
α-Glucosidase Inhibitory Phytochemical Components of Chinese Endemic Plant Whitfordiodendron filipes var. tomentosum
 and
and 
Abstract
:1. Introduction
2. Results and Discussion
2.1. Phytochemical Components
2.2. Chemotaxonomic Significance
2.3. α-Glucosidase Inhibitory Activity
2.4. Strucuture–Activity Relationship Analysis
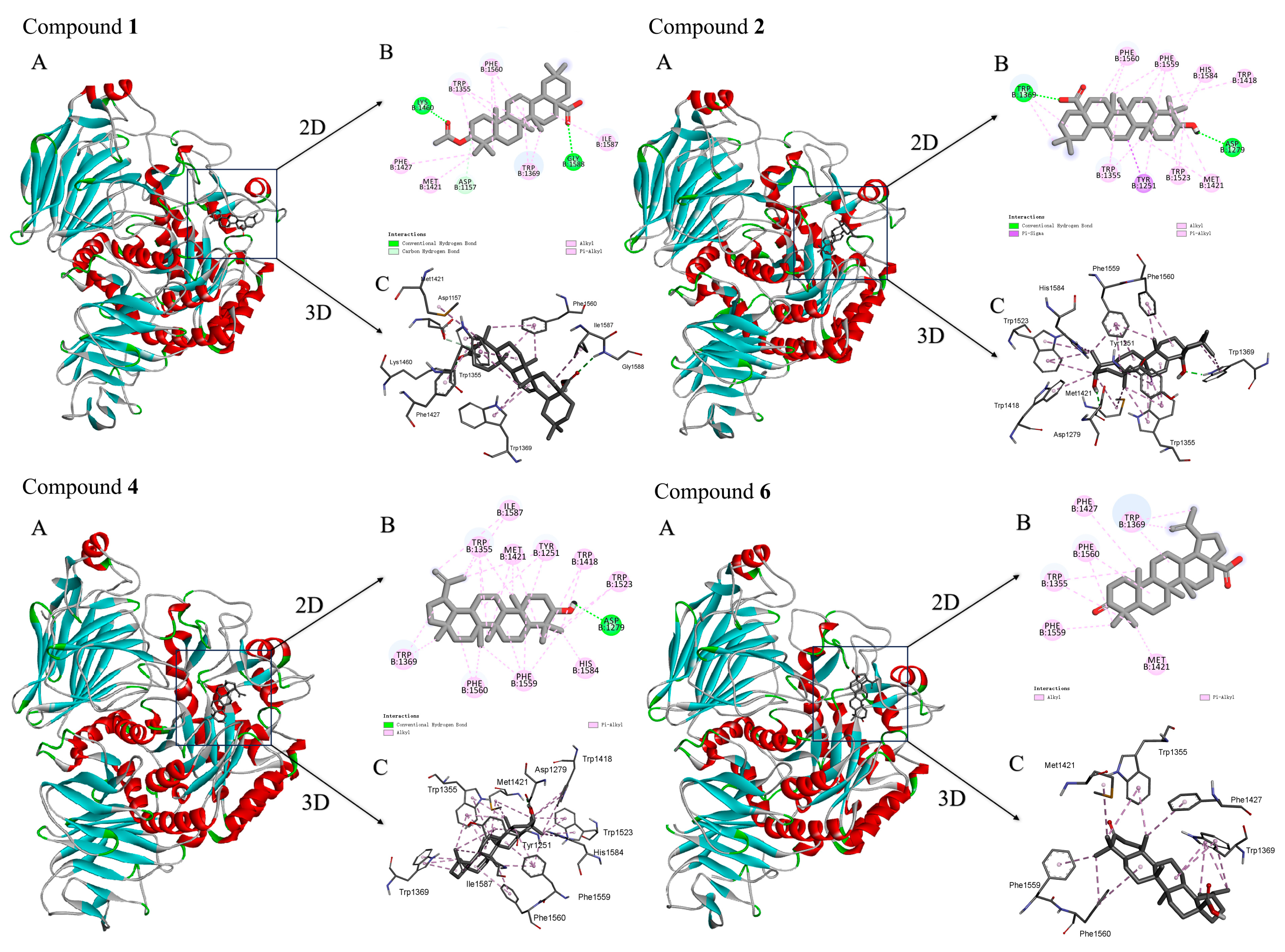
2.5. In Silico Study of Binding Modes
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Plant Material
3.3. Extraction and Isolation
3.4. Characteristic 1H and 13C NMR Spectral Data
3.5. In Vitro α-Glucosidase Inhibitory Activity Assay
3.6. Molecular Docking
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Yao, C.-L.; Zhang, J.-Q.; Li, J.-Y.; Wei, W.-L.; Wu, S.-F.; Guo, D.-A. Traditional Chinese medicine (TCM) as a source of new anticancer drugs. Nat. Prod. Rep. 2021, 38, 1618–1633. [Google Scholar] [CrossRef]
- Kong, L.Y.; Tan, R.X. Artemisinin, a miracle of traditional Chinese medicine. Nat. Prod. Rep. 2015, 32, 1617–1621. [Google Scholar] [CrossRef]
- Tu, Y. Artemisinin—A gift from traditional Chinese medicine to the world (Nobel lecture). Angew. Chem. Int. Ed. 2016, 55, 10210–10226. [Google Scholar] [CrossRef]
- Newman, D.J. Natural products and drug discovery. Natl. Sci. Rev. 2022, 9, nwac206. [Google Scholar] [CrossRef]
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs over the nearly four decades from 01/1981 to 09/2019. J. Nat. Prod. 2020, 83, 770–803. [Google Scholar] [CrossRef]
- Khayat, M.T.; Alharbi, M.; Ghazawi, K.F.; Mohamed, G.A.; Ibrahim, S.R.M. Ferula sinkiangensis (Chou-AWei, Chinese Ferula): Traditional uses, phytoconstituents, biosynthesis, and pharmacological activities. Plants 2023, 12, 902. [Google Scholar] [CrossRef]
- Zhao, J.-X.; Yue, J.-M. Frontier studies on natural products: Moving toward paradigm shifts. Sci. China Chem. 2023, 66, 928–942. [Google Scholar] [CrossRef]
- Li, L.; Chen, Z.; Zhang, X.; Jia, Y. Divergent strategy in natural product total synthesis. Chem. Rev. 2018, 118, 3752–3832. [Google Scholar] [CrossRef] [PubMed]
- Flora of China Editorial Committee. Whitfordiodendron. In Flora of China; Science Press: Beijing, China, 1994; Volume 40, p. 130. [Google Scholar]
- Lu, A.; Yan, H.; Hu, J.; Fu, X.; Qin, Z.; Guo, J.; Yang, M. Study on chemical constituents of Whitfordiodendron filipes. J. Dali Univ. 2017, 2, 45–48. [Google Scholar] [CrossRef]
- Liu, Y.-J. Studies on the Chemical Constituents of Whitfordiodendron filipes, Boschniakia himalaica and the Application of IBX in the Transformation of Plant Constituents. Master’s Dissertation, Yunnan Normal University, Kunming, China, 2015. [Google Scholar]
- Liu, Y.-J.; Wu, J.-C.; Li, H.-L.; Ma, Q.; Chen, Y.-G. Alkaloid and flavonoids from the seeds of Whitfordiodendron filipes. Chem. Nat. Compd. 2016, 52, 188–190. [Google Scholar] [CrossRef]
- Lu, A.-M. Studies on Chemical Constituents and Antioxidant Activities of Whitfordiodendron filips and Crotalaria ferruginea. Master’s Dissertation, Dali University, Dali, China, 2018. [Google Scholar]
- Lu, A.-m.; Fu, H.; Qin, Z.-m.; Zhu, X.-Y.; Guo, J.; Yang, M.-H. Chemical constituents from caulis of Whitfordiodendron filipes. J. Chin. Med. Mater. 2019, 42, 324–326. [Google Scholar] [CrossRef]
- Nguyen, H.T.; Nguyen, T.-T.; Duong, T.-H.; Tran, N.-M.-A.; Nguyen, C.H.; Nguyen, T.-H.-A.; Sichaem, J. α-Glucosidase inhibitory and antimicrobial benzoylphloroglucinols from Garcinia schomburgakiana fruits: In vitro and in silico studies. Molecules 2022, 27, 2574. [Google Scholar] [CrossRef] [PubMed]
- Hoang, M.H.; Nguyen, T.A.T.; Pham, N.K.T.; Dang, V.S.; Vo, T.N. A new oleanane-skeleton triterpene isolated from Coffea canephora. Nat. Prod. Res. 2022, 36, 5161–5167. [Google Scholar] [CrossRef] [PubMed]
- Derosa, G.; Maffioli, P. Efficacy and safety profile evaluation of acarbose alone and in association with other antidiabetic drugs: A systematic review. Clin. Ther. 2012, 34, 1221–1236. [Google Scholar] [CrossRef] [PubMed]
- Yin, Z.; Zhang, W.; Feng, F.; Zhang, Y.; Kang, W. α-Glucosidase inhibitors isolated from medicinal plants. Food Sci. Human Well. 2014, 3, 136–174. [Google Scholar] [CrossRef]
- Bu, Q.; Jin, Y.; Xu, M.-J.; Wu, L.; Liang, L.-F. Structurally diverse metabolites from the Ophiorrhiza japonica Bl. and their antioxidant activities in vitro and PPARα agonistic activities in silico. Molecules 2022, 27, 5301. [Google Scholar] [CrossRef]
- Bu, Q.; Yang, M.; Yan, X.-Y.; Li, S.-W.; Ge, Z.-Y.; Zhang, L.; Yao, L.-G.; Guo, Y.-W.; Liang, L.-F. Mililatensols A–C, new records of sarsolenane and capnosane diterpenes from soft coral Sarcophyton mililatensis. Mar. Drugs 2022, 20, 566. [Google Scholar] [CrossRef]
- Zhang, L.; Yang, M.; Chen, Z.-H.; Ge, Z.-Y.; Li, S.-W.; Yan, X.-Y.; Yao, L.-G.; Liang, L.-F.; Guo, Y.-W. Cembrane diterpenes possessing nonaromatic oxacycles from the Hainan soft coral Sarcophyton mililatensis. Int. J. Mol. Sci. 2023, 24, 1979. [Google Scholar] [CrossRef]
- Na, Z.; Xu, Y. Chemical constituents from twigs of Garcinia xipshuanbannaensis. China J. Chin. Mat. Med. 2009, 34, 2338–2342. [Google Scholar] [CrossRef]
- You, R.-R.; Chen, X.-Q.; He, D.-D.; Huang, C.-G.; Jin, Y.; Qian, S.-H.; Ju, J.-M.; Fan, J.-T. Chemical constituents from petroleum ether fraction of Swertia chirayita and their activities in vitro. China J. Chin. Mat. Med. 2017, 21, 3764–3769. [Google Scholar] [CrossRef]
- Ulubelen, A.; Topcu, G.; Lotter, H.; Wagner, H.; Eriş, C. Triterpenoids from the aerial parts of Salvia montbretii. Phytochemistry 1994, 36, 413–415. [Google Scholar] [CrossRef]
- Dantanarayana, A.P.; Kumar, N.S.; Muthukuda, P.M.; Mohamed, I.; Wazeer, M. A lupane derivative and the 13C NMR chemical shifts of some lupanols from Pleurostylia opposita. Phytochemistry 1982, 21, 2065–2068. [Google Scholar] [CrossRef]
- Khan, S.; Kazmi, M.H.; Ahmed, E.; Malik, A.; Sharif, A. Phytochemical studies on Sorbus cashmiriana. J. Chem. Soc. Pak. 2013, 35, 130–134. [Google Scholar]
- Lee, C.-K.; Chang, M.-H. Four new triterpenes from the heartwood of Melaleuca leucadendron. J. Nat. Prod. 1999, 62, 1003–1005. [Google Scholar] [CrossRef]
- Baker, P.M.; Barreiro, E.J.L.; Gilbert, B. Tetracyclic triterpenes of Barbacenia bicolor. Phytochemistry 1976, 15, 785–787. [Google Scholar] [CrossRef]
- Kontiza, I.; Abatis, D.; Malakate, K.; Vagias, C.; Roussis, V. 3-Keto steroids from the marine organisms Dendrophyllia cornigera and Cymodocea nodosa. Steroids 2006, 71, 177–181. [Google Scholar] [CrossRef] [PubMed]
- Hong, Q.-H.; Li, J.; Deng, L.-L.; Hao, X.-J.; Mu, S.-Z. Active constituents with α-glucosidase inhibitory activities from Sabia parviflora. Fitoterapia 2023, 167, 105516. [Google Scholar] [CrossRef] [PubMed]
- Chukwujekwu, J.C.; Rengasamy, K.R.R.; de Kock, C.A.; Smith, P.J.; Slavětínská, L.P.; van Staden, J. Alpha-glucosidase inhibitory and antiplasmodial properties of terpenoids from the leaves of Buddleja saligna Willd. J. Enzyme Inhib. Med. Chem. 2016, 31, 63–66. [Google Scholar] [CrossRef]
- Zhu, F.; Asada, T.; Sato, A.; Koi, Y.; Nishiwaki, H.; Tamura, H. Rosmarinic acid extract for antioxidant, antiallergic, and α-glucosidase inhibitory activities, isolated by supramolecular technique and solvent extraction from Perilla leaves. J. Agric. Food Chem. 2014, 62, 885–892. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Compound | IC50 (μM) |
|---|---|
| 1 | 16.8 ± 0.23 |
| 2 | 16.6 ± 0.25 |
| 3 | <100 |
| 4 | 19.2 ± 0.17 |
| 5 | <100 |
| 6 | 17.2 ± 0.27 |
| 7 | <100 |
| 8 | <100 |
| 9 | <100 |
| 10 | <100 |
| acarbose | 11.5 ± 0.05 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, J.-K.; Ge, Z.-Y.; Liao, X.-W.; Xue, J.; Wu, L.; Liang, L.-F. α-Glucosidase Inhibitory Phytochemical Components of Chinese Endemic Plant Whitfordiodendron filipes var. tomentosum. Plants 2024, 13, 692. https://doi.org/10.3390/plants13050692
Chen J-K, Ge Z-Y, Liao X-W, Xue J, Wu L, Liang L-F. α-Glucosidase Inhibitory Phytochemical Components of Chinese Endemic Plant Whitfordiodendron filipes var. tomentosum. Plants. 2024; 13(5):692. https://doi.org/10.3390/plants13050692
Chicago/Turabian StyleChen, Jun-Kun, Zeng-Yue Ge, Xiao-Wen Liao, Jun Xue, Lei Wu, and Lin-Fu Liang. 2024. "α-Glucosidase Inhibitory Phytochemical Components of Chinese Endemic Plant Whitfordiodendron filipes var. tomentosum" Plants 13, no. 5: 692. https://doi.org/10.3390/plants13050692
APA StyleChen, J. -K., Ge, Z. -Y., Liao, X. -W., Xue, J., Wu, L., & Liang, L. -F. (2024). α-Glucosidase Inhibitory Phytochemical Components of Chinese Endemic Plant Whitfordiodendron filipes var. tomentosum. Plants, 13(5), 692. https://doi.org/10.3390/plants13050692