on domestication genes questions the hypothesis that Tibet is one of the centers of barley domestication. Plant J. 2018, 94, 525–534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Civáň, P.; Drosou, K.; Armisen-Gimenez, D.; Duchemin, W.; Brown, T.A. Episodes of gene flow and selection during the evolutionary history of domesticated barley. BMC Genom. 2021, 22, 1–17. [Google Scholar] [CrossRef]
- Badr, A.; Müller, K.; Schäfer-Pregl, R.; El Rabey, H.; Effgen, S.; Ibrahim, H.; Pozzi, C.; Rohde, W.; Salamini, F. On the origin and domestication history of barley (Hordeum vulgare). Mol. Biol. Evol. 2000, 17, 499–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hicks, T. Genetic Diversity of the DREB1 Gene in Hordeum vulgare and Hordeum spontaneum. Ph.D. Dissertation, Saint Mary’s University, Halifax, NS, Canada, 2019. [Google Scholar]
- Bonchev, G.; Dušinský, R.; Hauptvogel, P.; Gaplovská-Kyselá, K.; Švec, M. On the diversity and origin of the barley complex agriocrithon inferred by iPBS transposon markers. Genet. Resour. Crop Evol. 2019, 66, 1573–1586. [Google Scholar] [CrossRef]
- Konishi, T. Genetic diversity in Hordeum agriocrithon E. Åberg, six-rowed barley with brittle rachis, from Tibet. Genet. Resour. Crop Evol. 2001, 48, 27–34. [Google Scholar] [CrossRef]
- Tanno, K.; Takeda, K. On the origin of six-rowed barley with brittle rachis, agriocrithon [Hordeum vulgare ssp. vulgare f. agriocrithon (Åberg) Bowd.], based on a DNA marker closely linked to the vrs1 (six-row gene) locus. Theor. Appl. Genet. 2004, 110, 145–150. [Google Scholar] [CrossRef]
- Badr, A.; El-Shazly, H. Molecular approaches to origin, ancestry and domestication history of crop plants: Barley and clover as examples. J. Genet. Eng. Biotechnol. 2012, 10, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Morrell, P.L.; Clegg, M.T. Genetic evidence for a second domestication of barley (Hordeum vulgare) east of the Fertile Crescent. Proc. Natl. Acad. Sci. USA 2007, 104, 3289–3294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morrell, P.L.; Lundy, K.E.; Clegg, M.T. Distinct geographic patterns of genetic diversity are maintained in wild barley (Hordeum vulgare ssp. spontaneum) despite migration. Proc. Natl. Acad. Sci. USA 2003, 100, 10812–10817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pourkheirandish, M.; Hensel, G.; Kilian, B.; Senthil, N.; Chen, G.; Sameri, M.; Azhaguvel, P.; Sakuma, S.; Dhanagond, S.; Sharma, R.; et al. Evolution of the grain dispersal system in barley. Cell 2015, 162, 527–539. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.L.; Ye, H.; Liu, L.; Wu, J.H.; Ru, W.M.; Sun, G.L. Molecular insights on the domestication of barley (Hordeum vulgare L.). Crit. Rev. Plant Sci. 2019, 38, 280–294. [Google Scholar] [CrossRef]
- Shang, Y.; Ma, Y.; Zhou, Y.; Zhang, H.; Duan, L.; Chen, H.; Zeng, J.; Zhou, Q.; Wang, S.; Gu, W.; et al. Biosynthesis, regulation, and domestication of bitterness in cucumber. Science 2014, 346, 1084–1088. [Google Scholar] [CrossRef] [PubMed]
- Beleggia, R.; Rau, D.; Laidò, G.; Platani, C.; Nigro, F.; Fragasso, M.; De Vita, P.; Scossa, F.; Fernie, A.R.; Nikoloski, Z.; et al. Evolutionary metabolomics reveals domestication-associated changes in tetraploid wheat kernels. Mol. Biol. Evol. 2016, 33, 1740–1753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Ma, Y.; Zeng, J.; Duan, L.; Xue, X.; Wang, H.; Lin, T.; Liu, Z.; Zeng, K.; Zhong, Y.; et al. Convergence and divergence of bitterness biosynthesis and regulation in Cucurbitaceae. Nat. Plants 2016, 2, 16183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, G.; Wang, S.; Huang, Z.; Zhang, S.; Liao, Q.; Zhang, C.; Lin, T.; Qin, M.; Peng, M.; Yang, C.; et al. Rewiring of the fruit metabolome in tomato breeding. Cell 2018, 172, 249–261. [Google Scholar] [CrossRef] [Green Version]
- Xu, G.H.; Cao, J.J.; Wang, X.F.; Chen, Q.Y.; Li, Z.; Tian, F. Evolutionary metabolomics identifies substantial metabolic divergence between maize and its wild ancestor, teosinte. Plant Cell 2019, 31, 1990–2009. [Google Scholar] [CrossRef] [Green Version]
- Durand, E.Y.; Patterson, N.; Reich, D.; Slatkin, M. Testing for ancient admixture between closely related populations. Mol. Biol. Evol. 2011, 28, 2239–2252. [Google Scholar] [CrossRef] [Green Version]
- Purugganan, M.D.; Fuller, D.Q. The nature of selection during plant domestication. Nature 2009, 457, 843–848. [Google Scholar] [CrossRef]
- Riehl, S.; Zeidi, M.; Conard, N.J. Emergence of agriculture in the foothills of the Zagros Mountains of Iran. Science 2013, 341, 65–67. [Google Scholar] [CrossRef]
- Pankin, A.; Altmüller, J.; Becker, C.; Von Korff, M. Targeted resequencing reveals genomic signatures of barley domestication. New Phytol. 2018, 218, 1247–1259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pankin, A.; Von Korff, M. Co-evolution of methods and thoughts in cereal domestication studies: A tale of barley (Hordeum vulgare). Curr. Opin. Plant Biol. 2017, 36, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Harlan, J.R. On the origin of barley: A second look. In International Barley Genetics Symposium Proceedings; Washington State University Press: Pullman, WA, USA, 1971; pp. 45–49. [Google Scholar]
- Harlan, J.R. Barley: Hordeum vulgare. In Evolution of Crop Plants; Simmonds, N.W., Ed.; Academic Press: New York, NY, USA, 1976; pp. 93–98. [Google Scholar]
- Zhang, Q.; Yang, G.P.; Dai, X.; Sun, J.Z. A comparative analysis of genetic polymorphism in wild and cultivated barley from Tibet using isozyme and ribosomal DNA markers. Genome 1994, 37, 631–638. [Google Scholar] [CrossRef]
- Feng, Z.Y.; Liu, X.J.; Zhang, Y.Z.; Ling, H.Q. Genetic diversity analysis of Tibetan wild barley using SSR markers. Acta. Genet. Sin. 2006, 33, 917–928. [Google Scholar] [CrossRef]
- Dai, F.; Chen, Z.-H.; Wang, X.; Li, Z.; Jin, G.; Wu, D.; Cai, S.; Wang, N.; Wu, F.; Nevo, E.; et al. Transcriptome profiling reveals mosaic genomic origins of modern cultivated barley. Proc. Natl. Acad. Sci. USA 2014, 111, 13403–13408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, W.; Xin, M.; Wang, Z.; Yao, Y.; Hu, Z.; Song, W.; Yu, K.; Chen, Y.; Wang, X.; Guan, P.; et al. Origin and adaptation to high altitude of Tibetan semi-wild wheat. Nat. Commun. 2020, 11, 5085. [Google Scholar] [CrossRef]
- Qiu, J.; Jia, L.; Wu, D.; Weng, X.; Chen, L.; Sun, J.; Chen, M.; Mao, L.; Jiang, B.; Ye, C.; et al. Diverse genetic mechanisms underlie worldwide convergent rice feralization. Genome Biol. 2020, 21, 70. [Google Scholar] [CrossRef]
- Jones, H.; Leigh, F.J.; Mackay, I.; Bower, M.A.; Smith, L.M.J.; Charles, M.P.; Jones, G.; Jones, M.K.; Brown, T.A.; Powell, W. Population based re-sequencing reveals that the flowering time adaptation of cultivated barley originated east of the Fertile Crescent. Mol. Biol. Evol. 2008, 25, 2211–2219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poets, A.M.; Mohammadi, M.; Seth, K.; Wang, H.; Kono, T.J.Y.; Fang, Z.; Muehlbauer, G.J.; Smith, K.P.; Morrell, P.L. The effects of both recent and long-term selection and genetic drift are readily evident in North American barley breeding populations. G3-Genes Genom. Genet. 2016, 6, 609–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russell, J.; Mascher, M.; Dawson, I.K.; Kyriakidis, S.; Calixto, C.; Freund, F.; Bayer, M.; Milne, I.; Marshall-Griffiths, T.; Heinen, S.; et al. Exome sequencing of geographically diverse barley landraces and wild relatives gives insights into environmental adaptation. Nat. Genet. 2016, 48, 1024–1030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Norsang, G.; Kocbach, L.; Tsoja, W.; Stamnes, J.J.; Dahlback, A.; Nema, P. Ground-based measurements and modeling of solar UV-B radiation in Lhasa. Tibet Atmos. Environ. 2009, 43, 1498–1502. [Google Scholar] [CrossRef]
- Zhang, R.-B.; Yuan, Y.-J.; Wei, W.-S.; Gou, X.-H.; Yu, S.-L.; Shang, H.-M.; Chen, F.; Zhang, T.-W.; Qin, L. Dendroclimatic reconstruction of autumn-winter mean minimum temperature in the eastern Tibetan Plateau since 1600 AD. Dendrochronologia 2015, 33, 1–7. [Google Scholar] [CrossRef]
- Ferl, R.J.; Schuerger, A.C.; Paul, A.L.; Gurley, W.B.; Corey, K.; Bucklin, R. Plant adaptation to low atmospheric pressures: Potential molecular responses. Life Support Biosph. Sci. 2002, 8, 93–101. [Google Scholar]
- Liu, D.L.; Lovett, J.V. Biologically active secondary metabolites of barley. I. Developing techniques and assessing allelopathy in barley. J. Chem. Ecol. 1993, 19, 2217–2230. [Google Scholar] [CrossRef] [PubMed]
- Corcuera, L.J.; Argandona, V.H.; Zúiga, G.E. Allelochemicals in wheat and barley: Role in plant-insect interactions. In Allelopathy: Basic and Applied Aspects; Springer: Amsterdam, The Netherlands, 1992; pp. 119–127. [Google Scholar] [CrossRef]
- Lovett, J.V.; Hoult, A.H.C. Allelopathy and self-defense in barley. In Allelopathy; ACS Symposium Series; American Chemical Society: Washington, DC, USA, 1994; pp. 170–183. [Google Scholar]
- Liu, J.; Osbourn, A.; Ma, P. MYB transcription factors as regulators of phenylpropanoid metabolism in plants. Mol. Plant 2015, 8, 689–708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, X.; Yuan, H.; Dong, X.; Peng, M.; Jing, X.; Xu, Q.; Tang, T.; Wang, Y.; Zha, S.; Gao, M.; et al. Genome-wide dissection of co-selected UV-B responsive pathways in the UV-B adaptation of qingke. Mol. Plant 2019, 13, 112–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Markham, K.R.; Ryan, K.G.; Bloor, S.J.; Mitchell, K.A. An increase in the luteolin: Apigenin ratio in Marchantia polymorpha on UV-B enhancement. Phytochemistry 1998, 48, 791–804. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef] [Green Version]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef] [PubMed]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; De Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Tamura, K.; Nei, M. MEGA: Molecular evolutionary genetics analysis software for microcomputers. Comput. Appl. Biosci. 1994, 10, 189–191. [Google Scholar] [CrossRef] [Green Version]
- Mascher, M.; Richmond, T.A.; Gerhardt, D.J.; Himmelbach, A.; Clissold, L.; Sampath, D.; Ayling, S.; Steuernagel, B.; Pfeifer, M.; D’Ascenzo, M.; et al. Barley whole exome capture: A tool for genomic research in the genus Hordeum and beyond. Plant J. 2013, 76, 494–505. [Google Scholar] [CrossRef] [Green Version]
- Rubin, C.-J.; Zody, M.C.; Eriksson, J.; Meadows, J.R.S.; Sherwood, E.; Webster, M.T.; Jiang, L.; Ingman, M.; Sharpe, T.; Ka, S.; et al. Whole-genome resequencing reveals loci under selection during chicken domestication. Nature 2010, 464, 587–591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, Z.; Gu, L.; Eils, R.; Schlesner, M.; Brors, B. Circlize implements and enhances circular visualization in R. Bioinformatics 2014, 30, 2811–2812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terhorst, J.; Kamm, J.A.; Song, Y.S. Robust and scalable inference of population history from hundreds of unphased whole genomes. Nat. Genet. 2017, 49, 303–309. [Google Scholar] [CrossRef] [PubMed]
- Pickrell, J.K.; Pritchard, J.K. Inference of population splits and mixtures from genome-wide allele frequency data. PLoS Genet. 2012, 8, e1002967. [Google Scholar] [CrossRef] [Green Version]
- Leinonen, T.; McCairns, R.J.S.; O’Hara, R.B.; Merilä, J. Qst—Fst comparisons: Evolutionary and ecological insights from genomic heterogeneity. Nat. Rev. Genet. 2013, 14, 179–190. [Google Scholar] [CrossRef]
- Excoffier, L.; Lischer, H.E.L. Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 2010, 10, 564–567. [Google Scholar] [CrossRef]
- Evans, L.M.; Slavov, G.T.; Rodgers-Melnick, E.; Martin, J.; Ranjan, P.; Muchero, W.; Brunner, A.M.; Schackwitz, W.; Gunter, L.; Chen, J.-G.; et al. Population genomics of Populus trichocarpa identifies signatures of selection and adaptive trait associations. Nat. Genet. 2014, 46, 1089–1096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malinsky, M.; Matschiner, M.; Svardal, H. Dsuite—Fast D-statistics and related admixture evidence from VCF files. Mol. Ecol. Resour. 2020, 21, 584–595. [Google Scholar] [CrossRef] [PubMed]
- Yin, C.B.; Li, H.H.; Li, S.S.; Xu, L.D.; Zhao, Z.G.; Wang, J.K. Genetic dissection on rice grain shape by the two-dimensional image analysis in one japonica × indica population consisting of recombinant inbred lines. Theor. Appl. Genet. 2015, 128, 1969–1986. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | π | θW | MAF | PIC |
|---|---|---|---|---|
| Wb-NE | 0.23036 | 0.21255 | 0.1745 | 0.2176 |
| Wb-T | 0.18906 | 0.15944 | 0.1453 | 0.1796 |
| Cb-C | 0.11034 | 0.10183 | 0.1120 | 0.1048 |
| Gene ID | KEGG ID | Function |
|---|---|---|
| HORVU1Hr1G010130 | K00001 | alcohol dehydrogenase |
| HORVU4Hr1G013370 | K01557 | FAHD1, acylpyruvate hydrolase |
| HORVU3Hr1G073220 | K14454 | GOT1, aspartate aminotransferase |
| HORVU6Hr1G027650 | K00457 | HPD, 4-hydroxyphenylpyruvate dioxygenase |
| Gene ID | KEGG ID | Function |
|---|---|---|
| HORVU3Hr1G080830 | K00487 | C4H, CYP73A, trans-cinnamate 4-monooxygenase |
| HORVU4Hr1G072150 | K01904 | 4CL, 4-coumarate-CoA ligase |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, Y.; Lu, G.; Sun, G.; Sun, D.; Ren, X. Transcriptome and Metabolite Insights into Domestication Process of Cultivated Barley in China. Plants 2022, 11, 209. https://doi.org/10.3390/plants11020209
Zhou Y, Lu G, Sun G, Sun D, Ren X. Transcriptome and Metabolite Insights into Domestication Process of Cultivated Barley in China. Plants. 2022; 11(2):209. https://doi.org/10.3390/plants11020209
Chicago/Turabian StyleZhou, Yu, Guang Lu, Genlou Sun, Daokun Sun, and Xifeng Ren. 2022. "Transcriptome and Metabolite Insights into Domestication Process of Cultivated Barley in China" Plants 11, no. 2: 209. https://doi.org/10.3390/plants11020209
APA StyleZhou, Y., Lu, G., Sun, G., Sun, D., & Ren, X. (2022). Transcriptome and Metabolite Insights into Domestication Process of Cultivated Barley in China. Plants, 11(2), 209. https://doi.org/10.3390/plants11020209