
The Dynamic Changes of Brassica napus Seed Microbiota across the Entire Seed Life in the Field

 ,
,  ,
,  , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
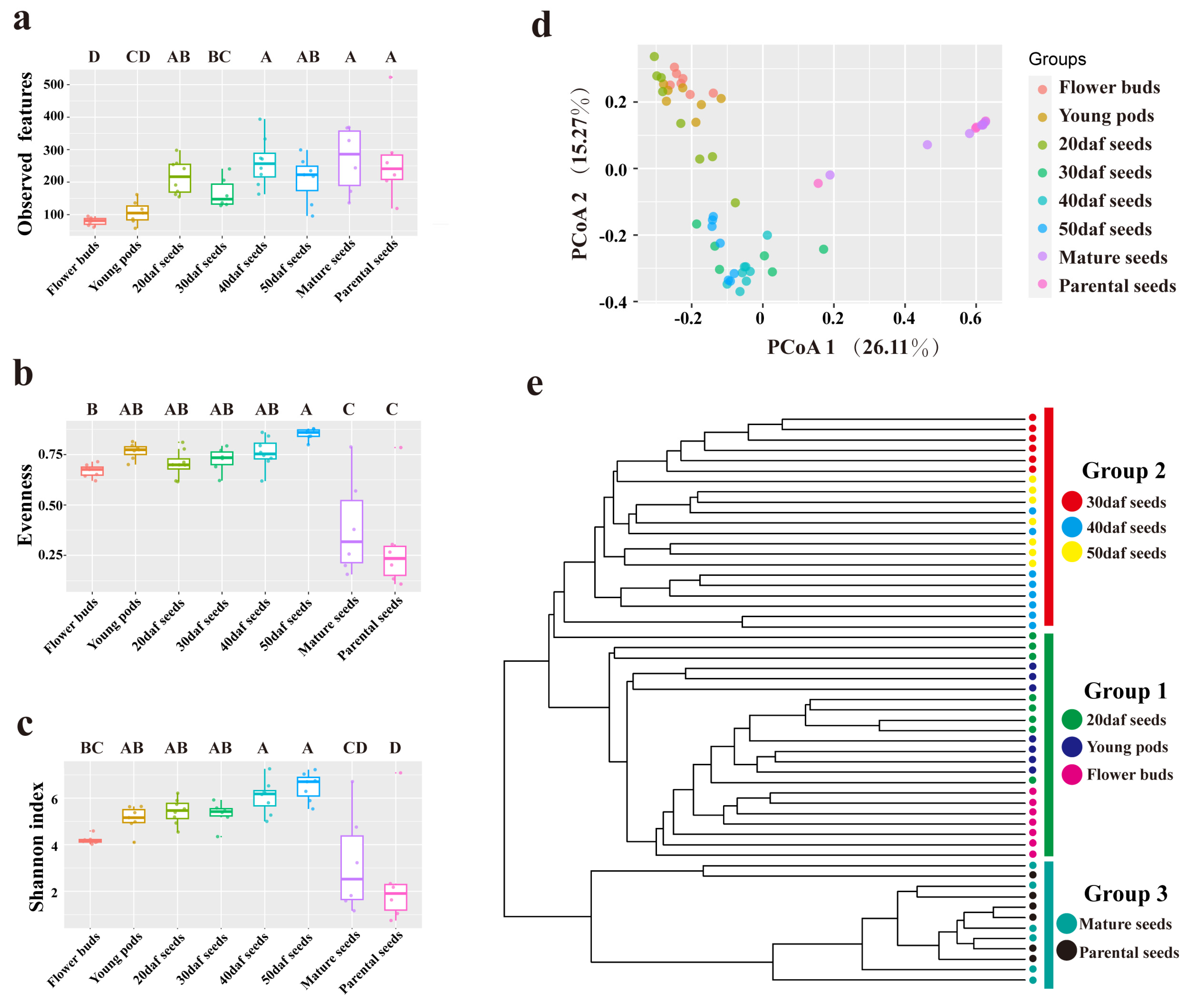
2.1. Alpha and Beta Diversity among All Samples
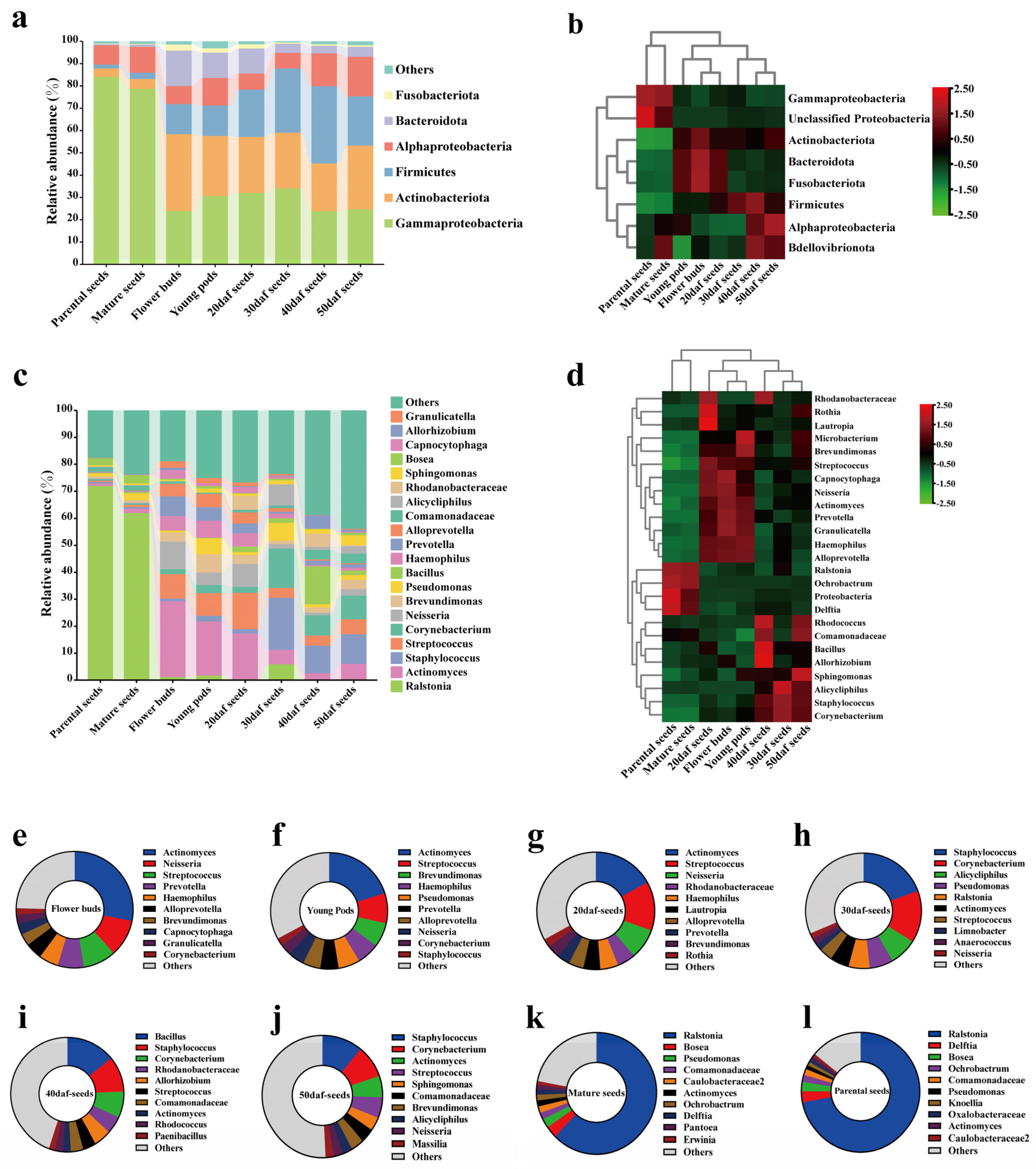
2.2. Composition and Differences in Seed Microbiota at the Phylum and Genus Levels
2.3. Functions of Seed Microbiota during Seed Development of Rapeseed
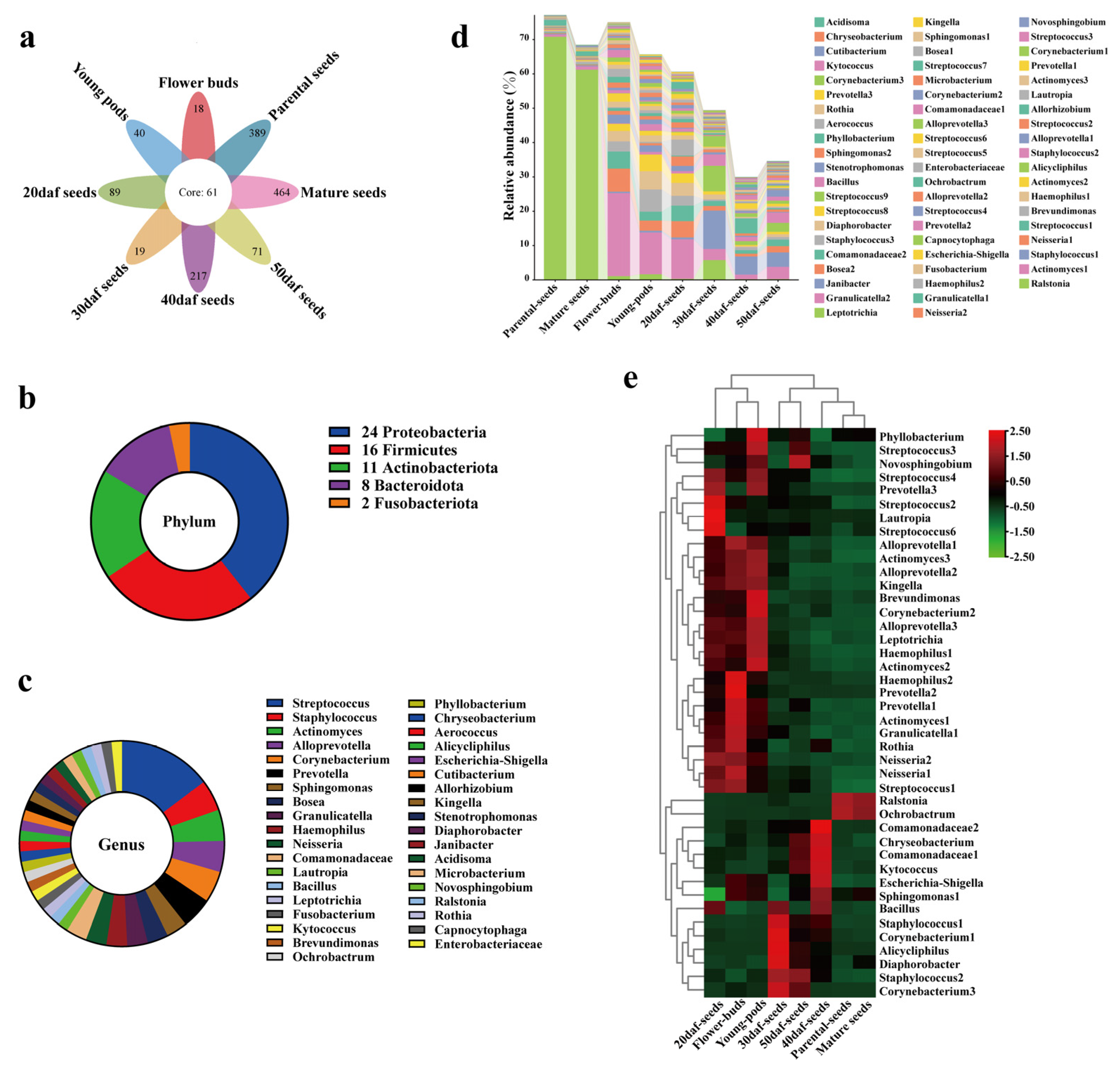
2.4. Identification of Seed Core Microbiota from Seed Sowing to Maturation
2.5. The Differences in Seed Microbiota between Developmental and Maturity Stages
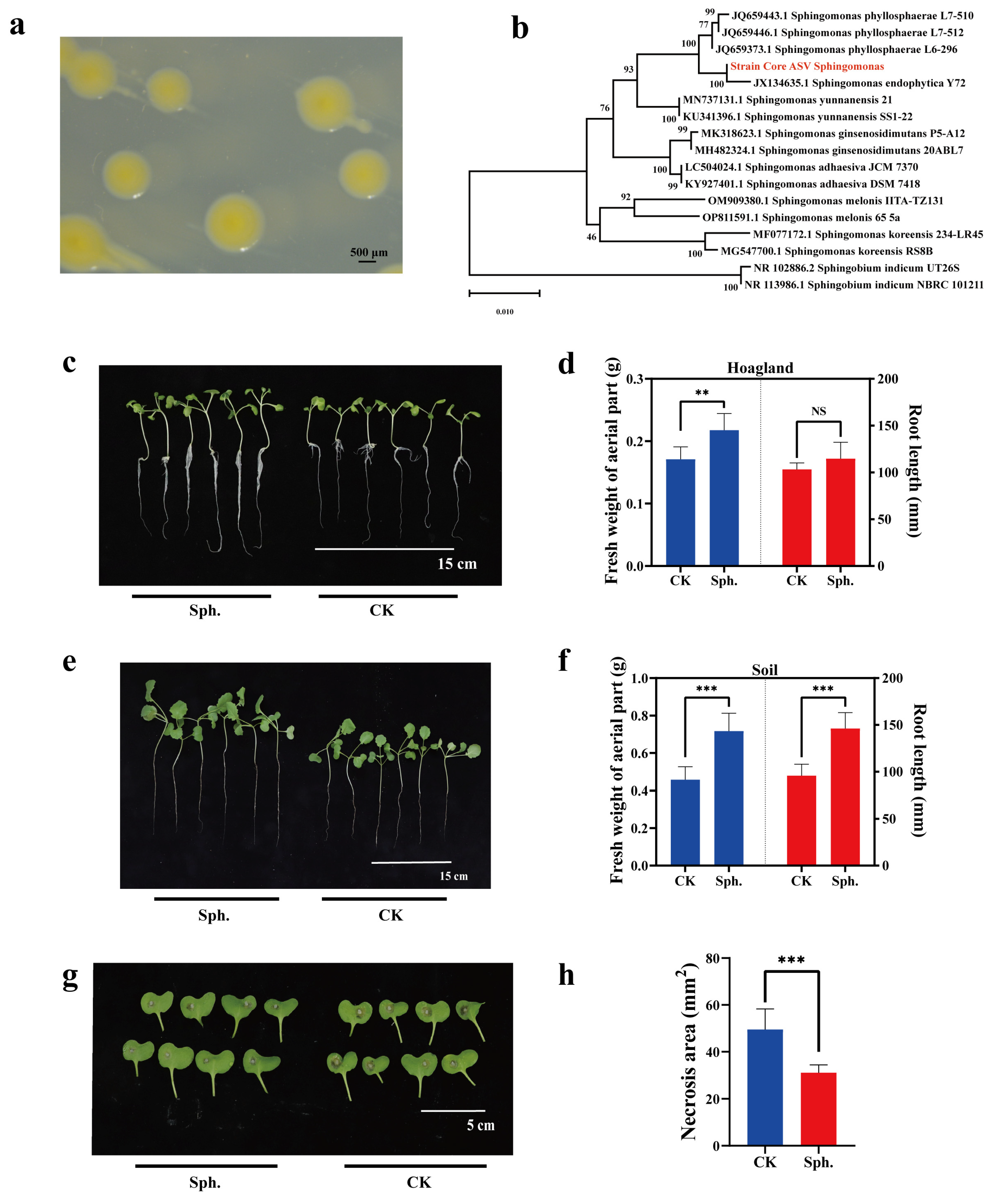
2.6. Exploring Microbial Resources of Rapeseed Seeds
3. Discussion
3.1. Dynamic Changes in the Composition of Seed Microbiota of Rapeseed, from Sowing to Harvesting
3.2. The Function of Microbiota Is Consistent with the Accumulation of Seed Metabolites during Seed Development
3.3. Core Microbiota from Sowing to Maturation of Rapeseed Seeds
3.4. Unique and Missing ASVs during Seed Development and Maturation
3.5. Providing a Basis for the Application Period of Microbes Sufficient to Establish Healthy Seed Microbiota
3.6. Application of Core Microbes
4. Methods
4.1. Plant Growth and Sample Collection
4.2. DNA Extraction, PCR Amplification and Sequencing
4.3. Bioinformatics Analysis of 16S rRNA Gene Profiling
4.4. Isolation and Identification of Seed Bacteria
4.5. Plant Treatments and Assays
4.6. Plant Growth Promotion Assay
4.7. Antagonistic Activities of Bacteria against Sc. sclerotiorum
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Singh, B.K.; Trivedi, P. Microbiome and the future for food and nutrient security. Microb. Biotechnol. 2017, 10, 50–53. [Google Scholar] [CrossRef]
- Vandenkoornhuyse, P.; Quaiser, A.; Duhamel, M.; Le Van, A.; Dufresne, A. The importance of the microbiome of the plant holobiont. New Phytol. 2015, 206, 1196–1206. [Google Scholar] [CrossRef]
- Kim, H.; Lee, Y.-H. The rice microbiome: A model platform for crop holobiome. Phytobiomes J. 2020, 4, 5–18. [Google Scholar] [CrossRef]
- Berg, G.; Raaijmakers, J.M. Saving seed microbiomes. ISME J. 2018, 12, 1167–1170. [Google Scholar] [CrossRef]
- Van Der Fels-Klerx, H.J.; Klemsdal, S.; Hietaniemi, V.; Lindblad, M.; Ioannou-Kakouri, E.; Van Asselt, E.D. Mycotoxin contamination of cereal grain commodities in relation to climate in North West Europe. Food Addit. Contam. Part A 2012, 29, 1581–1592. [Google Scholar] [CrossRef]
- Nelson, E.B. The seed microbiome: Origins, interactions, and impacts. Plant Soil. 2018, 422, 7–34. [Google Scholar] [CrossRef]
- Shahzad, R.; Waqas, M.; Khan, A.L.; Al-Hosni, K.; Kang, S.-M.; Seo, C.-W.; Lee, I.-J. Indoleacetic acid production and plant growth promoting potential of bacterial endophytes isolated from rice (Oryza sativa L.) seeds. Acta Biol. Hung. 2017, 68, 175–186. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-López, A.S.; Thijs, S.; Beckers, B.; González-Chávez, M.C.; Weyens, N.; Carrillo-González, R.; Vangronsveld, J. Community structure and diversity of endophytic bacteria in seeds of three consecutive generations of Crotalaria pumila growing on metal mine residues. Plant Soil 2018, 422, 51–66. [Google Scholar] [CrossRef]
- Rahman, M.D.M.; Flory, E.; Koyro, H.-W.; Abideen, Z.; Schikora, A.; Suarez, C.; Schnell, S.; Cardinale, M. Consistent associations with beneficial bacteria in the seed endosphere of barley (Hordeum vulgare L.). Syst. Appl. Microbiol. 2018, 41, 386–398. [Google Scholar] [CrossRef] [PubMed]
- Puente, M.E.; Li, C.Y.; Bashan, Y. Endophytic bacteria in cacti seeds can improve the development of cactus seedlings. Environ. Exp. Bot. 2009, 66, 402–408. [Google Scholar] [CrossRef]
- Shahzad, R.; Waqas, M.; Khan, A.L.; Asaf, S.; Khan, M.A.; Kang, S.-M.; Yun, B.-W.; Lee, I.-J. Seed-borne endophytic Bacillus amyloliquefaciens RWL-1 produces gibberellins and regulates endogenous phytohormones of Oryza sativa. Plant Physiol. Biochem. 2016, 106, 236–243. [Google Scholar] [CrossRef] [PubMed]
- Ruiza, D.; Agaras, B.; de Werrab, P.; Wall, L.G.; Valverde, C. Characterization and screening of plant probiotic traits of bacteria isolated from rice seeds cultivated in Argentina. J. Microbiol. 2011, 49, 902–912. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.; White, J. Indigenous endophytic seed bacteria promote seedling development and defend against fungal disease in browntop millet (Urochloa ramosa L.). J. Appl. Microbiol. 2018, 124, 764–778. [Google Scholar] [CrossRef] [PubMed]
- Jing, R.; Li, N.; Wang, W.; Liu, Y. An endophytic strain JK of genus bacillus isolated from the seeds of super hybrid rice (Oryza sativa L., Shenliangyou 5814) has antagonistic activity against rice blast pathogen. Microb. Pathog 2020, 147, 104422. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, H.; Fan, X.; Wang, Y.; Kusstatscher, P.; Duan, J.; Wu, S.; Chen, S.; Qiao, K.; Wang, Y.; Ma, B.; et al. Bacterial seed endophyte shapes disease resistance in rice. Nat. Plants 2021, 7, 60–72. [Google Scholar] [CrossRef] [PubMed]
- Qu, Z.; Zhao, H.; Zhang, H.; Wang, Q.; Yao, Y.; Cheng, J.; Lin, Y.; Xie, J.; Fu, Y.; Jiang, D. Bio-priming with a hypovirulent phytopathogenic fungus enhances the connection and strength of microbial interaction network in rapeseed. NPJ Biofilms Microbiomes 2020, 6, 45. [Google Scholar] [CrossRef] [PubMed]
- Tian, B.; Xie, J.; Fu, Y.; Cheng, J.; Li, B.; Chen, T.; Zhao, Y.; Gao, Z.; Yang, P.; Barbetti, M.J.; et al. A cosmopolitan fungal pathogen of dicots adopts an endophytic lifestyle on cereal crops and protects them from major fungal diseases. ISME J. 2020, 14, 3120–3135. [Google Scholar] [CrossRef]
- Zhang, H.; Xie, J.; Fu, Y.; Cheng, J.; Qu, Z.; Zhao, Z.; Cheng, S.; Chen, T.; Li, B.; Wang, Q.; et al. A 2-kb Mycovirus Converts a Pathogenic Fungus into a Beneficial Endophyte for Brassica Protection and Yield Enhancement. Mol. Plant 2020, 13, 1420–1433. [Google Scholar] [CrossRef]
- Bziuk, N.; Maccario, L.; Straube, B.; Wehner, G.; Sørensen, S.J.; Schikora, A.; Smalla, K. The treasure inside barley seeds: Microbial diversity and plant beneficial bacteria. Environ. Microbiome 2021, 16, 20. [Google Scholar] [CrossRef]
- Rybakova, D.; Mancinelli, R.; Wikström, M.; Birch-Jensen, A.-S.; Postma, J.; Ehlers, R.-U.; Goertz, S.; Berg, G. The structure of the Brassica napus seed microbiome is cultivar-dependent and affects the interactions of symbionts and pathogens. Microbiome 2017, 5, 104. [Google Scholar] [CrossRef]
- Wassermann, B.; Abdelfattah, A.; Wicaksono, W.A.; Kusstatscher, P.; Müller, H.; Cernava, T.; Goertz, S.; Rietz, S.; Abbadi, A.; Berg, G. The Brassica napus seed microbiota is cultivar-specific and transmitted via paternal breeding lines. Microb. Biotechnol. 2022, 15, 2379–2390. [Google Scholar] [CrossRef]
- Liu, Y.; Zuo, S.; Xu, L.; Zou, Y.; Song, W. Study on diversity of endophytic bacterial communities in seeds of hybrid maize and their parental lines. Arch. Microbiol. 2012, 194, 1001–1012. [Google Scholar] [CrossRef] [PubMed]
- Eyre, A.; Wang, M.; Oh, Y.; Dean, R. Identification and Characterization of the Core Rice Seed Microbiome. Phytobiomes J. 2019, 3, 148–157. [Google Scholar] [CrossRef]
- Zhang, X.; Ma, Y.N.; Wang, X.; Liao, K.; He, S.; Zhao, X.; Guo, H.; Zhao, D.; Wei, H.L. Dynamics of rice microbiomes reveal core vertically transmitted seed endophytes. Microbiome 2022, 10, 216. [Google Scholar] [CrossRef]
- Rezki, S.; Campion, C.; Simoneau, P.; Jacques, M.-A.; Shade, A.; Barret, M. Assembly of seed-associated microbial communities within and across successive plant generations. Plant Soil 2018, 422, 67–79. [Google Scholar] [CrossRef]
- Bergna, A.; Cernava, T.; Rändler, M.; Grosch, R.; Zachow, C.; Berg, G. Tomato Seeds Preferably Transmit Plant Beneficial Endophytes. Phytobiomes J. 2018, 2, 183–193. [Google Scholar] [CrossRef]
- Jin, J.; Krohn, C.; Franks, A.E.; Wang, X.; Wood, J.L.; Petrovski, S.; McCaskill, M.; Batinovic, S.; Xie, Z.; Tang, C. Elevated atmospheric CO2 alters the microbial community composition and metabolic potential to mineralize organic phosphorus in the rhizosphere of wheat. Microbiome 2022, 10, 12. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.G.; Xiong, C.; Wei, Z.; Chen, Q.L.; Ma, B.; Zhou, S.Y.; Tan, J.; Zhang, L.M.; Cui, H.L.; Duan, G.L. Impacts of global change on the phyllosphere microbiome. New Phytol. 2022, 234, 1977–1986. [Google Scholar] [CrossRef] [PubMed]
- Rudgers, J.A.; Afkhami, M.E.; Bell-Dereske, L.; Chung, Y.A.; Crawford, K.M.; Kivlin, S.N.; Mann, M.A.; Nuñez, M.A. Climate Disruption of Plant-Microbe Interactions. Annu. Rev. Ecol. Evol. Syst. 2020, 51, 561–586. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, Y.-X.; Zhang, N.; Hu, B.; Jin, T.; Xu, H.; Qin, Y.; Yan, P.; Zhang, X.; Guo, X.; et al. NRT1.1B is associated with root microbiota composition and nitrogen use in field-grown rice. Nat. Biotechnol. 2019, 37, 676–684. [Google Scholar] [CrossRef]
- Malacrinò, A.; Mosca, S.; Li Destri Nicosia, M.G.; Agosteo, G.E.; Schena, L. Plant Genotype Shapes the Bacterial Microbiome of Fruits, Leaves, and Soil in Olive Plants. Plants 2022, 11, 613. [Google Scholar] [CrossRef]
- Kwak, M.-J.; Kong, H.G.; Choi, K.; Kwon, S.-K.; Song, J.Y.; Lee, J.; Lee, P.A.; Choi, S.Y.; Seo, M.; Lee, H.J.; et al. Rhizosphere microbiome structure alters to enable wilt resistance in tomato. Nat. Biotechnol. 2018, 36, 1100–1109. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Abdelfattah, A.; Norelli, J.; Burchard, E.; Schena, L.; Droby, S.; Wisniewski, M. Apple endophytic microbiota of different rootstock/scion combinations suggests a genotype-specific influence. Microbiome 2018, 6, 18. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Waghmode, T.R.; Sun, R.; Kuramae, E.E.; Hu, C.; Liu, B. Root-associated microbiomes of wheat under the combined effect of plant development and nitrogen fertilization. Microbiome 2019, 7, 136. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhang, N.; Liu, Y.-X.; Zhang, X.; Hu, B.; Qin, Y.; Xu, H.; Wang, H.; Guo, X.; Qian, J.; et al. Root microbiota shift in rice correlates with resident time in the field and developmental stage. Sci. China Life Sci. 2018, 61, 613–621. [Google Scholar] [CrossRef] [PubMed]
- Xiong, C.; Singh, B.K.; He, J.Z.; Han, Y.L.; Li, P.P.; Wan, L.H.; Meng, G.Z.; Liu, S.Y.; Wang, J.T.; Wu, C.F.; et al. Plant developmental stage drives the differentiation in ecological role of the maize microbiome. Microbiome 2021, 9, 171. [Google Scholar] [CrossRef] [PubMed]
- Chaparro, J.M.; Badri, D.V.; Vivanco, J.M. Rhizosphere microbiome assemblage is affected by plant development. ISME J. 2014, 8, 790–803. [Google Scholar] [CrossRef] [PubMed]
- Beckers, B.; Op De Beeck, M.; Weyens, N.; Boerjan, W.; Vangronsveld, J. Structural variability and niche differentiation in the rhizosphere and endosphere bacterial microbiome of field-grown poplar trees. Microbiome 2017, 5, 25. [Google Scholar] [CrossRef] [PubMed]
- Cregger, M.A.; Veach, A.M.; Yang, Z.K.; Crouch, M.J.; Vilgalys, R.; Tuskan, G.A.; Schadt, C.W. The Populus holobiont: Dissecting the effects of plant niches and genotype on the microbiome. Microbiome 2018, 6, 31. [Google Scholar] [CrossRef]
- Bulgarelli, D.; Rott, M.; Schlaeppi, K.; Ver Loren van Themaat, E.; Ahmadinejad, N.; Assenza, F.; Rauf, P.; Huettel, B.; Reinhardt, R.; Schmelzer, E.; et al. Revealing structure and assembly cues for Arabidopsis root-inhabiting bacterial microbiota. Nature 2012, 488, 91–95. [Google Scholar] [CrossRef]
- Berendsen, R.L.; Vismans, G.; Yu, K.; Song, Y.; de Jonge, R.; Burgman, W.P.; Burmølle, M.; Herschend, J.; Bakker, P.A.; Pieterse, C.M. Disease-induced assemblage of a plant-beneficial bacterial consortium. ISME J. 2018, 12, 1496–1507. [Google Scholar] [CrossRef] [PubMed]
- Carrión, V.J.; Perez-Jaramillo, J.; Cordovez, V.; Tracanna, V.; de Hollander, M.; Ruiz-Buck, D.; Mendes, L.W.; van Ijcken, W.F.J.; Gomez-Exposito, R.; Elsayed, S.S.; et al. Pathogen-induced activation of disease-suppressive functions in the endophytic root microbiome. Science 2019, 366, 606–612. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Xiong, C.; Gao, C.; Tsui, C.K.M.; Wang, M.-M.; Zhou, X.; Zhang, A.-M.; Cai, L. Disease-induced changes in plant microbiome assembly and functional adaptation. Microbiome 2021, 9, 187. [Google Scholar] [CrossRef] [PubMed]
- Hou, S.; Thiergart, T.; Vannier, N.; Mesny, F.; Ziegler, J.; Pickel, B.; Hacquard, S. A microbiota–root–shoot circuit favours Arabidopsis growth over defence under suboptimal light. Nat. Plants 2021, 7, 1078–1092. [Google Scholar] [CrossRef]
- Trivedi, P.; Leach, J.E.; Tringe, S.G.; Sa, T.; Singh, B.K. Plant–microbiome interactions: From community assembly to plant health. Nat. Rev. Microbiol. 2020, 18, 607–621. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, M.; Huang, S.; Li, L.; Gao, Q.; Wang, Y.; Zhang, S.; Huang, S.; Yuan, L.; Wen, Y.; et al. A highly conserved core bacterial microbiota with nitrogen-fixation capacity inhabits the xylem sap in maize plants. Nat. Commun. 2022, 13, 3361. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, L.; George, T.S.; Feng, G. A core microbiome in the hyphosphere of arbuscular mycorrhizal fungi has functional significance in organic phosphorus mineralization. New Phytol. 2023, 238, 859–873. [Google Scholar] [CrossRef]
- Chang, J.; Tian, L.; Leite, M.F.A.; Sun, Y.; Shi, S.; Xu, S.; Wang, J.; Chen, H.; Chen, D.; Zhang, J.; et al. Nitrogen, manganese, iron, and carbon resource acquisition are potential functions of the wild rice Oryza rufipogon core rhizomicrobiome. Microbiome 2022, 10, 196. [Google Scholar] [CrossRef]
- Simonin, M.; Briand, M.; Chesneau, G.; Rochefort, A.; Marais, C.; Sarniguet, A.; Barret, M. Seed microbiota revealed by a large-scale meta-analysis including 50 plant species. New Phytol. 2022, 234, 1448–1463. [Google Scholar] [CrossRef]
- Toju, H.; Peay, K.G.; Yamamichi, M.; Narisawa, K.; Hiruma, K.; Naito, K.; Fukuda, S.; Ushio, M.; Nakaoka, S.; Onoda, Y.; et al. Core microbiomes for sustainable agroecosystems. Nat. Plants 2018, 4, 247–257. [Google Scholar] [CrossRef]
- Fukami, T. Historical Contingency in Community Assembly: Integrating Niches, Species Pools, and Priority Effects. Annu. Rev. Ecol. Evol. Syst. 2015, 46, 1–23. [Google Scholar] [CrossRef]
- Svoboda, P.; Lindström, E.S.; Ahmed Osman, O.; Langenheder, S. Dispersal timing determines the importance of priority effects in bacterial communities. ISME J. 2018, 12, 644–646. [Google Scholar] [CrossRef]
- Kim, H.; Jeon, J.; Lee, K.K.; Lee, Y.-H. Longitudinal transmission of bacterial and fungal communities from seed to seed in rice. Commun. Biol. 2022, 5, 772. [Google Scholar] [CrossRef]
- Abdelfattah, A.; Tack, A.J.M.; Lobato, C.; Wassermann, B.; Berg, G. From seed to seed: The role of microbial inheritance in the assembly of the plant microbiome. Trends Microbiol. 2023, 31, 346–355. [Google Scholar] [CrossRef]
- Johnston-Monje, D.; Raizada, M.N. Conservation and Diversity of Seed Associated Endophytes in Zea across Boundaries of Evolution, Ethnography and Ecology. PLoS ONE 2011, 6, e20396. [Google Scholar] [CrossRef] [PubMed]
- Hardoim, P.R.; Hardoim, C.C.P.; van Overbeek, L.S.; van Elsas, J.D. Dynamics of Seed-Borne Rice Endophytes on Early Plant Growth Stages. PLoS ONE 2012, 7, e30438. [Google Scholar] [CrossRef] [PubMed]
- Gundel, P.E.; Rudgers, J.A.; Ghersa, C.M. Incorporating the process of vertical transmission into understanding of host–symbiont dynamics. Oikos 2011, 120, 1121–1128. [Google Scholar] [CrossRef]
- Chesneau, G.; Torres-Cortes, G.; Briand, M.; Darrasse, A.; Preveaux, A.; Marais, C.; Jacques, M.A.; Shade, A.; Barret, M. Temporal dynamics of bacterial communities during seed development and maturation. FEMS Microbiol. Ecol. 2020, 96, fiaa190. [Google Scholar] [CrossRef] [PubMed]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.R.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G.I. PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 2020, 38, 685–688. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Liu, H.; Jin, C.; Li, Q.; Guo, L. An efficient and comprehensive plant glycerolipids analysis approach based on high-performance liquid chromatography–quadrupole time-of-flight mass spectrometer. Plant Direct 2019, 3, e00183. [Google Scholar] [CrossRef] [PubMed]
- Chia, T.Y.; Pike, M.J.; Rawsthorne, S. Storage oil breakdown during embryo development of Brassica napus (L.). J. Exp. Bot. 2005, 56, 1285–1296. [Google Scholar] [CrossRef]
- Gupta, M.; Bhaskar, P.B.; Sriram, S.; Wang, P.H. Integration of omics approaches to understand oil/protein content during seed development in oilseed crops. Plant Cell Rep. 2017, 36, 637–652. [Google Scholar] [CrossRef] [PubMed]
- Hua, S.; Chen, Z.-H.; Zhang, Y.; Yu, H.; Lin, B.; Zhang, D. Chlorophyll and carbohydrate metabolism in developing silique and seed are prerequisite to seed oil content of Brassica napus L. Bot. Stud. 2014, 55, 34. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Chao, H.; Yin, Y.; Li, H.; Wang, H.; Zhao, W.; Hou, D.; Zhang, L.; Zhang, C.; Li, M. New insight into the genetic basis of oil content based on noninvasive three-dimensional phenotyping and tissue-specific transcriptome in Brassica napus. Biotechnol. Biofuels Bioprod. 2023, 16, 88. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Zhu, S.; Yuan, Y.; Wang, Y.; Zeng, L.; Batley, J.; Wang, Y.P. Transcriptomic comparison between developing seeds of yellow- and black-seeded Brassica napus reveals that genes influence seed quality. BMC Plant Biol. 2019, 19, 203. [Google Scholar] [CrossRef] [PubMed]
- Seo, M.-S.; Kim, J.S. Understanding of MYB Transcription Factors Involved in Glucosinolate Biosynthesis in Brassicaceae. Molecules 2017, 22, 1549. [Google Scholar] [CrossRef] [PubMed]
- Bednarek, P. Chemical warfare or modulators of defence responses—The function of secondary metabolites in plant immunity. Curr. Opin. Plant Biol. 2012, 15, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Tian, Z.; Zhao, Y.; Havlickova, L.; He, Z.; Harper, A.L.; Bancroft, I.; Zou, X.; Zhang, X.; Lu, G. Dynamic and Associative Transcriptomic Analysis of Glucosinolate Content in Seeds and Silique Walls of Brassica napus. Sci. Agric. Sin. 2018, 51, 635–651. [Google Scholar] [CrossRef]
- Kueneman, J.G.; Bonadies, E.; Thomas, D.; Roubik, D.W.; Wcislo, W.T. Neotropical bee microbiomes point to a fragmented social core and strong species-level effects. Microbiome 2023, 11, 150. [Google Scholar] [CrossRef]
- Li, J.; Sauers, L.; Zhuang, D.; Ren, H.; Guo, J.; Wang, L.; Zhuang, M.; Guo, Y.; Zhang, Z.; Wu, J.; et al. Divergence and convergence of gut microbiomes of wild insect pollinators. mBio 2023, 14, e01270-23. [Google Scholar] [CrossRef]
- Tett, A.; Pasolli, E.; Masetti, G.; Ercolini, D.; Segata, N. Prevotella diversity, niches and interactions with the human host. Nat. Rev. Microbiol. 2021, 19, 585–599. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Brettell, L.E.; Singh, B. Linking the Phyllosphere Microbiome to Plant Health. Trends Plant Sci. 2020, 25, 841–844. [Google Scholar] [CrossRef] [PubMed]
- Bright, M.; Bulgheresi, S. A complex journey: Transmission of microbial symbionts. Nat. Rev. Microbiol. 2010, 8, 218–230. [Google Scholar] [CrossRef] [PubMed]
- Li, P.-D.; Zhu, Z.-R.; Zhang, Y.; Xu, J.; Wang, H.; Wang, Z.; Li, H. The phyllosphere microbiome shifts toward combating melanose pathogen. Microbiome 2022, 10, 56. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Zhang, Z.; Zhu, C.; Wang, T.; Wang, R.; Ruan, L. Heritability of tomato rhizobacteria resistant to Ralstonia solanacearum. Microbiome 2022, 10, 227. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Stirling, E.; Xie, H.; Li, W.; Lv, X.; Matsumoto, H.; Cheng, H.; Xu, A.; Lai, W.; Wang, Y.; et al. Continental scale deciphering of microbiome networks untangles the phyllosphere homeostasis in tea plant. J. Adv. Res. 2023, 44, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Shen, D.; Xiong, Q.; Bao, B.; Zhang, W.; Dai, T.; Zhao, Y.; Borriss, R.; Fan, B. The plant-beneficial rhizobacterium Bacillus velezensis FZB42 Controls the Soybean Pathogen Phytophthora sojae Due to Bacilysin Production. Appl. Environ. Microbiol. 2021, 87, e01601–e01621. [Google Scholar] [CrossRef]
- Berlanga-Clavero, M.V.; Molina-Santiago, C.; Caraballo-Rodríguez, A.M.; Petras, D.; Díaz-Martínez, L.; Pérez-García, A.; de Vicente, A.; Carrión, V.J.; Dorrestein, P.C.; Romero, D. Bacillus subtilis biofilm matrix components target seed oil bodies to promote growth and anti-fungal resistance in melon. Nat. Microbiol. 2022, 7, 1001–1015. [Google Scholar] [CrossRef]
- Santoyo, G. How plants recruit their microbiome? New insights into beneficial interactions. J. Adv. Res. 2022, 40, 45–58. [Google Scholar] [CrossRef]
- Tang, S.; Guo, N.; Tang, Q.; Peng, F.; Liu, Y.; Xia, H.; Lu, S.; Guo, L. Pyruvate transporter BnaBASS2 impacts seed oil accumulation in Brassica napus. Plant Biotechnol. J. 2022, 20, 2406–2417. [Google Scholar] [CrossRef] [PubMed]
- Durán, P.; Thiergart, T.; Garrido-Oter, R.; Agler, M.; Kemen, E.; Schulze-Lefert, P.; Hacquard, S. Microbial Interkingdom Interactions in Roots Promote Arabidopsis Survival. Cell 2018, 175, 973–983. [Google Scholar] [CrossRef] [PubMed]
- Wen, T.; Yuan, J.; He, X.; Lin, Y.; Huang, Q.; Shen, Q. Enrichment of beneficial cucumber rhizosphere microbes mediated by organic acid secretion. Hortic. Res. 2020, 7, 154. [Google Scholar] [CrossRef] [PubMed]
- Xun, W.; Liu, Y.; Li, W.; Ren, Y.; Xiong, W.; Xu, Z.; Zhang, N.; Miao, Y.; Shen, Q.; Zhang, R. Specialized metabolic functions of keystone taxa sustain soil microbiome stability. Microbiome 2021, 9, 35. [Google Scholar] [CrossRef] [PubMed]
- Brachi, B.; Filiault, D.; Whitehurst, H.; Darme, P.; Le Gars, P.; Le Mentec, M.; Morton, T.C.; Kerdaffrec, E.; Rabanal, F.; Anastasio, A.; et al. Plant genetic effects on microbial hubs impact host fitness in repeated field trials. Proc. Natl. Acad. Sci. USA 2022, 119, e2201285119. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Gao, L.; Gao, Z.; Tian, B.; Chen, T.; Xie, J.; Cheng, J.; Fu, Y.; Li, Y.; Xiao, S.; et al. Exploring a rhizobium to fix nitrogen in non-leguminous plants by using a tumor-formation root pathogen. Phytopathol. Res. 2022, 4, 48. [Google Scholar] [CrossRef]
- Li, Y.; Yang, R.; Häggblom, M.M.; Li, M.; Guo, L.; Li, B.; Kolton, M.; Cao, Z.; Soleimani, M.; Chen, Z.; et al. Characterization of diazotrophic root endophytes in Chinese silvergrass (Miscanthus sinensis). Microbiome 2022, 10, 186. [Google Scholar] [CrossRef]
- Garrido-Oter, R.; Nakano, R.T.; Dombrowski, N.; Ma, K.W.; McHardy, A.C.; Schulze-Lefert, P. Modular Traits of the Rhizobiales Root Microbiota and Their Evolutionary Relationship with Symbiotic Rhizobia. Cell Host Microbe 2018, 24, 155–167. [Google Scholar] [CrossRef]
- Li, Z.; Bai, X.; Jiao, S.; Li, Y.; Li, P.; Yang, Y.; Zhang, H.; Wei, G. A simplified synthetic community rescues Astragalus mongholicus from root rot disease by activating plant-induced systemic resistance. Microbiome 2021, 9, 217. [Google Scholar] [CrossRef]
- Liu, H.; Li, J.; Carvalhais, L.C.; Percy, C.D.; Prakash Verma, J.; Schenk, P.M.; Singh, B.K. Evidence for the plant recruitment of beneficial microbes to suppress soil-borne pathogens. New Phytol. 2021, 229, 2873–2885. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Xu, Z.; Xie, J.; Hesselberg-Thomsen, V.; Tan, T.; Zheng, D.; Strube, M.L.; Dragoš, A.; Shen, Q.; Zhang, R.; et al. Bacillus velezensis stimulates resident rhizosphere Pseudomonas stutzeri for plant health through metabolic interactions. ISME J. 2022, 16, 774–787. [Google Scholar] [CrossRef] [PubMed]
- Ye, M.; Tang, X.; Yang, R.; Zhang, H.; Li, F.; Tao, F.; Li, F.; Wang, Z. Characteristics and Application of a Novel Species of Bacillus: Bacillus velezensis. ACS Chem. Biol. 2018, 13, 500–505. [Google Scholar] [CrossRef]
- Unemo, M.; Shafer, W.M. Antimicrobial resistance in Neisseria gonorrhoeae in the 21st century: Past, evolution, and future. Clin. Microbiol. Rev. 2014, 27, 587–613. [Google Scholar] [CrossRef] [PubMed]
- Prado, A.; Marolleau, B.; Vaissière, B.E.; Barret, M.; Torres-Cortes, G. Insect pollination: An ecological process involved in the assembly of the seed microbiota. Sci. Rep. 2020, 10, 3575. [Google Scholar] [CrossRef]
- Banerjee, S.; Van Der Heijden, M.G. Soil microbiomes and one health. Nat. Rev. Microbiol. 2023, 21, 6–20. [Google Scholar] [CrossRef]
- De Filippis, F.; Pasolli, E.; Tett, A.; Tarallo, S.; Naccarati, A.; De Angelis, M.; Neviani, E.; Cocolin, L.; Gobbetti, M.; Segata, N.; et al. Distinct Genetic and Functional Traits of Human Intestinal Prevotella copri Strains Are Associated with Different Habitual Diets. Cell Host Microbe 2019, 25, 444–453.e3. [Google Scholar] [CrossRef] [PubMed]
- Hayward, A.C. Biology and epidemiology of bacterial wilt caused by pseudomonas solanacearum. Annu. Rev. Phytopathol. 1991, 29, 65–87. [Google Scholar] [CrossRef]
- Jiang, G.; Wei, Z.; Xu, J.; Chen, H.; Zhang, Y.; She, X.; Macho, A.P.; Ding, W.; Liao, B. Bacterial Wilt in China: History, Current Status, and Future Perspectives. Front. Plant Sci. 2017, 8, 1549. [Google Scholar] [CrossRef]
- Wei, Z.; Huang, J.; Tan, S.; Mei, X.; Shen, Q.; Xu, Y. The congeneric strain Ralstonia pickettii QL-A6 of Ralstonia solanacearum as an effective biocontrol agent for bacterial wilt of tomato. Biol. Control 2013, 65, 278–285. [Google Scholar] [CrossRef]
- Menekşe, Ş.; Hacıseyitoğlu, D.; Süzük Yıldız, S.; Bayrakdar, F. An outbreak of Ralstonia pickettii bloodstream infection and clinical outcomes. J. Infect. Dev. Ctries. 2022, 16, 705–711. [Google Scholar] [CrossRef]
- Fang, Q.; Feng, Y.; Feng, P.; Wang, X.; Zong, Z. Nosocomial bloodstream infection and the emerging carbapenem-resistant pathogen Ralstonia insidiosa. BMC Infect. Dis. 2019, 19, 334. [Google Scholar] [CrossRef]
- Steinbüchel, A.; Schlegel, H.G. Physiology and molecular genetics of poly(β-hydroxyalkanoic acid) synthesis in Alcaligenes eutrophus. Mol. Mircobiol 1991, 5, 535–542. [Google Scholar] [CrossRef]
- Valentin, H.E.; Broyles, D.L.; Casagrande, L.A.; Colburn, S.M.; Creely, W.L.; DeLaquil, P.A.; Felton, H.M.; Gonzalez, K.A.; Houmiel, K.L.; Lutke, K.; et al. PHA production, from bacteria to plants. Int. J. Biol. Macromol. 1999, 25, 303–306. [Google Scholar] [CrossRef]
- Houmiel, K.L.; Slater, S.; Broyles, D.; Casagrande, L.; Colburn, S.; Gonzalez, K.; Mitsky, T.A.; Reiser, S.E.; Shah, D.; Taylor, N.B.; et al. Poly(beta-hydroxybutyrate) production in oilseed leukoplasts of Brassica napus. Planta 1999, 209, 547–550. [Google Scholar] [CrossRef] [PubMed]
- Hughes, A.; Mott, I.; Dunnill, P. Studies with natural rapeseed and microbially derived polyhydroxybutyrate to simulate extraction of plastic from transgenic material. Bioprocess. Eng. 2000, 23, 257–263. [Google Scholar] [CrossRef]
- Nasir, N.; Sayeed, M.A.; Jamil, B. Ralstonia pickettii Bacteremia: An Emerging Infection in a Tertiary Care Hospital Setting. Cureus 2019, 11, e5084. [Google Scholar] [CrossRef] [PubMed]
- Zhong, J.; Wu, D.; Zeng, Y.; Wu, G.; Zheng, N.; Huang, W.; Li, Y.; Tao, X.; Zhu, W.; Sheng, L.; et al. The Microbial and Metabolic Signatures of Patients with Stable Coronary Artery Disease. Microbiol. Spectr. 2022, 10, e02467-22. [Google Scholar] [CrossRef]
- Seyyedi, S.M.; Tavakkol Afshari, R.; Daneshmandi, M.S. The relationships between fatty acids and heterotrophic seedling growth in winter canola cultivars during accelerated seed aging process. S. Afr. J. Bot. 2018, 119, 353–361. [Google Scholar] [CrossRef]
- Tyler, H.L.; Triplett, E.W. Plants as a Habitat for Beneficial and/or Human Pathogenic Bacteria. Annu. Rev. Phytopathol. 2008, 46, 53–73. [Google Scholar] [CrossRef]
- Cooley Michael, B.; Miller William, G.; Mandrell Robert, E. Colonization of Arabidopsis thaliana with Salmonella enterica and Enterohemorrhagic Escherichia coli O157:H7 and Competition by Enterobacter asburiae. Appl. Environ. Microbiol. 2003, 69, 4915–4926. [Google Scholar] [CrossRef] [PubMed]
- Mitter, B.; Pfaffenbichler, N.; Flavell, R.; Compant, S.; Antonielli, L.; Petric, A.; Berninger, T.; Naveed, M.; Sheibani-Tezerji, R.; von Maltzahn, G.; et al. A New Approach to Modify Plant Microbiomes and Traits by Introducing Beneficial Bacteria at Flowering into Progeny Seeds. Front. Microbiol. 2017, 8, 11. [Google Scholar] [CrossRef]
- Wei, Z.; Jousset, A. Plant breeding goes microbial. Trends Plant Sci. 2017, 22, 555–558. [Google Scholar] [CrossRef]
- Singh, B.K.; Trivedi, P.; Egidi, E.; Macdonald, C.A.; Delgado-Baquerizo, M. Crop microbiome and sustainable agriculture. Nat. Rev. Microbiol. 2020, 18, 601–602. [Google Scholar] [CrossRef]
- Qiu, Z.; Egidi, E.; Liu, H.; Kaur, S.; Singh, B.K. New frontiers in agriculture productivity: Optimised microbial inoculants and in situ microbiome engineering. Biotechnol. Adv. 2019, 37, 107371. [Google Scholar] [CrossRef]
- de Muinck, E.J.; Trosvik, P.; Gilfillan, G.D.; Hov, J.R.; Sundaram, A.Y. A novel ultra high-throughput 16S rRNA gene amplicon sequencing library preparation method for the Illumina HiSeq platform. Microbiome 2017, 5, 68. [Google Scholar] [CrossRef] [PubMed]
- Chelius, M.; Triplett, E. The Diversity of Archaea and Bacteria in Association with the Roots of Zea mays L. Microb. Ecol. 2001, 41, 252–263. [Google Scholar] [CrossRef] [PubMed]
- Lundberg, D.S.; Lebeis, S.L.; Paredes, S.H.; Yourstone, S.; Gehring, J.; Malfatti, S.; Tremblay, J.; Engelbrektson, A.; Kunin, V.; Rio, T.G.d. Defining the core Arabidopsis thaliana root microbiome. Nature 2012, 488, 86–90. [Google Scholar] [CrossRef] [PubMed]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef] [PubMed]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, P.; Parfrey, L.W.; Yarza, P.; Gerken, J.; Pruesse, E.; Quast, C.; Schweer, T.; Peplies, J.; Ludwig, W.; Glöckner, F.O. The SILVA and “All-species Living Tree Project (LTP)” taxonomic frameworks. Nucleic Acids Res. 2014, 42, D643–D648. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Nomura, K.; Wang, X.; Sohrabi, R.; Xu, J.; Yao, L.; Paasch, B.C.; Ma, L.; Kremer, J.; Cheng, Y.; et al. A plant genetic network for preventing dysbiosis in the phyllosphere. Nature 2020, 580, 653–657. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Müller, D.B.; Srinivas, G.; Garrido-Oter, R.; Potthoff, E.; Rott, M.; Dombrowski, N.; Münch, P.C.; Spaepen, S.; Remus-Emsermann, M.; et al. Functional overlap of the Arabidopsis leaf and root microbiota. Nature 2015, 528, 364–369. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yao, Y.; Liu, C.; Zhang, Y.; Lin, Y.; Chen, T.; Xie, J.; Chang, H.; Fu, Y.; Cheng, J.; Li, B.; et al. The Dynamic Changes of Brassica napus Seed Microbiota across the Entire Seed Life in the Field. Plants 2024, 13, 912. https://doi.org/10.3390/plants13060912
Yao Y, Liu C, Zhang Y, Lin Y, Chen T, Xie J, Chang H, Fu Y, Cheng J, Li B, et al. The Dynamic Changes of Brassica napus Seed Microbiota across the Entire Seed Life in the Field. Plants. 2024; 13(6):912. https://doi.org/10.3390/plants13060912
Chicago/Turabian StyleYao, Yao, Changxing Liu, Yu Zhang, Yang Lin, Tao Chen, Jiatao Xie, Haibin Chang, Yanping Fu, Jiasen Cheng, Bo Li, and et al. 2024. "The Dynamic Changes of Brassica napus Seed Microbiota across the Entire Seed Life in the Field" Plants 13, no. 6: 912. https://doi.org/10.3390/plants13060912
APA StyleYao, Y., Liu, C., Zhang, Y., Lin, Y., Chen, T., Xie, J., Chang, H., Fu, Y., Cheng, J., Li, B., Yu, X., Lyu, X., Feng, Y., Bian, X., & Jiang, D. (2024). The Dynamic Changes of Brassica napus Seed Microbiota across the Entire Seed Life in the Field. Plants, 13(6), 912. https://doi.org/10.3390/plants13060912