

Analysis of Genetic Diversity and Population Structure of Cowpea (Vigna unguiculata (L.) Walp) Genotypes Using Single Nucleotide Polymorphism Markers

 ,
,  , and
, and 
Abstract
:1. Introduction
2. Results
2.1. Allele Polymorphism
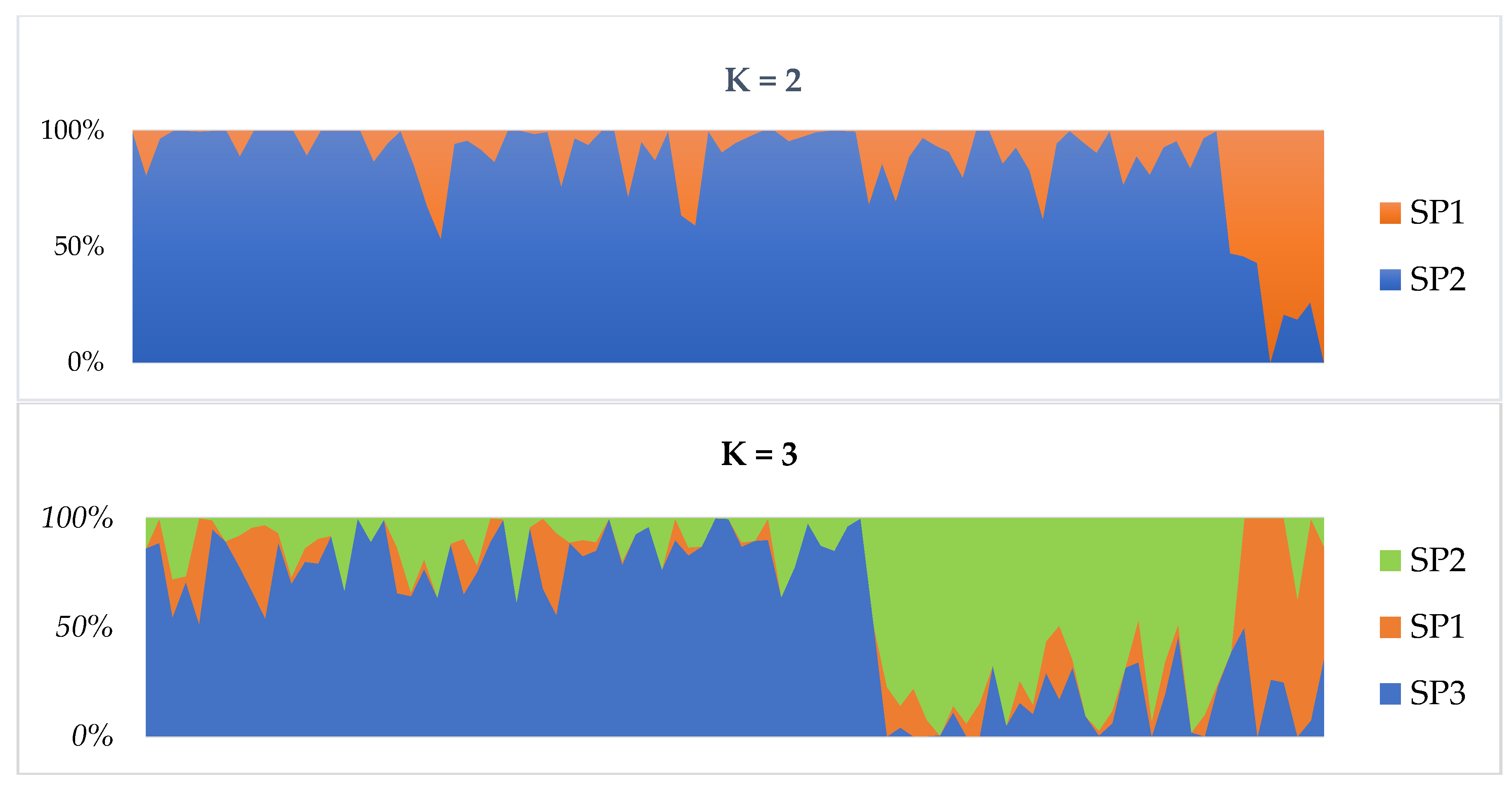
2.2. Population Structure and Clustering
2.3. Genetic Diversity among Subpopulations
2.4. Analysis of Molecular Variance (AMOVA)
3. Discussion
4. Materials and Methods
4.1. Plant Materials
4.2. DNA Extraction and Sequencing
4.3. Data Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Badiane, F.A.; Diouf, M.; Diouf, D. Cowpea. In Broadening the Genetic Base of Grain Legumes; Singh, M., Bisht, I.S., Dutta, M., Eds.; Springer: Berlin/Heidelberg, Germany, 2014; Volume 79, pp. 95–114. [Google Scholar]
- Coulibaly, S.; Pasquet, R.S.; Papa, R. Gepts PAFLP analysis of the phenetic organization genetic diversity of Vigna unguiculata, L. Walp. reveals extensive gene flow between wild and domesticated types. Theor. Appl. Genet. 2002, 104, 358–366. [Google Scholar] [CrossRef] [PubMed]
- Gbedevi, K.M.; Boukar, O.; Ishikawa, H.; Abe, A.; Ongom, P.O.; Unachukwu, N.; Rabbi, I.; Fatokun, C. Genetic diversity and population structure of cowpea [Vigna unguiculata (L.) Walp.] germplasm collected from Togo based on DArT markers. Genes 2021, 12, 1451. [Google Scholar] [CrossRef] [PubMed]
- Ehlers, J.D.; Hall, A.E. Cowpea (Vigna unguiculata L. walp.). Field Crops Res. 1997, 53, 187–204. [Google Scholar] [CrossRef]
- Seo, E.; Kim, K.; Jun, T.; Choi, J.; Kim, S.; Muñoz-Amatriaín, M.; Sun, H.; Ha, B. Population structure and genetic diversity in Korean cowpea germplasm based on SNP markers. Plants 2020, 9, 1190. [Google Scholar] [CrossRef]
- Carvalho, M.; Muñoz-Amatriaín, M.; Castro, I.; Lino-Neto, T.; Matos, M.; Egea-Cortines, M.; Rosa, E.; Close, T.; Carnide, V. Genetic diversity and structure of Iberian Peninsula cowpeas compared to world-wide cowpea accessions using high density SNP markers. BMC Genom. 2017, 18, 891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gbaye, O.A.; Holloway, G.J. Varietal effects of cowpea, Vigna unguiculata, on tolerance to malathion in Callosobruchus maculatus (Coleoptera: Bruchidae). J. Stored Prod. Res. 2011, 47, 365–371. [Google Scholar] [CrossRef]
- Kondwakwenda, A.; Sibiya, J.; Amelework, A.B.; Zengeni, R. Diversity analysis of provitamin A maize inbred lines using single nucleotide polymorphism markers. Acta Agric. Scand. B Soil Plant Sci. 2020, 70, 265–271. [Google Scholar] [CrossRef]
- Bhandari, H.R.; Bhanu, A.N.; Srivastava, K.; Singh, M.N.; Shreya, H.A. Assessment of genetic diversity in crop plants-an overview. Adv. Plants Agric. Res. 2017, 7, 279–286. [Google Scholar]
- Campos, A.; Tedesco, S.; Vasconcelos, V.; Cristobal, S. Proteomic research in bivalves: Towards the identification of molecular markers of aquatic pollution. J. Proteom. 2012, 75, 4346–4359. [Google Scholar] [CrossRef]
- Horn, L.N.; Shimelis, H. Production constraints and breeding approaches for cowpea improvement for drought prone agro-ecologies in Sub-Saharan Africa. Ann. Agric. Sci. 2020, 66, 83–91. [Google Scholar] [CrossRef]
- De Vicente, M.C.; Lopez, C.; Fulton, T. Genetic Diversity Analysis with Molecular Marker Data: Learning Module; International Plant Genetic Resources Institute (IPGRI): Rome, Italy, 2004. [Google Scholar] [CrossRef]
- Bucheyeki, T.L.; Gwanama, C.; Mgonja, M.; Chisi, M.; Folkertsma, R.; Mutegi, R. Genetic variability characterisation of Tanzania sorghum landraces based on simple sequence repeats (SSRs) molecular and morphological markers. Afr. Crop Sci. J. 2009, 17, 71–86. [Google Scholar] [CrossRef]
- Sami, R.A.; Yeye, M.Y.; Ishiyaku, M.F.; Usman, I.S. Heritability studies in some sweet sorghum (Sorghum Bicolor. L. Moench) genotypes. J. Biol. Agric. Healthc. 2013, 3, 49–51.s. [Google Scholar]
- Sejake, T.; Shargie, N.; Christian, R.; Amelework, A.B.; Tsilo, T.J. Genetic diversity in sorghum (‘Sorghum bicolor’ L. Moench) accessions using SNP based Kompetitive allele-specific (KASP) markers. Aust. J. Crop. Sci. 2021, 15, 890–898. [Google Scholar] [CrossRef]
- Cui, C.; Mei, H.; Liu, Y.; Zhang, H.; Zheng, Y. Genetic diversity, population structure, and linkage disequilibrium of an association-mapping panel revealed by genome-wide SNP markers in sesame. Front. Plant Sci. 2017, 8, 1189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malviya, N.; Sarangi, B.K.; Yadav, M.K.; Yadav, D. Analysis of genetic diversity in cowpea (Vigna unguiculata L. Walp.) cultivars with random amplified polymorphic DNA markers. Plant Syst. Evol. 2012, 298, 523–526. [Google Scholar] [CrossRef]
- Mafakheri, K.; Bihamta, M.R.; Abbasi, A.R. Assessment of genetic diversity in cowpea (Vigna unguiculata L.) germplasm using morphological and molecular characterisation. Cogent Food Agric. 2017, 3, 1327092. [Google Scholar] [CrossRef]
- Nkhoma, N.; Shimelis, H.; Laing, M.; Shayanowako, A.; Mathew, I. Assessing the genetic diversity of cowpea [Vigna unguiculata (L.) Walp.] germplasm collections using phenotypic traits and SNP markers. BMC Genome 2020, 21, 00914-7. [Google Scholar] [CrossRef]
- Fang, J.; Chao, C.; Roberts, P.A.; Ehlers, J.D. Genetic diversity of cowpea [Vigna unguiculata (L.) Walp.] in four West African and USA breeding programs as determined by AFLP analysis. Genet. Resour. Crop Evol. 2007, 54, 1197–1209. [Google Scholar] [CrossRef]
- Ba, F.S.; Pasquet, R.S.; Gepts, P. Genetic diversity in cowpea [Vigna unguiculata (L.) Walp.] as revelead by RAPD markers. Genet. Resour. Crop Evol. 2004, 51, 539–550. [Google Scholar] [CrossRef]
- Rafalski, A. Applications of single nucleotide polymorphisms in crop genetics. Plant Biol. 2002, 5, 94–100. [Google Scholar] [CrossRef]
- Kujur, A.; Bajaj, D.; Upadhyaya, H.D.; Das, S.; Ranjan, R.; Shree, T.; Saxena, M.S.; Badoni, S.; Kumar, V.; Tripathi, S. Employing genome-wide SNP discovery and genotyping strategy to extrapolate the natural allelic diversity and domestication patterns in chickpea. Front. Plant Sci. 2015, 6, 162. [Google Scholar] [CrossRef] [PubMed]
- Desalegne, B.A.; Dagne, K.; Melaku, G.; Boukar, O.; Fatokun, C.A. Efficiency of SNP and SSR-based analysis of genetic diversity, population structure, and relationships among cowpea (Vigna unguiculata (L.) Walp.) germplasm from East Africa and IITA inbred lines. J. Crop Sci. Biotech. 2017, 20, 107–128. [Google Scholar] [CrossRef]
- Elshire, R.J.; Glaubitz, J.C.; Sun, Q.; Poland, J.A.; Kawamoto, K.; Buckler, E.S.; Mitchell, S.E. A robust, simple sequence genotyping-by-sequence (GBS) approach for high diversity species. PLoS ONE 2011, 6, e19379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bastien, M.; Sonah, H.; Belzile, F. Genome wide association mapping of resistance in soybean with a genotyping-by-sequencing approach. Plant Genom. 2014, 7, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Ketema, S.; Tesfaye, B.; Keneni, G.; Fenta, B.A.; Assefa, E.; Greliche, N.; Machuka, E.; Yao, N. DArTSeq SNP-based markers revealed high genetic diversity and structured population in Ethiopian cowpea [Vigna unguiculata (L.) Walp] germplasms. PLoS ONE 2020, 15, e0239122. [Google Scholar] [CrossRef]
- Farahani, S.; Maleki, M.; Mehrabi, R.; Kanouni, H.; Scheben, A.; Batley, J.; Talebi, R. Whole genome diversity, population structure, and linkage disequilibrium analysis of chickpea (Cicer arietinum L.) genotypes using genome-wide DArTseq-based SNP markers. Genes 2019, 10, 676. [Google Scholar] [CrossRef] [Green Version]
- Valdisser, P.A.; Pereira, W.J.; Filho, J.E.; Müller, B.S.; Coelho, G.R.; de Menezes, I.P.; Vianna, J.P.; Zucchi, M.I.; Lanna, A.C.; Coelho, A.S. In-depth genome characterization of a Brazilian common bean core collection using DArTseq high-density SNP genotyping. BMC Genom. 2017, 18, 3805-4. [Google Scholar] [CrossRef] [Green Version]
- Uba, C.U.; Oselebe, H.O.; Tesfay, A.A.; Abtew, W.G. Genetic diversity and population structure analysis of Bambara groundnut (Vigna subterrenea L.) landraces using DArT SNP markers. PLoS ONE 2021, 16, e0253600. [Google Scholar] [CrossRef]
- Wright, S. Evolution and the Genetics of Populations: Variability within and among Natural Populations; University of Chicago Press: Chicago, IL, USA, 1984; Volume 4. [Google Scholar]
- Ebert, A.W.; Engels, J.M.M. Plant biodiversity and genetic resources matter. Plants 2020, 9, 1706. [Google Scholar] [CrossRef]
- Serba, D.D.; Muleta, K.T.; Amand, P.; Bernado, A.; Bai, G.; Perumal, R.; Bashir, E. Genetic diversity, population structure and linkage disequilibrium of pearl millet. Plant Genome 2019, 12, 180091. [Google Scholar] [CrossRef] [Green Version]
- Kumar, D.; Chhokar, V.; Sheoran, S.; Singh, R.; Sharma, P.; Jaiswal, S.; Iquebal, M.A.; Jaiswar, A.; Jaisri, J.; Angadi, U.B.; et al. Characterization of genetic diversity and population structure in wheat using array based SNP markers. Mol. Biol. 2020, 47, 293–306. [Google Scholar] [CrossRef]
- Eltaher, S.; Sallam, A.; Belamkar, V. Genetic diversity and population structure of F3:6 Nebraska winter wheat genotypes using genotyping-by-sequence. Front. Genet. 2018, 9, 11–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sodedji, F.A.; Agbahoungba, S.; Agoyi, E.E.; Kafoutchoni, K.M.; Kim, H.; Nguetta, S.A.; Assogbadjo, A.E. Diversity, population structure, and linkage disequilibrium among cowpea accessions. Plant Genome 2021, 14, e20113. [Google Scholar] [CrossRef] [PubMed]
- Fatokun, C.; Girma, G.; Abberton, M.; Gedil, M.; Unachukwu, N.; Oyatomi, O.; Yusuf, M.; Rabbi, I.; Boukar, O. Genetic diversity and population structure of a mini-core subset from the world cowpea (Vigna unguiculata (L.) Walp.) germplasm collection. Sci. Rep. 2018, 8, 16035. [Google Scholar] [CrossRef] [Green Version]
- Xiong, H.; Shi, A.; Mou, B.; Qin, J.; Motes, D.; Lu, W.; Ma, J.; Weng, Y.; Yang, W.; Wu, D. Genetic diversity and population structure of cowpea (Vigna unguiculata L. Walp). PLoS ONE 2016, 11, e0160941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Govindaraj, M.; Vetriventha, M.; Srinivasan, M. Importance of genetic diversity assessment in crop plants and its recent advances: An overview of its analytical perspectives. Genet. Res Int. 2015, 15, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fatokun, C.A.; Ng, Q. Outcrossing in cowpea. J. Food Agric. Environ. 2007, 5, 334–338. [Google Scholar]
- Wamalwa, E.N.; Muoma, J.; Wekesa, C. Genetic diversity of cowpea (Vigna unguiculata (L.) Walp.) accession in Kenya gene bank based on simple sequence repeat markers. Int. J. Genom. 2016, 2016, 8956412. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Li, Y.; Li, X.; Li, C.; Shi, Y.; Song, Y.; Zheng, Z.; Li, Y.; Wang, T. Analysis of genetic differentiation and genomic variation to reveal potential regions of importance during maize improvement. BMC Plant Biol. 2015, 15, 0646-7. [Google Scholar] [CrossRef] [Green Version]
- Sarr, A.; Bodian, A.; Gbedevi, K.M.; Ndir, K.N.; Ajewole, O.O.; Gueye, B.; Foncéka, D.; Diop, E.A.; Diop, B.M.; Cissé, N. Genetic diversity and population structure analyses of wild relatives and cultivated cowpea (Vigna unguiculata (L.) Walp.) from Senegal using simple sequence repeat markers. Plant Mol. Biol. Rep. 2020, 39, 112–124. [Google Scholar] [CrossRef]
- Ravelombola, W.; Qin, J.; Shi, A.; Weng, Y.; Bhattarai, G.; Dong, L.; Morris, J.B. A SNP-based association analysis for plant growth habit in worldwide cowpea (Vigna unguiculata (L.) Walp) germplasm. Plant Genet. Resour. 2017, 213, 284. [Google Scholar] [CrossRef]
- Nkhata, W.; Shimelis, H.; Melis, R.; Chirwa, R.; Mzengeza, T.; Mathew, I.; Shayanowako, A. Population structure and genetic diversity analyses of common beans germplasm collections of East and Southern Africa using morphological traits and high-density SNP markers. PLoS ONE 2010, 12, e0243238. [Google Scholar] [CrossRef] [PubMed]
- Hellens, R.P.; Moreau, C.; Lin-Wang, K.; Schwinn, K.E.; Thomson, S.J.; Fiers, M.; Frew, T.J.; Murray, S.R.; Hofer, J.M.; Jacobs, J.M. Identification of Mendel’s white flower character. PLoS ONE 2010, 5, e13230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Cui, S.; Dang, P. GWAS and bulked segregant analysis reveal the Loci controlling growth habit-related traits in cultivated Peanut (Arachis hypogaea L.). BMC Genom. 2022, 23, 403. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.; Rafii, M.; Ramlee, S.; Jusoh, M.; Mamun, M. Genetic analysis and selection of Bambara groundnut (Vigna subterranea [L.] Verdc.) landraces for high yield revealed by qualitative and quantitative traits. Sci. Rep. 2021, 11, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Gepts, P.; Papa, R. Possible effects of (trans) gene flow from crops on the genetic diversity from landraces and wild relatives. Environ. Biosaf. Res. 2003, 2, 89–103. [Google Scholar] [CrossRef] [Green Version]
- Wright, S. The interpretation of population structure by F-statistics with special regard to systems of mating. Evolution 1965, 19, 395–420. [Google Scholar]
- Kilian, A.; Wenzl, P.; Huttner, E.; Carling, J.; Xia, L.; Blois, H.; Caig, V.; Heller-Uszynska, K.; Jaccoud, D.; Hopper, C. Diversity arrays technology: A generic genome profiling technology on open platforms, in: Data production and analysis in population genomics. Methods Mol. Biol. 2012, 888, 67–89. [Google Scholar] [CrossRef]
- Peakall, R.; Smouse, P.E. Genalex 6.5: Genetic analysis in excel. Population genetic software for teaching and research-an update. Bioinformatics 2012, 28, 2537–2539. [Google Scholar] [CrossRef] [Green Version]
- Nei, M.; Li, W. Mathematical model for studying genetic variation in terms of restriction endonucleases. Proc. Natl. Acad. Sci. USA 1979, 76, 5269–5273. [Google Scholar] [CrossRef] [Green Version]
- Debener, T.; Salamini, F.; Gebhardt, C. Phylogeny of wild and cultivated Solanum species based on nuclear restriction fragment length polymorphisms (RFLPs). Theor. Appl. Genet. 1990, 79, 360–368. [Google Scholar] [CrossRef] [PubMed]
- Perrier, X.; Jacquemoud-Collet, J.P. DARwin Software. Dissimilarity Analysis and Representation for Windows. 2006. Available online: http://www.darwin.cirad.fr/darwin.html (accessed on 1 March 2006).
- Pritchard, J.K.; Stephens, M.; Rosenberg, N.A.; Donnelly, P. Association mapping in structured populations. Am. J. Human Genet. 2000, 67, 170–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef] [PubMed]
- Earl, D.A.; VonHoldt, B.M. STRUCTURE HARVESTER: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Gene Res. 2012, 4, 359–361. [Google Scholar] [CrossRef]
- Jakobsson, M.; Rosenberg, N. ACLUMPP: A cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 2007, 23, 1801–1806. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Chromosome | NSU | NPS | %P | Ne | Ho | uHe | FIS | PIC |
|---|---|---|---|---|---|---|---|---|
| 1 | 670 | 345 | 51.49 | 1.43 (0.017) | 0.088 (0.004) | 0.269 (0.008) | 0.66 (0.012) | 0.27 (0.008) |
| 2 | 725 | 377 | 52.00 | 1.41 (0.016) | 0.077 (0.003) | 0.258 (0.008) | 0.68 (0.011) | 0.26 (0.008) |
| 3 | 1222 | 668 | 54.66 | 1.46 (0.012) | 0.082 (0.003) | 0.288 (0.006) | 0.70 (0.008) | 0.29 (0.005) |
| 4 | 1123 | 533 | 47.46 | 1.47 (0.013) | 0.094 (0.004) | 0.290 (0.006) | 0.67 (0.010) | 0.29 (0.006) |
| 5 | 965 | 453 | 46.94 | 1.42 (0.015) | 0.081 (0.004) | 0.264 (0.007) | 0.68 (0.011) | 0.26 (0.007) |
| 6 | 915 | 503 | 54.97 | 1.42 (0.015) | 0.075 (0.003) | 0.259 (0.007) | 0.68 (0.009) | 0.26 (0.007) |
| 7 | 1066 | 580 | 54.41 | 1.48 (0.013) | 0.079 (0.003) | 0.295 (0.006) | 0.72 (0.009) | 0.29 (0.006) |
| 8 | 823 | 459 | 55.77 | 1.38 (0.014) | 0.079 (0.004) | 0.246 (0.007) | 0.67 (0.011) | 0.25 (0.007) |
| 9 | 762 | 437 | 57.35 | 1.34 (0.014) | 0.076 (0.004) | 0.221 (0.007) | 0.66 (0.013) | 0.22 (0.007) |
| 10 | 1110 | 538 | 48.47 | 1.47 (0.014) | 0.088 (0.003) | 0.289 (0.007) | 0.69 (0.009) | 0.29 (0.007) |
| 11 | 1110 | 510 | 45.95 | 1.42 (0.014) | 0.096 (0.004) | 0.265 (0.007) | 0.64 (0.011) | 0.26 (0.007) |
| UN | 1449 | 461 | 31.82 | 1.37 (0.015) | 0.086 (0.005) | 0.234 (0.008) | 0.59 (0.016) | 0.32 (0.008) |
| Overall mean | 11940 | 5864 | 49.11 | 1.43 (0.004) | 0.084 (0.001) | 0.267 (0.002) | 0.67 (0.003) | 0.27 (0.002) |
| Pop. | Na | Ne | I | Ho | He | FIS | %P | PA |
|---|---|---|---|---|---|---|---|---|
| Model-based population structure analysis | ||||||||
| SP1 | 5 | 1.189 (0.006) | 0.189 (0.004) | 0.035 (0.002) | 0.162 (0.003) | 0.684 (0.007) | 30.68 | 3 |
| SP2 | 18 | 1.368 (0.004) | 0.352 (0.003) | 0.116 (0.002) | 0.233 (0.002) | 0.398 (0.005) | 80.64 | 229 |
| SP3 | 40 | 1.397 (0.004) | 0.381 (0.003) | 0.083 (0.001) | 0.247 (0.002) | 0.617 (0.004) | 90.96 | 508 |
| SP4 | 27 | 1.258 (0.004) | 0.257 (0.003) | 0.068 (0.001) | 0.165 (0.002) | 0.437 (0.005) | 68.66 | 42 |
| Overall | 90 | 1.303 (0.002) | 0.295 (0.002) | 0.075 (0.001) | 0.202 (0.001) | 0.514 (0.003) | 67.74 | - |
| Distance-based population structure analysis | ||||||||
| C1 | 41 | 1.342 (0.004) | 0.340 (0.003) | 0.059 (0.001) | 0.218 (0.002) | 0.682 (0.004) | 86.77 | 65 |
| C2 | 24 | 1.404 (0.004) | 0.387 (0.003) | 0.108 (0.001) | 0.254 (0.002) | 0.512 (0.005) | 88.93 | 234 |
| C3 | 25 | 1.385 (0.004) | 0.366 (0.003) | 0.097 (0.001) | 0.240 (0.002) | 0.526 (0.005) | 85.44 | 177 |
| Overall | 90 | 1.377 (0.003) | 0.364 (0.002) | 0.088 (0.001) | 0.237 (0.001) | 0.573 (0.003) | 87.05 | - |
| Source | DF | SS | MS | Est. Var | Per. Var | F-Statistics |
|---|---|---|---|---|---|---|
| Model-based structure analysis | ||||||
| Among Population | 3 | 37,575.3 | 12,525.1 | 270.2 | 22.14 | FST = 0.221 (p < 0.001) |
| Among Individual | 86 | 144,053.5 | 1675.0 | 725.2 | 59.44 | FIS = 0.763 (p < 0.001) |
| Within Individual | 90 | 20,217.5 | 224.6 | 224.6 | 18.42 | FIT = 0.816 (p < 0.001) |
| Total | 179 | 201,846.2 | - | 1220.0 | 100.00 | - |
| Distance based structure analysis | ||||||
| Among Population | 2 | 20,380.1 | 10,190.1 | 143.8 | 12.16 | FST = 0.122 (p < 0.001) |
| Among Individual | 87 | 161,248.8 | 1853.4 | 814.4 | 68.85 | FIS = 0.784 (p < 0.001) |
| Within Individual | 90 | 20,217.5 | 224.64 | 224.6 | 18.99 | FIT = 0.810 (p < 0.001) |
| Total | 179 | 201,846.4 | - | 1182.8 | 100.0 | - |
| SP1 | SP2 | SP3 | SP4 | |
|---|---|---|---|---|
| SP1 | - | 0.239 (0.796) | 0.142 (1.511) | 0.192 (1.052) |
| SP2 | 0.214 (0.807) | - | 0.106 (2.108) | 0.149 (1.428) |
| SP3 | 0.112 (0.894) | 0.103 (0.902) | - | 0.103 (2.172) |
| SP4 | 0.134 (0.874) | 0.135 (0.874) | 0.089 (0.915) | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gumede, M.T.; Gerrano, A.S.; Amelework, A.B.; Modi, A.T. Analysis of Genetic Diversity and Population Structure of Cowpea (Vigna unguiculata (L.) Walp) Genotypes Using Single Nucleotide Polymorphism Markers. Plants 2022, 11, 3480. https://doi.org/10.3390/plants11243480
Gumede MT, Gerrano AS, Amelework AB, Modi AT. Analysis of Genetic Diversity and Population Structure of Cowpea (Vigna unguiculata (L.) Walp) Genotypes Using Single Nucleotide Polymorphism Markers. Plants. 2022; 11(24):3480. https://doi.org/10.3390/plants11243480
Chicago/Turabian StyleGumede, Mbali Thembi, Abe Shegro Gerrano, Assefa Beyene Amelework, and Albert Thembinkosi Modi. 2022. "Analysis of Genetic Diversity and Population Structure of Cowpea (Vigna unguiculata (L.) Walp) Genotypes Using Single Nucleotide Polymorphism Markers" Plants 11, no. 24: 3480. https://doi.org/10.3390/plants11243480
APA StyleGumede, M. T., Gerrano, A. S., Amelework, A. B., & Modi, A. T. (2022). Analysis of Genetic Diversity and Population Structure of Cowpea (Vigna unguiculata (L.) Walp) Genotypes Using Single Nucleotide Polymorphism Markers. Plants, 11(24), 3480. https://doi.org/10.3390/plants11243480