

Phytochemical Screening and Isolation of New Ent-Clerodane Diterpenoids from Croton guatemalensis Lotsy

 ,
,  , and
, and
Abstract
:1. Introduction
2. Results and Discussion
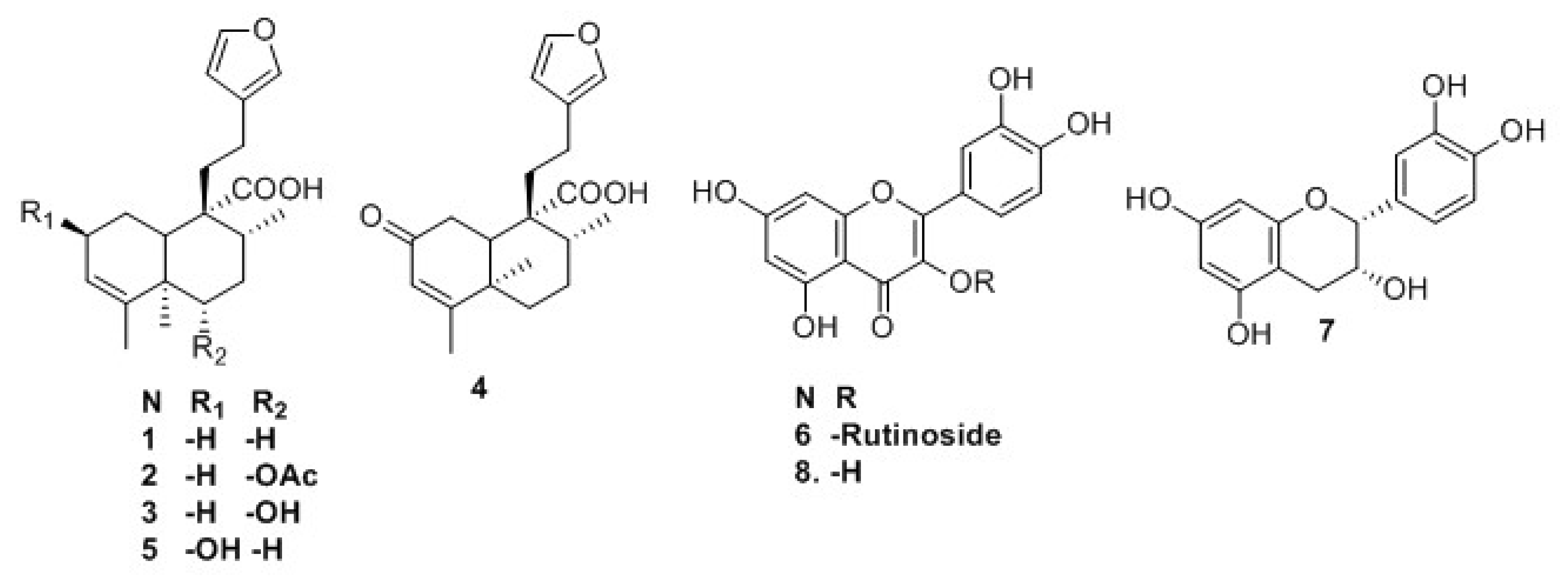
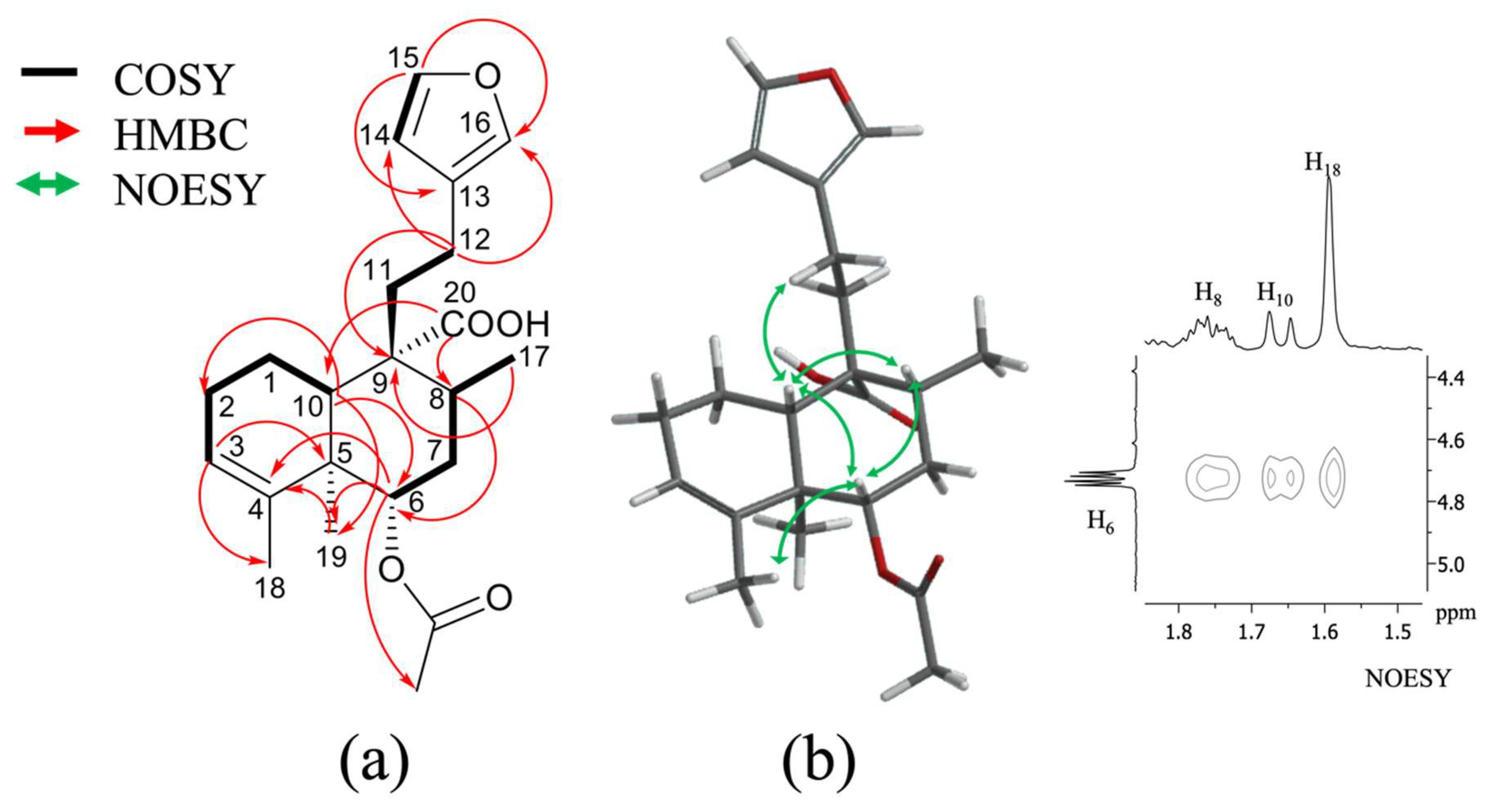
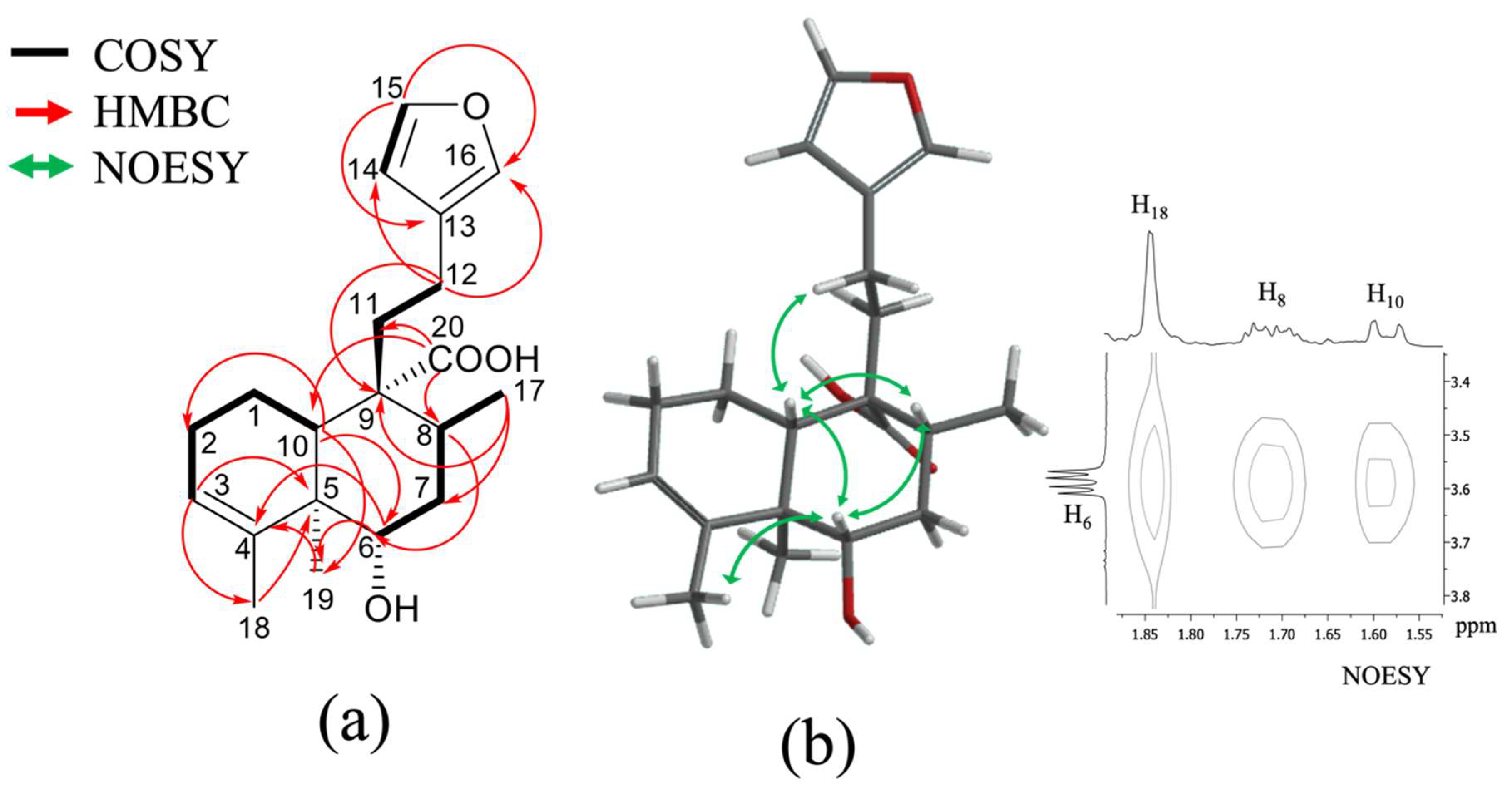
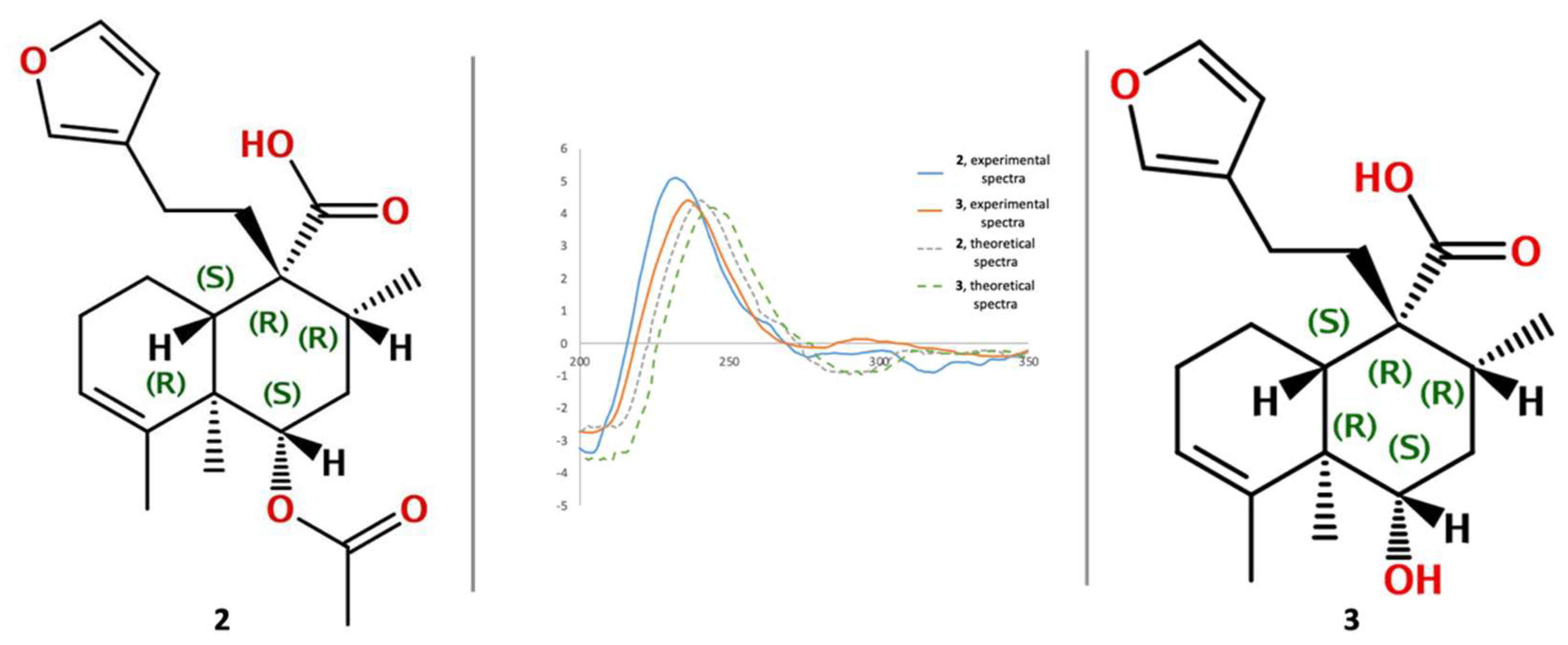
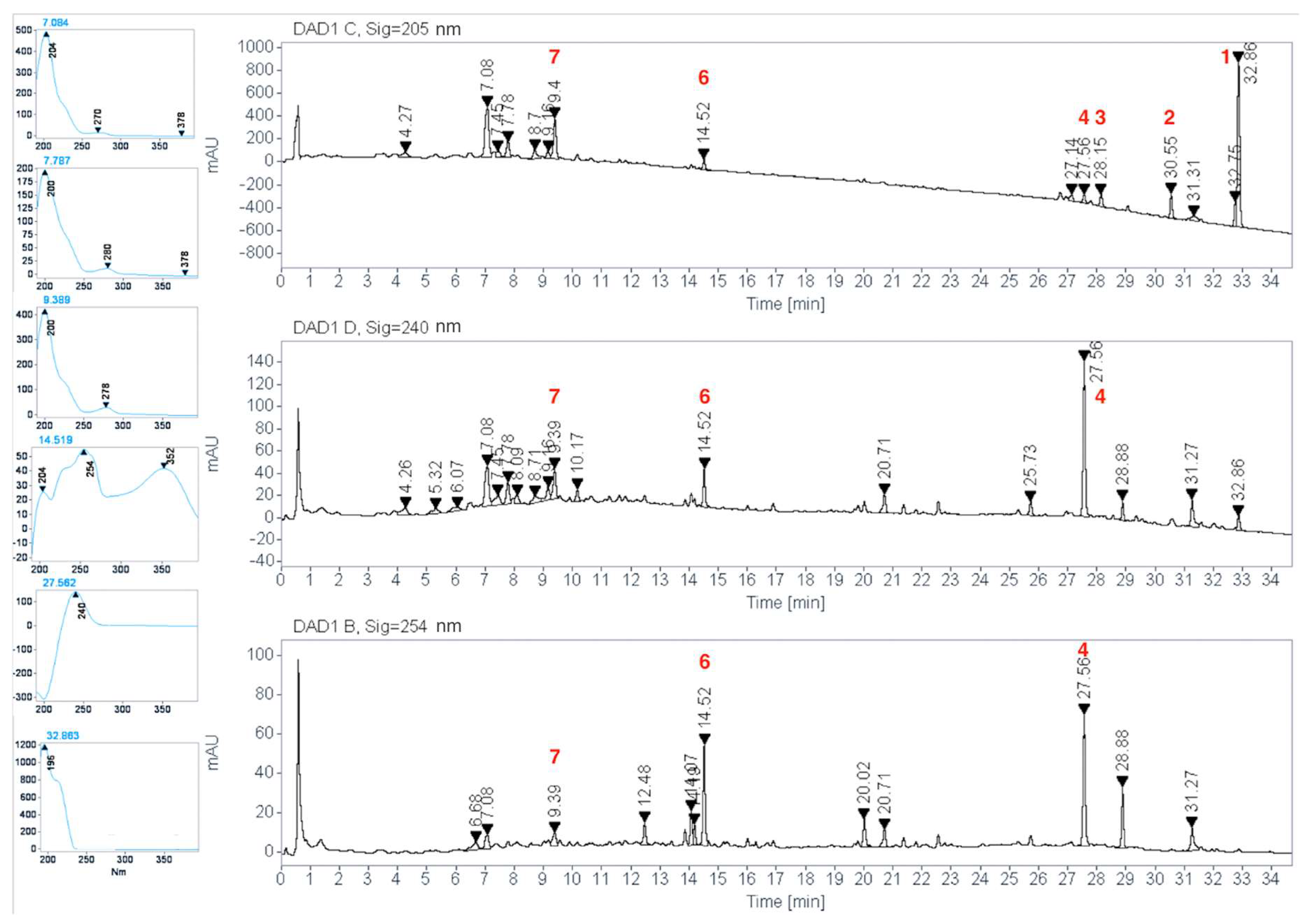
2.1. Isolation and Identification of Previously Undescribed Compounds
2.2. HPLC Phytochemical Profiling
2.3. Quantification of Rutin (6) in C. guatemalensis Extract
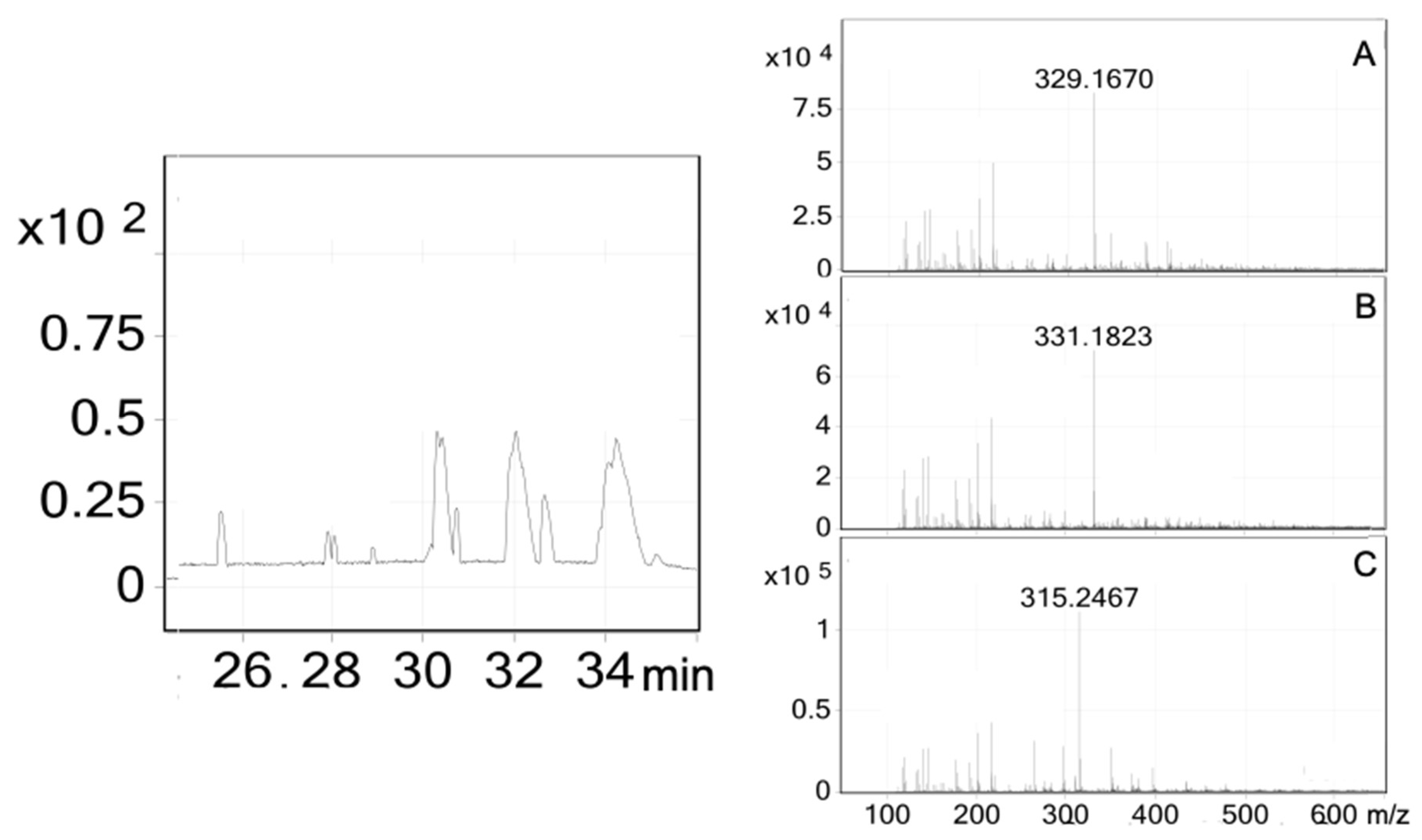
2.4. Affinity-Directed Fractionation
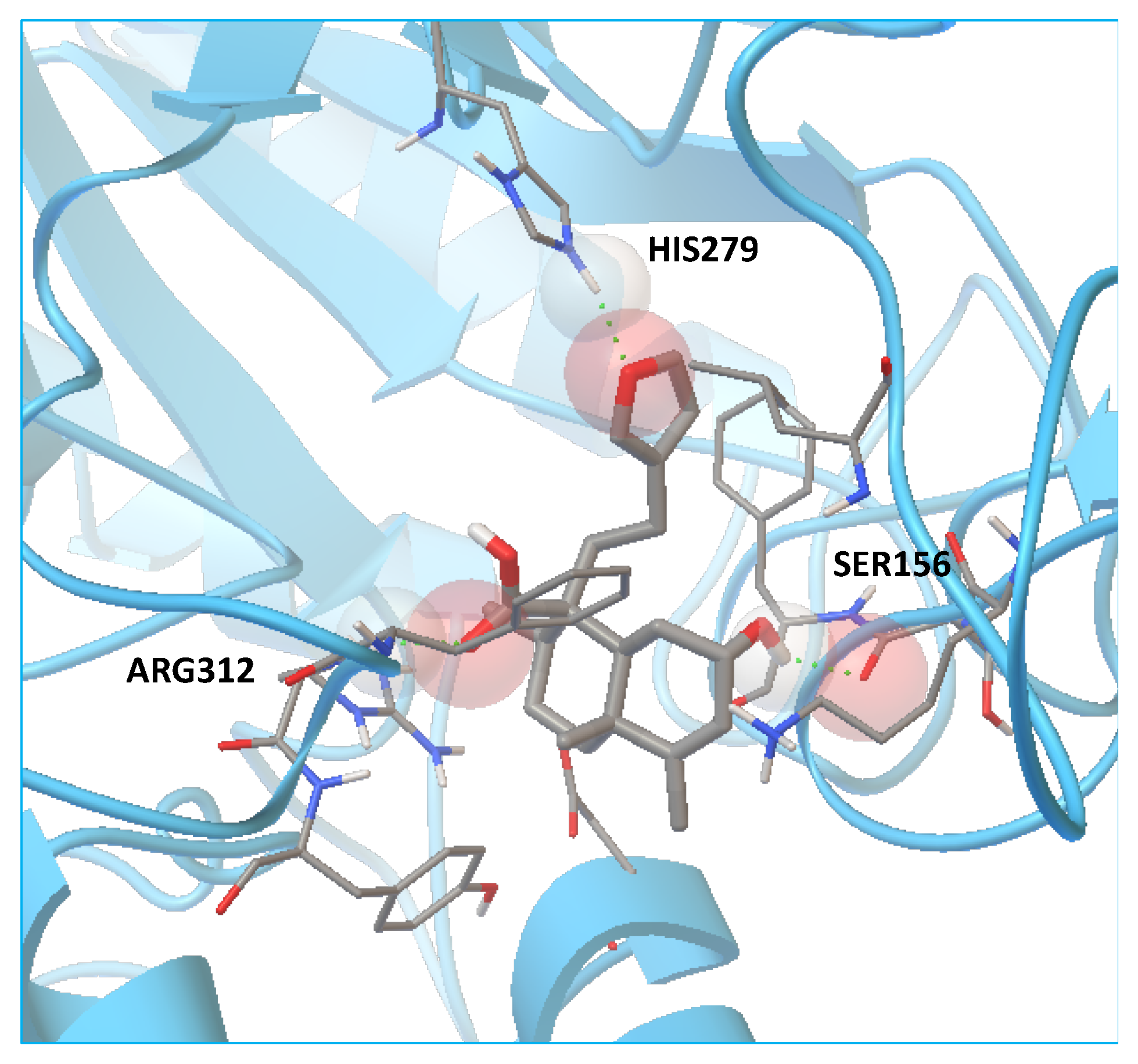
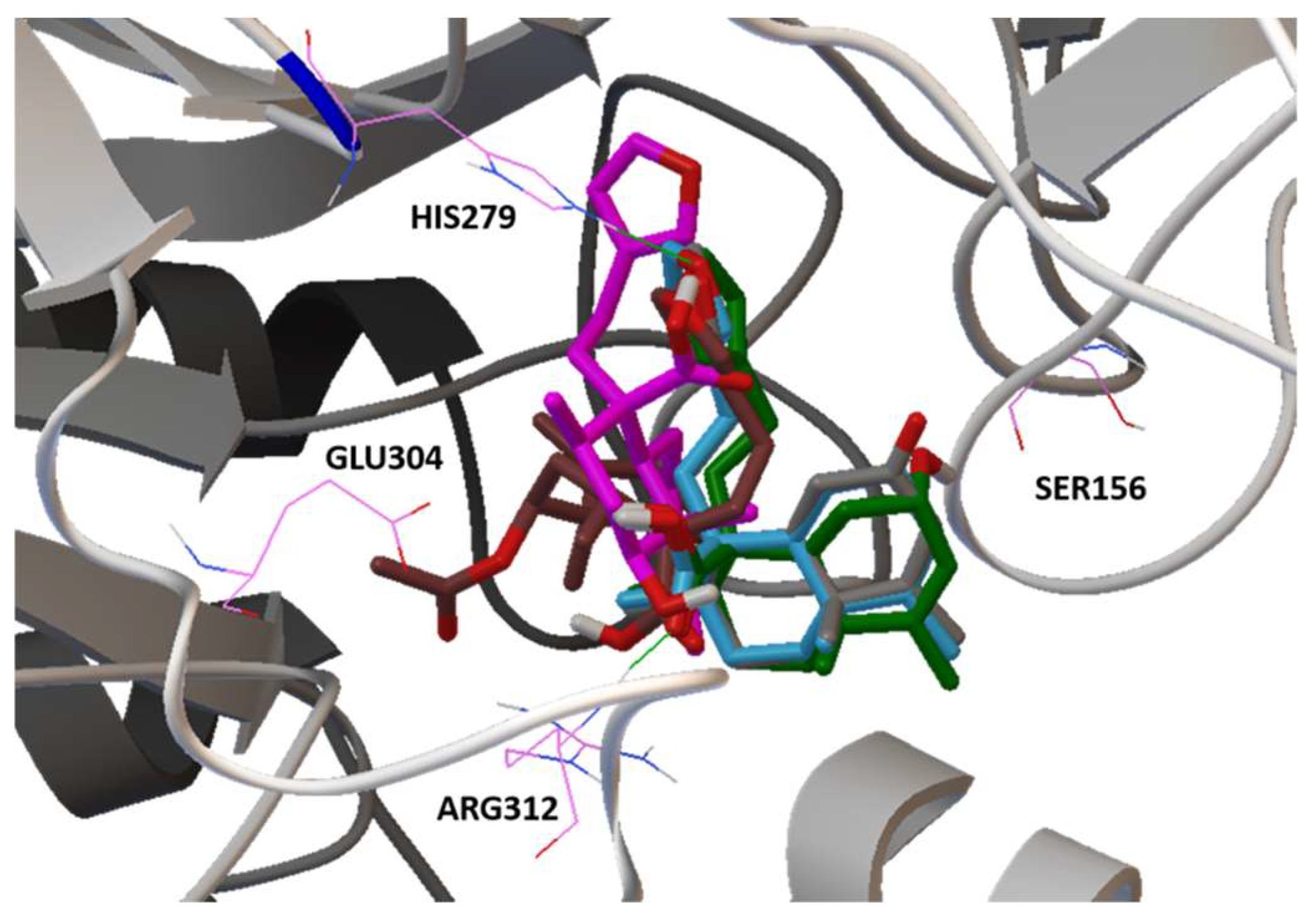
2.5. Molecular Docking
3. Conclusions
4. Materials and Methods
4.1. General Experimental Procedure
4.2. Plant Material and Extracts
4.3. Isolation Compounds
4.3.1. 6(s)-Acetoxy-15,16-diepoxy-ent-cleroda-3,13(16),14-trien-20-oic Acid (Crotoguatenoic Acid A; 2)
4.3.2. 6(s)-Hydroxy-15,16-diepoxy-ent-cleroda-3,13(16),14-trien-20-oic Acid (Crotoguatenoic Acid B; 3)
4.4. HPLC Analysis
4.5. HPLC Method Validation
4.6. Affinity-Directed Fractionation
4.7. Molecular Docking
4.8. Computational Details
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Xu, W.H.; Liu, W.Y.; Liang, Q. Chemical Constituents from Croton Species and Their Biological Activities. Molecules 2018, 23, 2333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salatino, A.; Faria Salatino, M.L.; Negri, G. Traditional Uses, Chemistry and Pharmacology of Croton Species (Euphorbiaceae). J. Braz. Chem. Soc. 2007, 18, 11–33. [Google Scholar] [CrossRef] [Green Version]
- Cruz, E.C.; Andrade-Cetto, A. Ethnopharmacological Field Study of the Plants Used to Treat Type 2 Diabetes among the Cakchiquels in Guatemala. J. Ethnopharmacol. 2015, 159, 238–244. [Google Scholar] [CrossRef] [PubMed]
- Okokon, J.E.; Nwafor, P.A. Antiplasmodial Activity of Root Extract and Fractions of Croton zambesicus. J. Ethnopharmacol. 2009, 121, 74–78. [Google Scholar] [CrossRef]
- Jünior, S.F.P.; Conserva, L.M.; Filho, J.M.B. Clerodane Diterpenes from Croton Species: Distribution and a Compilation of Their 13C NMR Spectral Data. Nat. Prod. Commun. 2006, 1, 1934578X0600100410. [Google Scholar] [CrossRef] [Green Version]
- Gomes, K.K.; MacEdo, G.E.; Rodrigues, N.R.; Ziech, C.C.; Martins, I.K.; Rodrigues, J.F.; De Brum Vieira, P.; Boligon, A.A.; De Brito Junior, F.E.; De Menezes, I.R.A.; et al. Croton campestris A. St.-Hill Methanolic Fraction in a Chlorpyrifos-Induced Toxicity Model in Drosophila melanogaster: Protective Role of Gallic Acid. Oxid. Med. Cell Longev. 2020, 2020, 3960170. [Google Scholar] [CrossRef] [Green Version]
- Ortiz, A.P.; Puebla, P.; Guerrero, M.F. Vascular Interactions of Croton Schiedeanus Major Flavonoids in Isolated Aortic Rings from Wistar Rats. Vitae 2021, 28, 1–14. [Google Scholar] [CrossRef]
- Cheila, C.B.; dos Anjos, G.L.; Nóbrega, R.S.A.; da Magaton, A.; de Miranda, F.M.; de Dias, F. Greener Ultrasound-Assisted Extraction of Bioactive Phenolic Compounds in Croton heliotropiifolius Kunth Leaves. Microchem. J. 2020, 159, 105525. [Google Scholar] [CrossRef]
- Oliani, J.; Ferreira, M.J.P.; Salatino, A.; Salatino, M.L.F. Leaf Flavonoids from Croton Urucurana and C. floribundus (Euphorbiaceae). Biochem. Syst. Ecol. 2021, 94, 104217. [Google Scholar] [CrossRef]
- Turiel, N.A.; Ribeiro, A.F.; Carvalho, E.E.N.; Domingos, V.D.; Lucas, F.C.A.; Carreira, L.M.M.; Andrade, E.H.A.; Maia, J.G.S. Essential Oils Composition of Croton Species from the Amazon. Nat. Prod. Commun. 2013, 8, 1471–1772. [Google Scholar] [CrossRef]
- Morais, S.M.; Cossolosso, D.S.; Silva, A.A.S.; de Moraes Filho, M.O.; Teixeira, M.J.; Campello, C.C.; Bonilla, O.H.; de Paula, V.F.; Vila-Nova, N.S. Essential Oils from Croton Species: Chemical Composition, in Vitro and in Silico Antileishmanial Evaluation, Antioxidant and Cytotoxicity Activities. J. Braz. Chem. Soc. 2019, 30, 2404–2412. [Google Scholar] [CrossRef]
- Castro, K.N.d.C.; Chagas, A.C.d.S.; Costa-Júnior, L.M.; Canuto, K.M.; Brito, E.S.d.; Rodrigues, T.H.S.; de Andrade, I.M. Acaricidal Potential of Volatile Oils from Croton Species on Rhipicephalus Microplus. Rev. Bras. Farmacogn. 2019, 29, 811–815. [Google Scholar] [CrossRef]
- Carlos Dantas da Cruz, R.; da Silva Carvalho, K.; Justino Oliveira Costa, R.; Alexandre da Silva, P.; ucia da Cunha Silva, S.L.; Andrade Gualberto, S.; Buarque de Gusm, N.; Antonia de Souza, I. Phytochemical and Toxicological Evaluation of a Blend of Essential Oils of Croton Species on Aedes Aegypti and Mus Musculus. S. Afr. J. Bot. 2020, 132, 188–195. [Google Scholar] [CrossRef]
- Sadgrove, N.J.; Madeley, L.G.; Van Wyk, B.E. Volatiles from African Species of Croton (Euphorbiaceae), Including New Diterpenes in Essential Oil from Croton gratissimus. Heliyon 2019, 5, e02677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franssen, F.F.J.; Smeijsters, L.J.J.W.; Berger, I.; Medinilla Aldana, B.E. In Vivo and in Vitro Antiplasmodial Activities of Some Plants Traditionally Used in Guatemala against Malaria. Antimicrob. Agents Chemother. 1997, 41, 1500–1503. [Google Scholar] [CrossRef] [Green Version]
- José Del Carmen, R.O.; Willam, H.M.J.; Del Carmen, G.M.A.; Nataly, J.G.; Stefany, C.O.S.; Anahi, C.A.; Domingo, P.T.J.; Leonardo, G.P.; De La Mora Miguel, P. Antinociceptive Effect of Aqueous Extracts from the Bark of Croton guatemalensis Lotsy in Mice. Res. Pharm. Sci. 2016, 11, 15–22. [Google Scholar]
- Andrade-Cetto, A.; Cruz, E.C.; Cabello-Hernández, C.A.; Cárdenas-Vázquez, R. Hypoglycemic Activity of Medicinal Plants Used among the Cakchiquels in Guatemala for the Treatment of Type 2 Diabetes. Evid.-Based Complement. Altern. Med. 2019, 2019, 2168603. [Google Scholar] [CrossRef] [Green Version]
- Henderson, M.S.; Murray, R.D.H.; McCrindle, R.; McMaster, D. Constituents of Solidago Species. Part III. The Constitution of Diterpenoids from Solidago juncea Ait. Can. J. Chem. 1973, 51, 1322–1331. [Google Scholar] [CrossRef]
- Lin, B.D.; Zhou, B.; Dong, L.; Wu, Y.; Yue, J.M. Formosins A–F: Diterpenoids with Anti-Microbial Activities from Excoecaria Formosana. Nat. Prod. Bioprospect. 2016, 6, 57–61. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Hernandez, D.; Oliveros-Bastidas, A.; Alonso-Amelot, M.E.; Calcagno-Pissarelli, M.P. Diterpene Foliar Exudates of Blakiella Bartsiifolia and Phytotoxicity of Clerodanes. Nat. Prod. Commun. 2014, 9, 1407–1412. [Google Scholar] [CrossRef] [Green Version]
- Seto, R.; Nakamura, H.; Nanjo, F.; Hara, Y. Preparation of Epimers of Tea Catechins by Heat Treatment. Biosci. Biotechnol. Biochem. 1997, 61, 1434–1439. [Google Scholar] [CrossRef]
- Li, R.; Morris-Natschke, S.L.; Lee, K.H. Clerodane Diterpenes: Sources, Structures, and Biological Activities. Nat. Prod. Rep. 2016, 33, 1166–1226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vihakas, M. Flavonoids and Other Phenolic Compounds: Characterization and Interactions with Lepidopteran and Sawfly Larvae; Birth Defects Orig. Art. Ser; Turun yliopisto: Turku, Finland, 1975; Volume 11, ISBN 9789512959051. [Google Scholar]
- Wolfender, J.L. HPLC in Natural Product Analysis: The Detection Issue. Planta Med. 2009, 75, 719–734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Connor, R.T.; Goldblatt, L.A. Correlation of Ultraviolet and Infrared Spectra of Terpene Hydrocarbons. Anal. Chem. 1954, 26, 1726–1737. [Google Scholar] [CrossRef]
- De Heluani, C.S.; Catalán, C.A.N.; Hernández, L.R.; Burgueño-Tapia, E.; Joseph-Nathan, P. 13C NMR Assignments and Conformational Evaluation of Diterpenes from Croton sarcopetalus Muell. Magn. Reson. Chem. 1998, 36, 947–950. [Google Scholar] [CrossRef]
- Aguilar-Guadarrama, A.B.; Rios, M.Y. Three New Sesquiterpenes from Croton arboreous. J. Nat. Prod. 2004, 67, 914–917. [Google Scholar] [CrossRef]
- Capasso, A.; Piacente, S.; De Tommasi, N.; Ragucci, M.; Pizza, C. Constituents of Croton menthodorus and Their Effects on Electrically Induced Contractions of the Guinea-Pig Isolated Ileum. Phyther. Res. 2000, 14, 156–159. [Google Scholar] [CrossRef]
- Zou, G.A.; Su, Z.H.; Zhang, H.W.; Wang, Y.; Yang, J.S.; Zou, Z.M. Flavonoids from the Stems of Croton caudatus Geisel. Var. Tomentosus Hook. Molecules 2010, 15, 1097–1102. [Google Scholar] [CrossRef] [Green Version]
- Dos Santos, K.P.; Motta, L.B.; Santos, D.Y.A.C.; Salatino, M.L.F.; Salatino, A.; Ferreira, M.J.P.; Lago, J.H.G.; Ruiz, A.L.T.G.; De Carvalho, J.E.; Furlan, C.M. Antiproliferative Activity of Flavonoids from Croton sphaerogynus Baill. (Euphorbiaceae). Biomed. Res. Int. 2015, 2015, 212809. [Google Scholar] [CrossRef] [Green Version]
- Aponte-Buitrago, R.; Mayorga-Wandurraga, H.; Moreno-Murillo, B. Flavonols and Sesquiterpenoids from Outer Bark and Leaves of Croton Polycarpus Benth. (Euphorbiaceae). Blacpma 2017, 16, 471–485. [Google Scholar]
- Li, S.; Han, Q.; Qiao, C.; Song, J.; Xu, H. Chinese Medicine Chemical Markers for the Quality Control of Herbal Medicines: An Overview. Chin. Med. 2008, 3, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yen, F.S.; Qin, C.S.; Tan, S.; Xuan, S.; Ying, P.J.; Le, H.Y.; Darmarajan, T.; Gunasekaran, B.; Salvamani, S. Hypoglycemic Effects of Plant Flavonoids: A Review. Evid.-Based Complement. Altern. Med. 2021, 2021, 2057333. [Google Scholar] [CrossRef]
- Escandón-Rivera, S.M.; Mata, R.; Andrade-Cetto, A. Molecules Isolated from Mexican Hypoglycemic Plants: A Review. Molecules 2020, 25, 4145. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Evans, F.J.; Roberts, M.F.; Phillipson, J.D.; Zenk, M.H.; Gleba, Y.Y. Polyphenolic Compounds from Croton lechleri. Phytochemistry 1991, 30, 2033–2040. [Google Scholar] [CrossRef]
- Cordeiro, K.W.; Felipe, J.L.; Malange, K.F.; Do Prado, P.R.; De Oliveira Figueiredo, P.; Garcez, F.R.; De Cássia Freitas, K.; Garcez, W.S.; Toffoli-Kadri, M.C. Anti-Inflammatory and Antinociceptive Activities of Croton urucurana Baillon Bark. J. Ethnopharmacol. 2016, 183, 128–135. [Google Scholar] [CrossRef] [PubMed]
- Qu, Z.; Liu, A.; Li, P.; Liu, C.; Xiao, W.; Huang, J.; Liu, Z.; Zhang, S. Advances in Physiological Functions and Mechanisms of (-)-Epicatechin. Crit. Rev. Food Sci. Nutr. 2021, 61, 211–233. [Google Scholar] [CrossRef] [PubMed]
- Mechchate, H.; Es-safi, I.; Haddad, H.; Bekkari, H.; Grafov, A.; Bousta, D. Combination of Catechin, Epicatechin, and Rutin: Optimization of a Novel Complete Antidiabetic Formulation Using a Mixture Design Approach. J. Nutr. Biochem. 2021, 88, 108520. [Google Scholar] [CrossRef]
- Aderogba, M.A.; Ndhlala, A.R.; Van Staden, J. Acetylcholinesterase Inhibitors from Croton Sylvaticus Ethyl Acetate Leaf Extract and Their Mutagenic Effects. Nat. Prod. Commun. 2013, 8, 795–798. [Google Scholar] [CrossRef] [Green Version]
- Salehi, B.; Machin, L.; Monzote, L.; Sharifi-Rad, J.; Ezzat, S.M.; Salem, M.A.; Merghany, R.M.; El Mahdy, N.M.; Klllç, C.S.; Sytar, O.; et al. Therapeutic Potential of Quercetin: New Insights and Perspectives for Human Health. ACS Omega 2020, 5, 11849–11872. [Google Scholar] [CrossRef]
- ICH. International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use. ICH Harmon. Tripart. Guidel. 2005, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Salgado-Garciglia, R.; Hernández-García, A.; Montiel-Montoya, J.; Valdez-Morales, M.; López-Valdez, L.G.; Herrera-Cabrera, B.E.; Zaragoza-Martínez, F.; Lucho Constantino, G.G.; Barrales-cureño, H.J. Flavonoids Quantification in Acer Negundo L., Extracts by Hplc Analysis. Agro Product. 2021, II, 1–8. [Google Scholar] [CrossRef]
- Saraf, A.; Sankhala, S. Simultaneous Determination of Rutin and Quercetin in Different Parts of Tecomella Undulata (Seem): An Endangered Medicinal Plant. Int. J. Pharmacogn. Phytochem. Res. 2014, 6, 434–439. [Google Scholar]
- Fan, M.; Chen, G.; Sun, B.; Wu, J.; Li, N.; Sarker, S.D.; Nahar, L.; Guo, M. Screening for Natural Inhibitors of Human Topoisomerases from Medicinal Plants with Bio-Affinity Ultrafiltration and LC–MS. Phytochem. Rev. 2020, 19, 1231–1261, ISBN 0123456789. [Google Scholar] [CrossRef]
- Rosas-Ramírez, D.; Pereda-Miranda, R.; Escandón-Rivera, S.; Arreguín-Espinosa, R. Identification of α-Glucosidase Inhibitors from Ipomoea Alba by Affinity-Directed Fractionation-Mass Spectrometry. Rev. Bras. Farmacogn. 2020, 30, 336–345. [Google Scholar] [CrossRef]
- Kimura, A.; Lee, J.H.; Lee, I.S.; Lee, H.S.; Park, K.H.; Chiba, S.; Kim, D. Two Potent Competitive Inhibitors Discriminating α-Glucosidase Family I from Family II. Carbohydr. Res. 2004, 339, 1035–1040. [Google Scholar] [CrossRef] [PubMed]
- Ren, L.; Qin, X.; Cao, X.; Wang, L.; Bai, F.; Bai, G.; Shen, Y. Structural Insight into Substrate Specificity of Human Intestinal Maltase-Glucoamylase. Protein Cell 2011, 2, 827–836. [Google Scholar] [CrossRef] [Green Version]
- Rosas-Ramírez, D.; Escandón-Rivera, S.; Pereda-Miranda, R. Morning Glory Resin Glycosides as α-Glucosidase Inhibitors: In Vitro and in Silico Analysis. Phytochemistry 2018, 148, 39–47. [Google Scholar] [CrossRef]
- Bruhn, T.; Schaumlöffel, A.; Hemberger, Y.; Bringmann, G. SpecDis: Quantifying the Comparison of Calculated and Experimental Electronic Circular Dichroism Spectra. Chirality 2013, 25, 243–249. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 2 a | 3 a | |||
|---|---|---|---|---|
| Position | δH | δC | δH | δC |
| 1 | α 1.85 b, m β 1.93 b, m | 19.8 | α 1.84 b, m β 1.93 b, m | 20.1 |
| 2 | α 1.99 b, m β 2.07 b, m | 27.1 | α 2.02 b, m β 2.10 b, m | 27.2 |
| 3 | 5.30, dd (2.87, 1.39) | 123.6 | 5.31, m | 123.1 |
| 4 | - | 141.5 | - | 143.0 |
| 5 | - | 42.9 | - | 44.7 |
| 6 | 4.73 dd (11.34, 5.00) | 78.2 | 3.59, dd (11.35, 4.97) | 76.1 |
| 7 | α 2.20 b, m β 1.76 b, m | 33.0 | α 2.20 b, dd (13.71, 2.26) β 1.70 b, m | 37.4 |
| 8 | 1.76 b, m | 34.3 | 1.71 ddd (14.06, 6.21, 3.58) | 34.7 |
| 9 | - | 49.3 | - | 49.4 |
| 10 | 1.66, d (11.50) | 47.1 | 1.58 dd (11.63, 1.30) | 47.2 |
| 11 | α 1.85 b, m β 2.26 b, m | 33.9 | α 1.93, m β 2.26 b, m | 34.0 |
| 12 | 2.33 b,c, m | 17.8 | 2.33 b,c, m | 17.8 |
| 13 | - | 124.4 | - | 124.5 |
| 14 | 6.26, dd (1.83, 0.90) | 110.9 | 6.26, dd (1.83, 0.95) | 110.9 |
| 15 | 7.35, t (1.69) | 143.1 | 7.35, t (1.69) | 143.0 |
| 16 | 7.23, dd (1.61, 0.88) | 138.7 | 7.23, dd (1.62, 0.91) | 138.7 |
| 17 | 1.14, d (6.69) | 16.1 | 1.15, d (6.71) | 16.3 |
| 18 | 1.59, br s | 21.2 | 1.84, br s | 22.6 |
| 19 | 1.09, s | 13.9 | 0.98, s | 12.8 |
| 20 | - | 182.3 | - | 181.9 |
| -OAc | 2.04 | 22.0 | - | - |
| 170.8 | ||||
| Rt | Linear Range (μg/mL) | Calibration Equation | R2 a | LOD (μg/mL) | LOQ (μg/mL) | Precision | Recovery (%mean) | |
|---|---|---|---|---|---|---|---|---|
| Intraday (%RSD) | Interday (%RSD) | |||||||
| 14.52 | 20–250 | Y = 9.29484284x + 17.083753 | 0.9996 | 0.19 | 0.57 | 0.79 | 0.22 | 100.74 |
| Batch | %EWE a | Content in mg/g b |
|---|---|---|
| 09–2013 | 20.5 | 0.6067 ± 0.0025 |
| 06–2015 | 19.3 | 0.5585 ± 0.0042 |
| 10–2019 | 18.7 | 0.6440 ± 0.0068 |
| Compound | Formula a | ESI-MS [M–H]– b | ESI-MS [M + H]+ b | MAL12 | MGAM | ||
|---|---|---|---|---|---|---|---|
| Theoretical Ki | Hydrogen Bond | Theoretical Ki | Hydrogen Bond | ||||
| 1 | C20H28O3 (316) | 315.2467 | - | 7.12 μM | His279, Arg312 | 3.02 μM | Gln1372, Arg1377 |
| 2 | C22H30O5 (374) | - | - | 17.1 μM | His279, Arg312 | 6.31 μM | Gln1372, Arg1377 |
| 3 | C20H28O4 (332) | 331.1823 | 333.2444 | 13 μM | Arg312 | 5.09 μM | Gln1372, Arg1377 |
| 4 | C20H26O4 (330) | 329.1670 | - | 6.73 μM | His279, Arg312 | 1.95 μM | Gln1372, Arg1377 |
| 5 | C20H28O4 (332) | 331.1823 | 333.2444 | 4.14 μM | Ser156, His279, Arg312 | 2.4 μM | Try1251, Gln1372, Arg1377 |
| Acarbose c | C25H43NO18 (645) | - | 646 | 51.4 nM | His279, Gln322, Glu304, Arg312 | 35.7 nM | Tyr1251, Gln1372, Arg1377, Gln1561, Gly1588 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Escandón-Rivera, S.M.; Andrade-Cetto, A.; Rosas-Ramírez, D.G.; Arreguín-Espinosa, R. Phytochemical Screening and Isolation of New Ent-Clerodane Diterpenoids from Croton guatemalensis Lotsy. Plants 2022, 11, 3159. https://doi.org/10.3390/plants11223159
Escandón-Rivera SM, Andrade-Cetto A, Rosas-Ramírez DG, Arreguín-Espinosa R. Phytochemical Screening and Isolation of New Ent-Clerodane Diterpenoids from Croton guatemalensis Lotsy. Plants. 2022; 11(22):3159. https://doi.org/10.3390/plants11223159
Chicago/Turabian StyleEscandón-Rivera, Sonia Marlen, Adolfo Andrade-Cetto, Daniel Genaro Rosas-Ramírez, and Roberto Arreguín-Espinosa. 2022. "Phytochemical Screening and Isolation of New Ent-Clerodane Diterpenoids from Croton guatemalensis Lotsy" Plants 11, no. 22: 3159. https://doi.org/10.3390/plants11223159
APA StyleEscandón-Rivera, S. M., Andrade-Cetto, A., Rosas-Ramírez, D. G., & Arreguín-Espinosa, R. (2022). Phytochemical Screening and Isolation of New Ent-Clerodane Diterpenoids from Croton guatemalensis Lotsy. Plants, 11(22), 3159. https://doi.org/10.3390/plants11223159