
Identification and Genetic Dissection of Resistance to Red Crown Rot Disease in a Diverse Soybean Germplasm Population

 ,
, 

 , and
, and
Abstract
:1. Introduction
2. Results
2.1. Response of Soybean Accessions to Red Crown Rot Strain
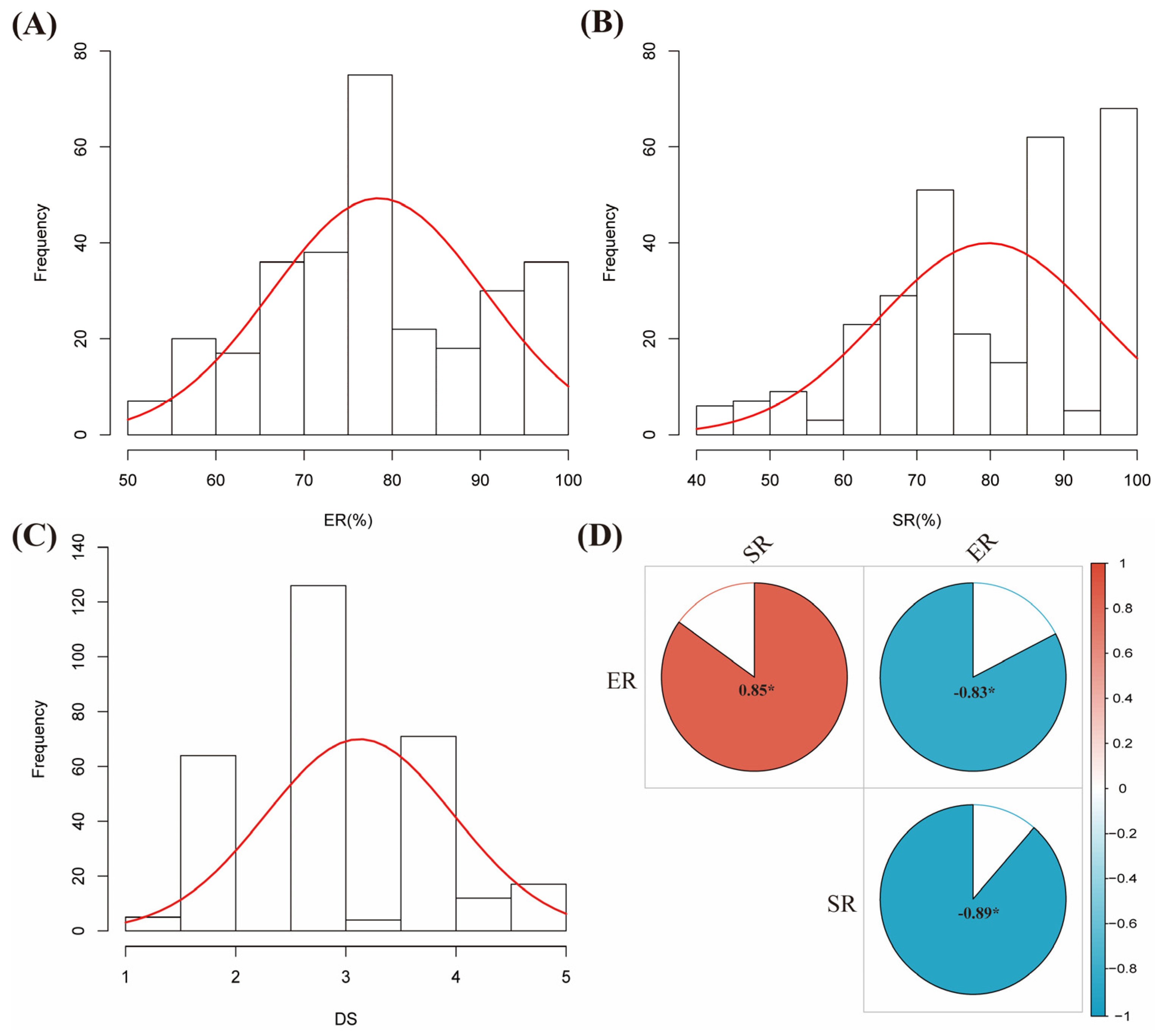
2.2. Identification of Resistance to Soybean Red Crown Rot Strain
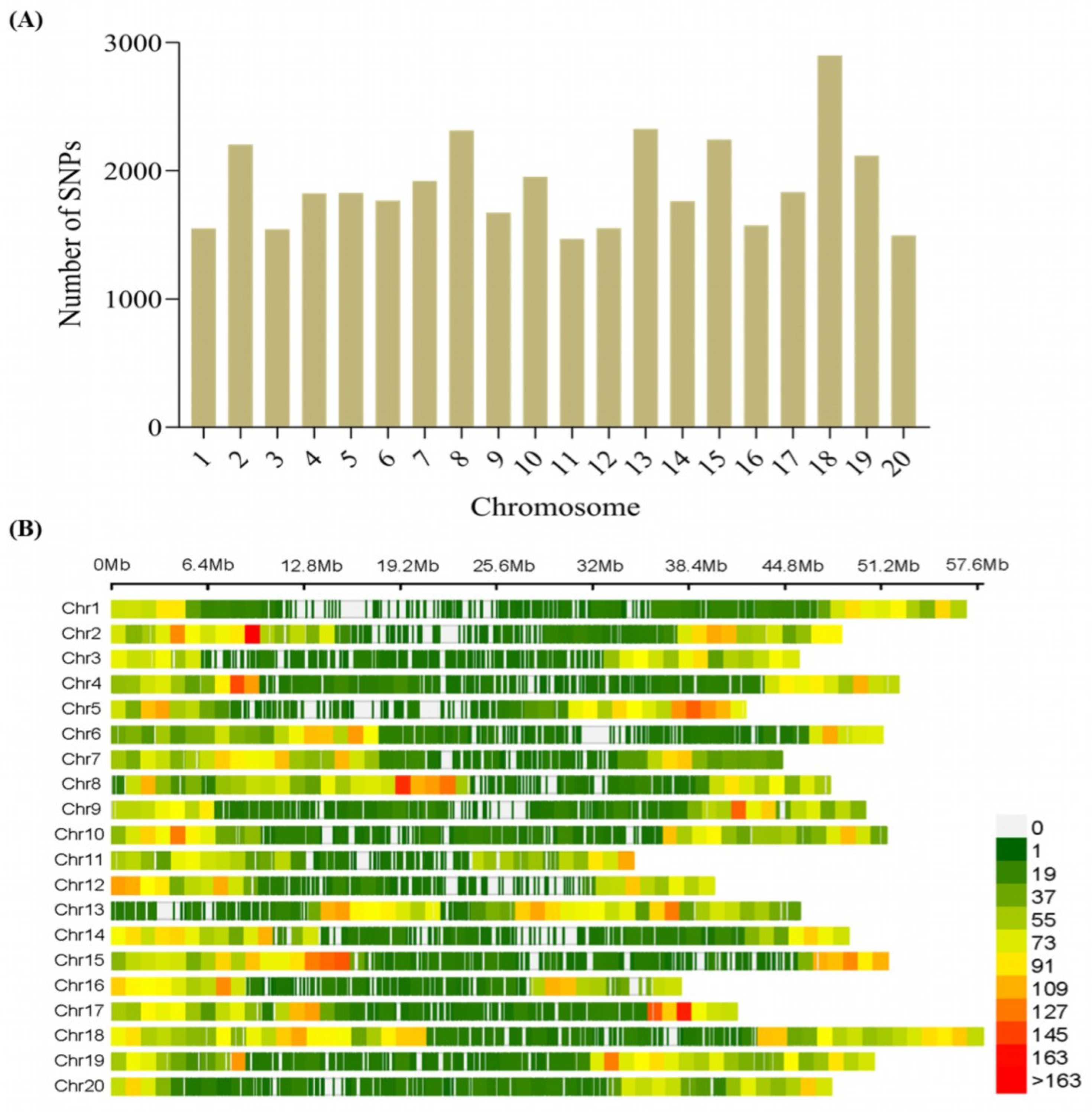
2.3. SNP Density and Distribution among the 20 Chromosomes of Soybean
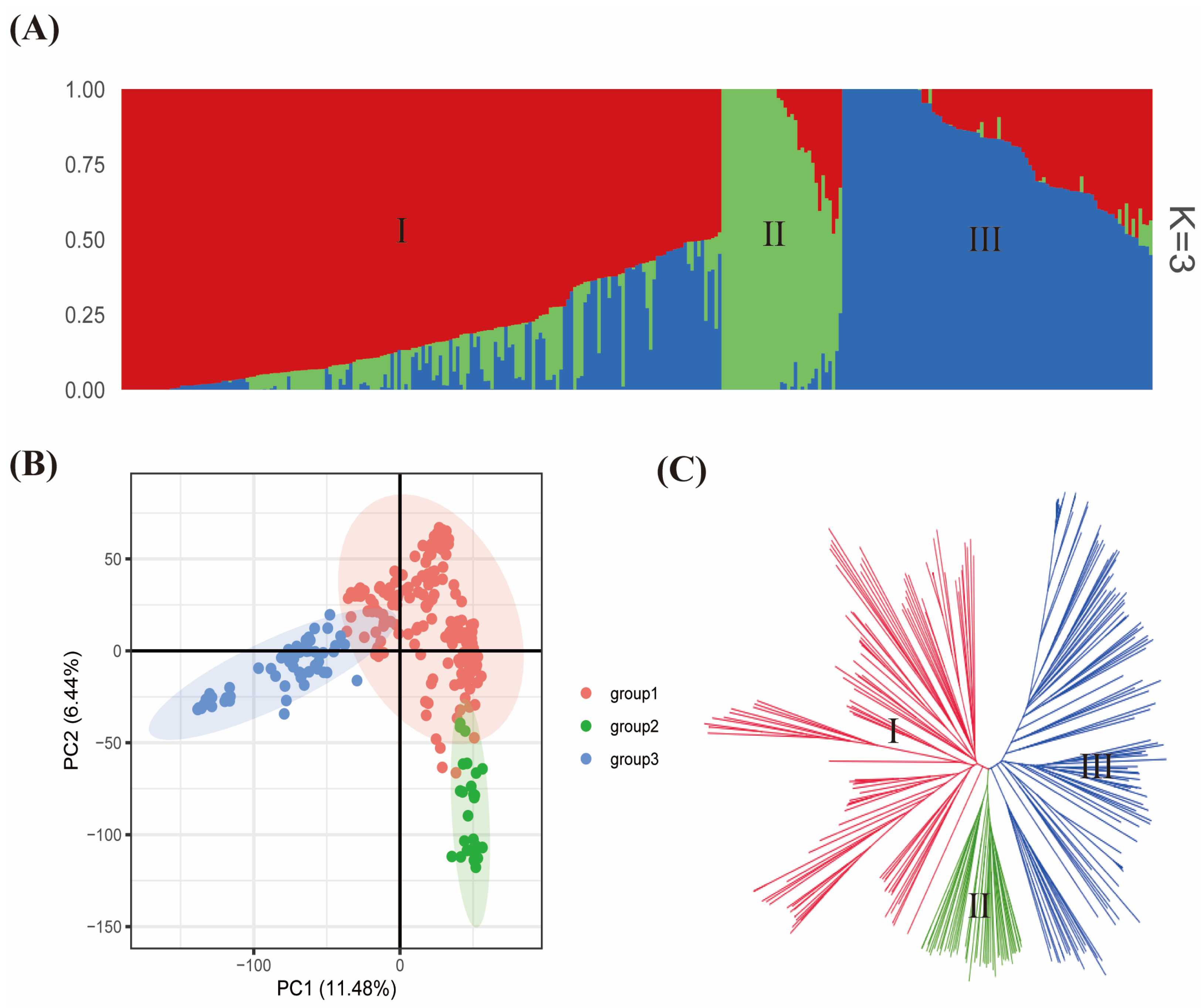
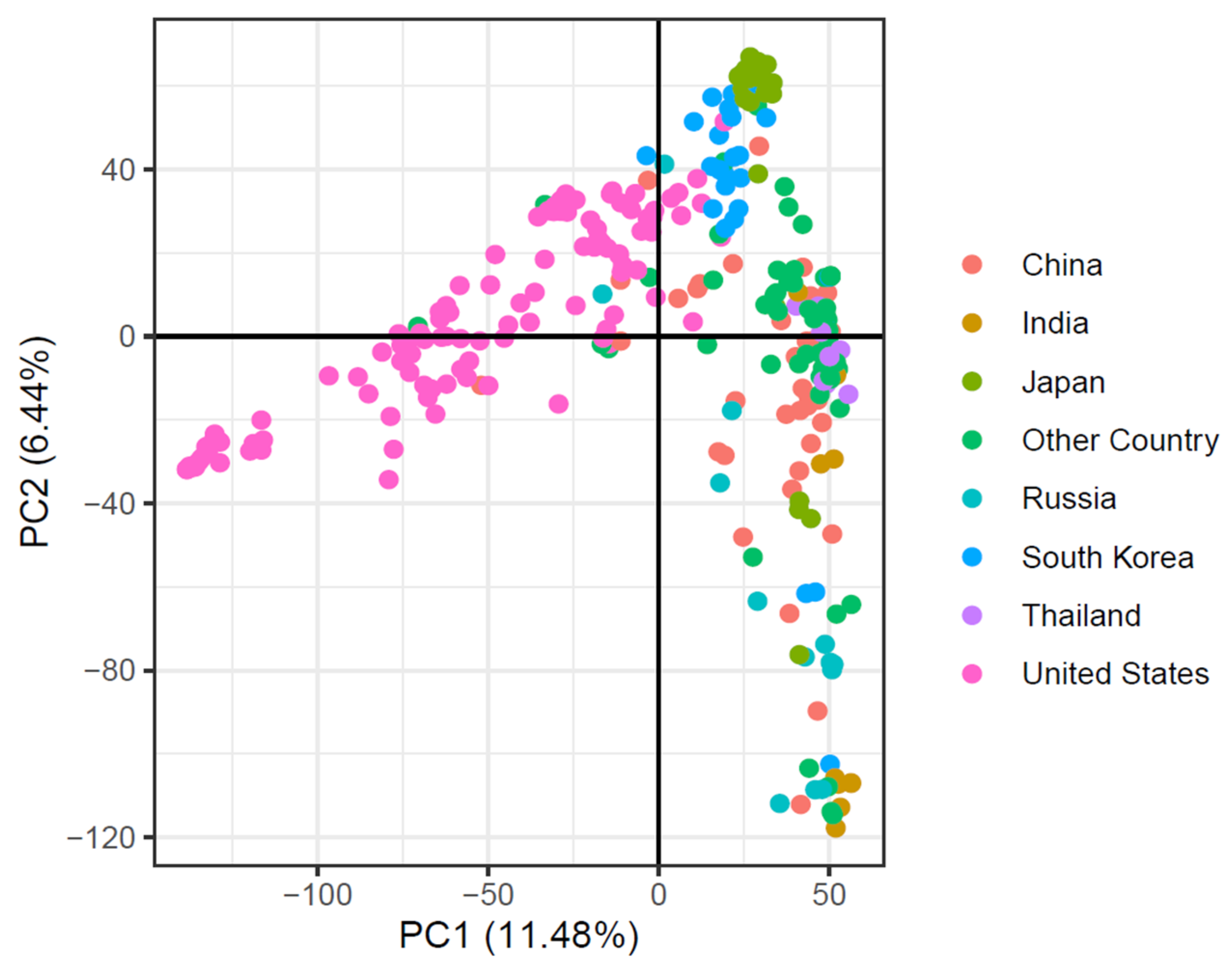
2.4. Worldwide Soybean Germplasm, Its Population Stratification, Genetic Diversity, and Population Structures Based on Their Origin
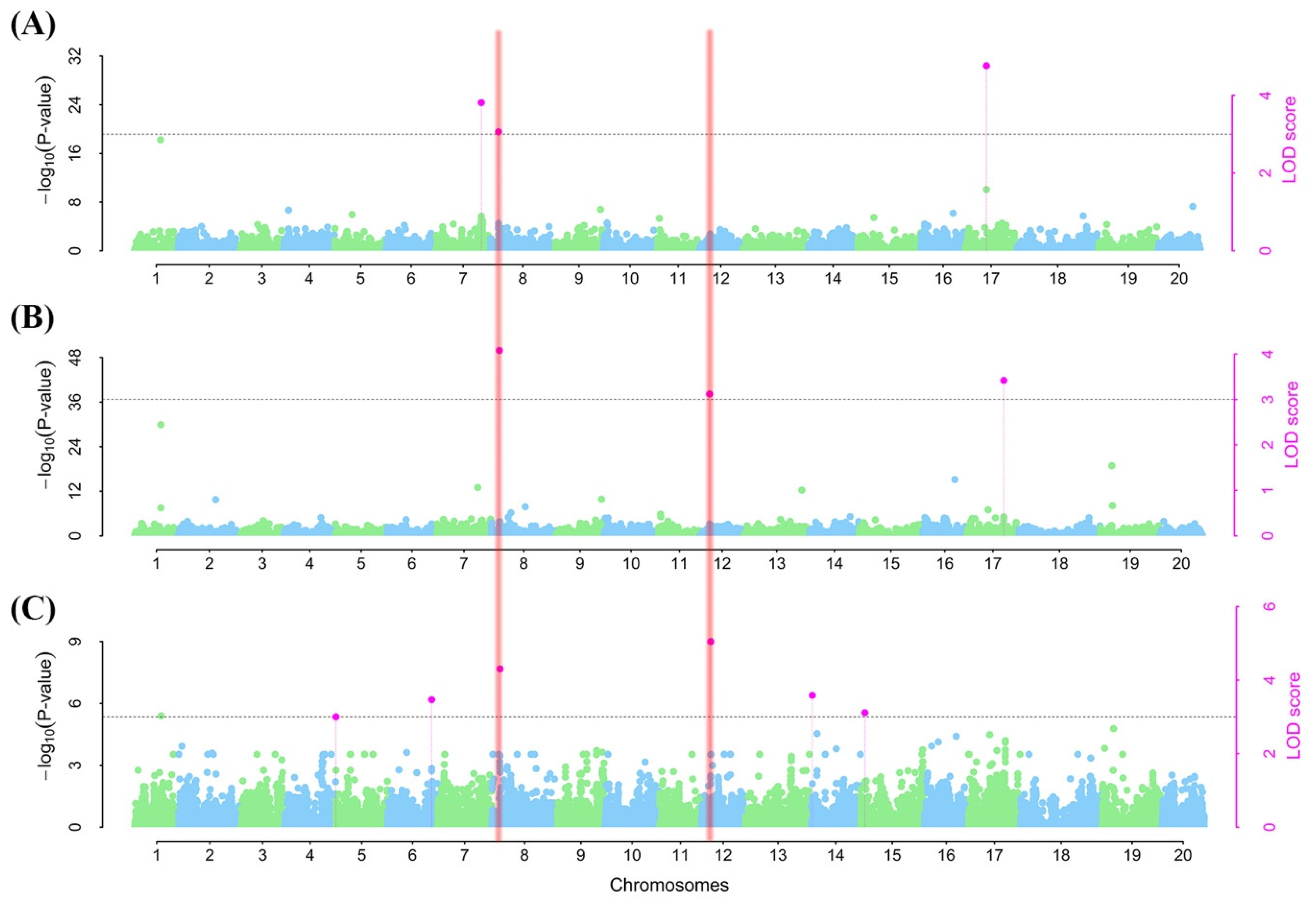
2.5. Marker–Trait Associations (MTAs)
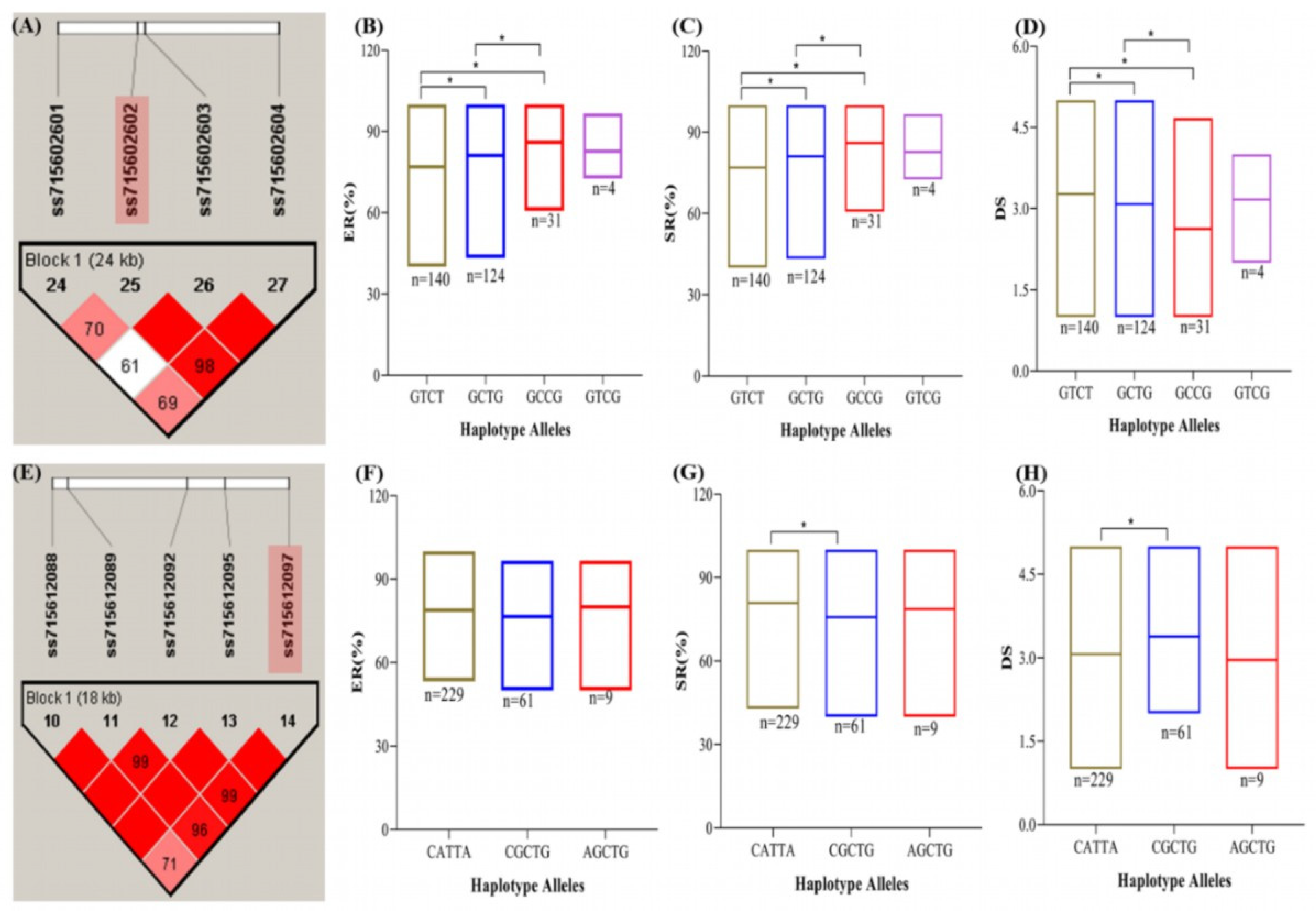
2.6. Haplotype Analysis for the Identification of Superior Haplotypes and Candidate Genes Mining
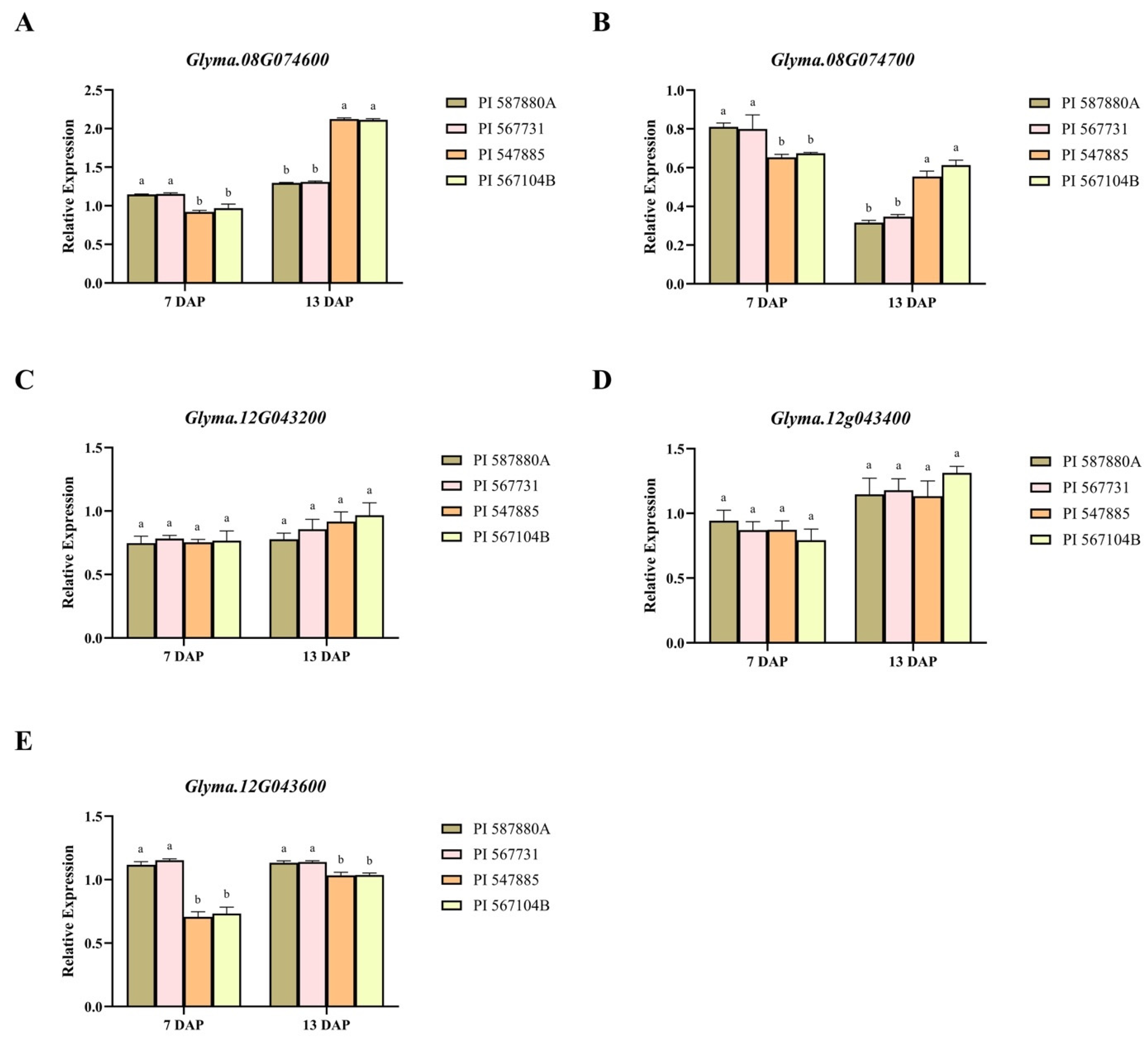
2.7. Analysis of Expression of Genes Associated with RCR Resistance
3. Discussion
3.1. Genetic Diversity among Soybean Germplasm
3.2. Evaluation and Identification of Resistance to Soybean Red Crown Rot Strain
3.3. Marker–Trait Associations (MTAs), Haplotype Analysis, and Candidate Gene Mining
4. Materials and Methods
4.1. Seed Source, Planting Preparation, and Growth Conditions
4.2. Pathogen Culture, Inoculation, Planting, and Growth Conditions
4.3. Data Collection and Analysis
4.3.1. Determination of Emergence Rate, Survival Rate, and Classification for Resistance to RCR
4.3.2. Evaluation of Soybean for Resistance to Calonectria ilicicola and Statistical Analysis
4.4. Genotyping, Quality Control, and Population Structure Analysis
4.5. Genetic Diversity among the Soybean Accession Based on Their Origin and Statistical Accessed Analysis
4.6. Multi-Locus Genome-Wide Association Analysis
4.7. Haplotype Analysis and Candidate Gene Analysis
4.8. RNA Extraction and qRT-PCR
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| ANOVA | Analysis of variance |
| DAP | Day after planting |
| DS | Disease severity |
| ER | emergence rate |
| GWAS | Genome-wide association studies |
| MATs | Marker–trait associations |
| qRT-PCR | Real-time quantitative polymerase chain reaction |
| QTL | Quantitative trait loci |
| QTNs | Quantitative trait nucleotides |
| RCR | Red crown rot |
| SNP | Single-nucleotide polymorphism |
| SR | survival rate |
References
- Duan, Z.; Li, Q.; Wang, H.; He, X.; Zhang, M. Genetic regulatory networks of soybean seed size, oil and protein contents. Front. Plant Sci. 2023, 14, 1160418. [Google Scholar] [CrossRef] [PubMed]
- Sekhon, J.K.; Maurer, D.; Wang, T.; Jung, S.; Rosentrater, K.A. Ethanol production by soy fiber treatment and simultaneous saccharification and co-fermentation in an integrated corn-soy biorefinery. Fermentation 2018, 4, 35. [Google Scholar] [CrossRef]
- Debnath, D.; Babu, S.C. Prospects for sustainable intensification of soybean production in sub-Saharan Africa. Afr. J. Agric. Resour. Econ. 2020, 15, 365–371. [Google Scholar] [CrossRef] [PubMed]
- Vedovatto, F.; Bonatto, C.; Bazoti, S.F.; Venturin, B.; Alves Jr, S.L.; Kunz, A.; Steinmetz, R.L.; Treichel, H.; Mazutti, M.A.; Zabot, G.L. Production of biofuels from soybean straw and hull hydrolysates obtained by subcritical water hydrolysis. Bioresour. Technol. 2021, 328, 124837. [Google Scholar] [CrossRef]
- Lin, F.; Chhapekar, S.S.; Vieira, C.C.; Da Silva, M.P.; Rojas, A.; Lee, D.; Liu, N.; Pardo, E.M.; Lee, Y.-C.; Dong, Z. Breeding for disease resistance in soybean: A global perspective. Theor. Appl. Genet. 2022, 135, 3773–3872. [Google Scholar] [CrossRef]
- Crous, P.; Wingfield, M.; Alfenas, A. Cylindrocladium parasiticum sp. nov., a new name for C. crotalariae. Mycol. Res. 1993, 97, 889–896. [Google Scholar] [CrossRef]
- Crous, P.W. Taxonomy and Pathology of Cylindrocladium (Calonectria) and Allied Genera; American Phytopathological Society (APS Press): St. Paul, MN, USA, 2002. [Google Scholar]
- Bel, D.; Sobers, E.K. A Peg, Pod, and Root Necrosis of Peanuts Caused By a Species of Calonectria. Phytopathology 1966, 56, 1361–1364. [Google Scholar]
- Berggren, G.; Snow, J. Red crown rot. In Compendium of Soybean Disease, 3rd ed.; Sinclair, J.B., Backman, P.A., Eds.; American Phytopathological Society: St. Paul, MN, USA, 1989; pp. 44–45. [Google Scholar]
- Hartman, G.; Rupe, J.; Sikora, E.; Domier, L.; Davis, J.; Steffey, K. Compendium of Soybean Diseases and Pests, 5th ed.; The American Phytopathological Society: St. Paul, MN, USA, 2015; p. 3. [Google Scholar]
- Wrather, J.; Anderson, T.; Arsyad, D.; Tan, Y.; Ploper, L.D.; Porta-Puglia, A.; Ram, H.; Yorinori, J. Soybean disease loss estimates for the top ten soybean-producing counries in 1998. Can. J. Plant Pathol. 2001, 23, 115–121. [Google Scholar] [CrossRef]
- Kleczewski, N.; Plewa, D.; Kangas, C.; Phillippi, E.; Kleczewski, V. First report of red crown rot of soybeans caused by Calonectria ilicicola (anamorph: Cylindrocladium parasiticum) in Illinois. Plant Dis. 2019, 103, 1777. [Google Scholar] [CrossRef]
- Liu, H.; Shen, Y.; Chang, H.; Tseng, M.; Lin, Y. First report of soybean red crown rot caused by Calonectria ilicicola in Taiwan. Plant Dis. 2020, 104, 979. [Google Scholar] [CrossRef]
- Neves, D.L.; Mehl, K.M.; Bradley, C.A. First report of red crown rot, caused by Calonectria ilicicola, and its effect on soybean in Kentucky. Plant Health Prog. 2023, 24, 303–305. [Google Scholar] [CrossRef]
- Akamatsu, H.; Fujii, N.; Saito, T.; Sayama, A.; Matsuda, H.; Kato, M.; Kowada, R.; Yasuta, Y.; Igarashi, Y.; Komori, H. Factors affecting red crown rot caused by Calonectria ilicicola in soybean cultivation. J. Gen. Plant Pathol. 2020, 86, 363–375. [Google Scholar] [CrossRef]
- Sugimoto, T.; Kato, M.; Yoshida, S.; Matsumoto, I.; Kobayashi, T.; Kaga, A.; Hajika, M.; Yamamoto, R.; Watanabe, K.; Aino, M. Pathogenic diversity of Phytophthora sojae and breeding strategies to develop Phytophthora-resistant soybeans. Breed. Sci. 2012, 61, 511–522. [Google Scholar] [CrossRef] [PubMed]
- Roy, K.; McLean, K.; Lawrence, G.; Patel, M.; Moore, W. First report of red crown rot on soybeans in Mississippi. Plant Dis. 1989, 73, 273. [Google Scholar] [CrossRef]
- Berner, D.; Berggren, G.; Snow, J.; White, E. Distribution and management of red crown rot of soybean in Louisiana. Appl. Agric. Res. 1988, 3, 160–166. [Google Scholar]
- Glenn, D.; Phipps, P.; Stipes, R. Incidence and survival of Cylindrocladium parasiticum in peanut seed. Plant Dis. 2003, 87, 867–871. [Google Scholar] [CrossRef]
- Randall-Schadel, B.; Bailey, J.; Beute, M. Seed transmission of Cylindrocladium parasiticum in peanut. Plant Dis. 2001, 85, 362–370. [Google Scholar] [CrossRef]
- Jiang, C.-J.; Sugano, S.; Ochi, S.; Kaga, A.; Ishimoto, M. Evaluation of Glycine max and Glycine soja for resistance to Calonectria ilicicola. Agronomy 2020, 10, 887. [Google Scholar] [CrossRef]
- Li, Y.-H.; Reif, J.C.; Ma, Y.-S.; Hong, H.-L.; Liu, Z.-X.; Chang, R.-Z.; Qiu, L.-J. Targeted association mapping demonstrating the complex molecular genetics of fatty acid formation in soybean. BMC Genom. 2015, 16, 841. [Google Scholar] [CrossRef]
- Varshney, R.K.; Terauchi, R.; McCouch, S.R. Harvesting the promising fruits of genomics: Applying genome sequencing technologies to crop breeding. PLoS Biol. 2014, 12, e1001883. [Google Scholar] [CrossRef] [PubMed]
- Jianan, Z.; Li, W.; Zhang, Y.; Song, W.; Jiang, H.; Zhao, J.; Zhan, Y.; Teng, W.; Qiu, L.; Zhao, X. Identification of glutathione transferase gene associated with partial resistance to Sclerotinia stem rot of soybean using genome-wide association and linkage mapping. Theor. Appl. Genet. 2021, 134, 2699–2709. [Google Scholar] [CrossRef]
- Zhao, X.; Han, Y.; Li, Y.; Liu, D.; Sun, M.; Zhao, Y.; Lv, C.; Li, D.; Yang, Z.; Huang, L. Loci and candidate gene identification for resistance to Sclerotinia sclerotiorum in soybean (Glycine max L. Merr.) via association and linkage maps. Plant J. 2015, 82, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.; Cheng, W.; Wang, Y.; Gao, X.; Huang, D.; Kong, J.; Antwi-Boasiako, A.; Zheng, L.; Yan, W.; Chang, F. Identification of novel genomic regions for bacterial leaf pustule (BLP) resistance in soybean (Glycine max L.) via integrating linkage mapping and association analysis. Int. J. Mol. Sci. 2022, 23, 2113. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Bao, D.; Wang, W.; Zhang, C.; Jing, Y.; Jiang, H.; Qiu, L.; Li, W.; Han, Y. Loci and candidate gene identification for soybean resistance to Phytophthora root rot race 1 in combination with association and linkage mapping. Mol. Breed. 2020, 40, 100. [Google Scholar] [CrossRef]
- Varshney, R.K.; Pandey, M.K.; Bohra, A.; Singh, V.K.; Thudi, M.; Saxena, R.K. Toward the sequence-based breeding in legumes in the post-genome sequencing era. Theor. Appl. Genet. 2019, 132, 797–816. [Google Scholar] [CrossRef]
- Song, Q.; Hyten, D.L.; Jia, G.; Quigley, C.V.; Fickus, E.W.; Nelson, R.L.; Cregan, P.B. Development and evaluation of SoySNP50K, a high-density genotyping array for soybean. PLoS ONE 2013, 8, e54985. [Google Scholar] [CrossRef]
- Gupta, P.K.; Kulwal, P.L.; Jaiswal, V. Association mapping in crop plants: Opportunities and challenges. Adv. Genet. 2014, 85, 109–147. [Google Scholar]
- Ibrahim, A.K.; Zhang, L.; Niyitanga, S.; Afzal, M.Z.; Xu, Y.; Zhang, L.; Zhang, L.; Qi, J. Principles and approaches of association mapping in plant breeding. Trop. Plant Biol. 2020, 13, 212–224. [Google Scholar] [CrossRef]
- Li, M.; Zhang, Y.-W.; Zhang, Z.-C.; Xiang, Y.; Liu, M.-H.; Zhou, Y.-H.; Zuo, J.-F.; Zhang, H.-Q.; Chen, Y.; Zhang, Y.-M. A compressed variance component mixed model for detecting QTNs and QTN-by-environment and QTN-by-QTN interactions in genome-wide association studies. Mol. Plant 2022, 15, 630–650. [Google Scholar] [CrossRef]
- Sarkozi, A. New Standards to Curb the Global Spread of Plant Pests and Diseases; Food and Agriculture Organization of the United Nations: Roma, Italy, 2019. [Google Scholar]
- Stukenbrock, E.; Gurr, S. Address the growing urgency of fungal disease in crops. Nature 2023, 617, 31–34. [Google Scholar] [CrossRef]
- Zhang, A.; Li, Y.; Wang, L.; Wang, J.; Liu, Y.; Luan, X.; Liu, S.; Zhang, J.; Liu, H.; Yao, D. Analysis of LncRNA43234-Associated ceRNA Network Reveals Oil Metabolism in Soybean. J. Agric. Food Chem. 2023, 71, 9815–9825. [Google Scholar] [CrossRef]
- Liu, Z.; Li, H.; Wen, Z.; Fan, X.; Li, Y.; Guan, R.; Guo, Y.; Wang, S.; Wang, D.; Qiu, L. Comparison of genetic diversity between Chinese and American soybean (Glycine max (L.)) accessions revealed by high-density SNPs. Front. Plant Sci. 2017, 8, 2014. [Google Scholar] [CrossRef]
- Chander, S.; Garcia-Oliveira, A.L.; Gedil, M.; Shah, T.; Otusanya, G.O.; Asiedu, R.; Chigeza, G. Genetic diversity and population structure of soybean lines adapted to sub-Saharan Africa using single nucleotide polymorphism (SNP) markers. Agronomy 2021, 11, 604. [Google Scholar] [CrossRef]
- Abe, J.; Xu, D.; Suzuki, Y.; Kanazawa, A.; Shimamoto, Y. Soybean germplasm pools in Asia revealed by nuclear SSRs. Theor. Appl. Genet. 2003, 106, 445–453. [Google Scholar] [CrossRef] [PubMed]
- Li, A.Q.; Zhao, C.Z.; Wang, X.J.; Liu, Z.J.; Zhang, L.F.; Song, G.Q.; Yin, J.; Li, C.S.; Xia, H.; Bi, Y.P. Identification of SSR markers using soybean (Glycine max) ESTs from globular stage embryos. Electron. J. Biotechnol. 2010, 13, 6–7. [Google Scholar] [CrossRef]
- Tripathi, N.; Tripathi, M.K.; Tiwari, S.; Payasi, D.K. Molecular breeding to overcome biotic stresses in soybean: Update. Plants 2022, 11, 1967. [Google Scholar] [CrossRef] [PubMed]
- Yao, D.; Zhou, J. Advances in CRISPR/Cas9-based research related to soybean [Glycine max (Linn.) Merr] molecular breeding. Front. Plant Sci. 2023, 14, 1247707. [Google Scholar] [CrossRef] [PubMed]
- Varshney, R.K.; Bohra, A.; Yu, J.; Graner, A.; Zhang, Q.; Sorrells, M.E. Designing future crops: Genomics-assisted breeding comes of age. Trends Plant Sci. 2021, 26, 631–649. [Google Scholar] [CrossRef] [PubMed]
- Poland, J.; Rutkoski, J. Advances and challenges in genomic selection for disease resistance. Annu. Rev. Phytopathol. 2016, 54, 79–98. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.D. Susceptibility in Soybean to Red Crown Rot and Characteristics of Virulence in Calonectria Crotalariae; Louisiana State University and Agricultural & Mechanical College: Baton Rouge, LA, USA, 1994. [Google Scholar]
- Nakajima, T.; Sakai, S.; Gomi, T.; Kikuchi, A. Development of Methods for Assessing Resistance to Black Root Rot Caused by Calonectria Crotalariae in Soybean [Glycine max] and Screening for Resistant Germplasm; Bulletin of the Tohoku National Agricultural Experiment Station: Morioka, Japan, 1994. [Google Scholar]
- Brown, J.K. Durable resistance of crops to disease: A Darwinian perspective. Annu. Rev. Phytopathol. 2015, 53, 513–539. [Google Scholar] [CrossRef]
- Lukanda, M.M.; Dramadri, I.O.; Adjei, E.A.; Badji, A.; Arusei, P.; Gitonga, H.W.; Wasswa, P.; Edema, R.; Ochwo-Ssemakula, M.; Tukamuhabwa, P. Genome-Wide Association Analysis for Resistance to Coniothyrium glycines Causing Red Leaf Blotch Disease in Soybean. Genes 2023, 14, 1271. [Google Scholar] [CrossRef] [PubMed]
- Xiong, H.; Chen, Y.; Shi, A. A genome-wide association study and genomic prediction for Phakopsora pachyrhizi resistance in soybean. Front. Plant Sci. 2023, 14, 1179357. [Google Scholar] [CrossRef]
- Wen, Z.; Tan, R.; Yuan, J.; Bales, C.; Du, W.; Zhang, S.; Chilvers, M.I.; Schmidt, C.; Song, Q.; Cregan, P.B. Genome-wide association mapping of quantitative resistance to sudden death syndrome in soybean. BMC Genom. 2014, 15, 809. [Google Scholar] [CrossRef]
- Liu, X.; Huang, M.; Fan, B.; Buckler, E.S.; Zhang, Z. Iterative usage of fixed and random effect models for powerful and efficient genome-wide association studies. PLoS Genet. 2016, 12, e1005767. [Google Scholar] [CrossRef] [PubMed]
- Jiang, G.-L.; Rajcan, I.; Zhang, Y.-M.; Han, T.; Mian, R. Soybean molecular breeding and genetics. Front. Plant Sci. 2023, 14, 1157632. [Google Scholar] [CrossRef]
- Chandra, S.; Choudhary, M.; Bagaria, P.K.; Nataraj, V.; Kumawat, G.; Choudhary, J.R.; Sonah, H.; Gupta, S.; Wani, S.H.; Ratnaparkhe, M.B. Progress and prospectus in genetics and genomics of Phytophthora root and stem rot resistance in soybean (Glycine max L.). Front. Genet. 2022, 13, 939182. [Google Scholar] [CrossRef]
- St. Clair, D.A. Quantitative disease resistance and quantitative resistance loci in breeding. Annu. Rev. Phytopathol. 2010, 48, 247–268. [Google Scholar] [CrossRef] [PubMed]
- Zou, S.; Tang, Y.; Xu, Y.; Ji, J.; Lu, Y.; Wang, H.; Li, Q.; Tang, D. TuRLK1, a leucine-rich repeat receptor-like kinase, is indispensable for stripe rust resistance of YrU1 and confers broad resistance to multiple pathogens. BMC Plant Biol. 2022, 22, 280. [Google Scholar] [CrossRef]
- Thapa, G.; Gunupuru, L.R.; Hehir, J.G.; Kahla, A.; Mullins, E.; Doohan, F.M. A pathogen-responsive leucine rich receptor like kinase contributes to Fusarium resistance in cereals. Front. Plant Sci. 2018, 9, 326624. [Google Scholar] [CrossRef]
- Hu, Y.; Gong, H.; Lu, Z.; Zhang, P.; Zheng, S.; Wang, J.; Tian, B.; Fang, A.; Yang, Y.; Bi, C. Variable Tandem Glycine-Rich Repeats Contribute to Cell Death-Inducing Activity of a Glycosylphosphatidylinositol-Anchored Cell Wall Protein That Is Associated with the Pathogenicity of Sclerotinia sclerotiorum. Microbiol. Spectr. 2023, 11, e00986-23. [Google Scholar] [CrossRef]
- Martin-Hernandez, A.; Dufresne, M.; Hugouvieux, V.; Melton, R.; Osbourn, A. Effects of targeted replacement of the tomatinase gene on the interaction of Septoria lycopersici with tomato plants. Mol. Plant-Microbe Interact. 2000, 13, 1301–1311. [Google Scholar] [CrossRef]
- Pan, S.; Tang, L.; Pan, X.; Qi, L.; Yang, J. A member of the glycoside hydrolase family 76 is involved in growth, conidiation, and virulence in rice blast fungus. Physiol. Mol. Plant Pathol. 2021, 113, 101587. [Google Scholar] [CrossRef]
- Sugano, S.; Maeda, S.; Hayashi, N.; Kajiwara, H.; Inoue, H.; Jiang, C.J.; Takatsuji, H.; Mori, M. Tyrosine phosphorylation of a receptor-like cytoplasmic kinase, BSR1, plays a crucial role in resistance to multiple pathogens in rice. Plant J. 2018, 96, 1137–1147. [Google Scholar] [CrossRef]
- Guzha, A.; McGee, R.; Scholz, P.; Hartken, D.; Lüdke, D.; Bauer, K.; Wenig, M.; Zienkiewicz, K.; Herrfurth, C.; Feussner, I. Cell wall-localized BETA-XYLOSIDASE4 contributes to immunity of Arabidopsis against Botrytis cinerea. Plant Physiol. 2022, 189, 1794–1813. [Google Scholar] [CrossRef]
- Bauer, K.; Nayem, S.; Vlot, A.C. β-D-XYLOSIDASE 4 modulates systemic immune signaling in Arabidopsis thaliana. Front. Plant Sci. 2023, 13, 1096800. [Google Scholar] [CrossRef] [PubMed]
- Win, K.T.; Jiang, C.-J. A fresh weight-based method for evaluating soybean resistance to red crown rot. Breed. Sci. 2021, 71, 384–389. [Google Scholar] [CrossRef]
- Kobayashi, M.; Win, K.T.; Jiang, C.-J. Soybean hypocotyls prevent Calonectria ilicicola invasion by multi-layered defenses. Front. Plant Sci. 2022, 12, 813578. [Google Scholar] [CrossRef]
- Win, K.T.; Maeda, S.; Kobayashi, M.; Jiang, C.-J. Silicon enhances resistance to red crown rot caused by Calonectria ilicicola in soybean. Agronomy 2021, 11, 899. [Google Scholar] [CrossRef]
- Nishi, K.; Takahashi, H. Influence of low temperature preservation on survival of Calonectria crotalariae. Proc. Kanto-Tosan Plant Prot. Soc. 1990, 37, 51–53. [Google Scholar]
- Wei, T.; Simko, V. R Package “corrplot”: Visualization of a Correlation Matrix (Version 0.84). 2017. Available online: https://github.com/taiyun/corrplot (accessed on 10 September 2023).
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.; Bender, D.; Maller, J.; Sklar, P.; De Bakker, P.I.; Daly, M.J. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed]
- Adamack, A.T.; Gruber, B. PopGenReport: Simplifying basic population genetic analyses in R. Methods Ecol. Evol. 2014, 5, 384–387. [Google Scholar] [CrossRef]
- Sui, M.; Jing, Y.; Li, H.; Zhan, Y.; Luo, J.; Teng, W.; Qiu, L.; Zheng, H.; Li, W.; Zhao, X. Identification of loci and candidate genes analyses for tocopherol concentration of soybean seed. Front. Plant Sci. 2020, 11, 539460. [Google Scholar] [CrossRef]
- Zhang, Y.-W.; Tamba, C.L.; Wen, Y.-J.; Li, P.; Ren, W.-L.; Ni, Y.-L.; Gao, J.; Zhang, Y.-M. mrMLM v4. 0.2: An R platform for multi-locus genome-wide association studies. Genom. Proteom. Bioinform. 2020, 18, 481–487. [Google Scholar] [CrossRef]
- Barrett, J.C.; Fry, B.; Maller, J.; Daly, M.J. Haploview: Analysis and visualization of LD and haplotype maps. Bioinformatics 2005, 21, 263–265. [Google Scholar] [CrossRef]
- Lyu, X.; Cheng, Q.; Qin, C.; Li, Y.; Xu, X.; Ji, R.; Mu, R.; Li, H.; Zhao, T.; Liu, J. GmCRY1s modulate gibberellin metabolism to regulate soybean shade avoidance in response to reduced blue light. Mol. Plant 2021, 14, 298–314. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Country | No. of Cultivars | MAF a | Heb | PIC c |
|---|---|---|---|---|
| China | 35 | 0.23 | 0.31 | 0.25 |
| India | 11 | 0.19 | 0.26 | 0.21 |
| Japan | 28 | 0.18 | 0.24 | 0.20 |
| Russia | 14 | 0.26 | 0.35 | 0.27 |
| South Korea | 26 | 0.20 | 0.27 | 0.22 |
| Thailand | 10 | 0.14 | 0.19 | 0.16 |
| United States | 115 | 0.22 | 0.28 | 0.23 |
| Trait a | SNP b | Chr c | SNP pos d | LOD e | PVE (%) f | QTN Effect g | MAF h | Genotype i |
|---|---|---|---|---|---|---|---|---|
| ER | ss715597632 | 7 | 37631217 | 3.81 | 7.64 | 4.81 | 0.14 | AA |
| ss715602602 | 8 | 5709053 | 3.06 | 5.44 | 2.82 | 0.48 | CC | |
| ss715625925 | 17 | 12730065 | 4.76 | 6.37 | −8.11 | 0.50 | TT | |
| SR | ss715602602 | 8 | 5709053 | 4.08 | 6.43 | 3.88 | 0.48 | CC |
| ss715612097 | 12 | 3146531 | 3.12 | 5.29 | 4.22 | 0.22 | AA | |
| ss715627013 | 17 | 36384272 | 3.42 | 7.56 | −4.33 | 0.39 | AA | |
| DS | ss715592629 | 5 | 37228 | 3.00 | 5.09 | 0.22 | 0.28 | AA |
| ss715594897 | 6 | 48464349 | 3.47 | 4.82 | −0.22 | 0.30 | GG | |
| ss715602602 | 8 | 5709053 | 4.30 | 6.80 | −0.23 | 0.48 | CC | |
| ss715612097 | 12 | 3146531 | 5.05 | 7.10 | −0.29 | 0.22 | AA | |
| ss715619777 | 14 | 652974 | 3.58 | 4.68 | 0.22 | 0.25 | AA | |
| ss715621431 | 15 | 2649998 | 3.11 | 4.31 | −0.20 | 0.34 | GG |
| Chr_SNP_Position | Gene ID/Name a | Position (bp) | Annotation Descriptions b |
|---|---|---|---|
| Chr08_ ss715602602_ 5709053 | Glyma.08G074500 | 5684546–5692295 | BRI1-associated receptor kinase, protein phosphorylation, leucine-rich repeat |
| Glyma.08G074600 | 5695514–5698741 | Splicing factor, arginine/serine-rich, nucleic acid binding | |
| Glyma.08G074700 | 5707293–5712830 | Carbohydrate metabolic process, xylan catabolic process | |
| Chr12_ ss715612097_ 3146531 | Glyma.12G043100 | 3128278–3132475 | Carbohydrate metabolic process, glycosyl hydrolase |
| Glyma.12G043200 | 3132968–3138908 | Phenylalanyl-tRNA synthetase; tRNA aminoacylation; tRNA aminoacylation for protein translation | |
| Glyma.12G043300 | 3142970–3144547 | Nucleic acid binding | |
| Glyma.12G043400 | 3143730–3147610 | Erythronate-4-phosphate dehydrogenase | |
| Glyma.12G043500 | 3148801–3150720 | BTB/POZ domain-containing protein | |
| Glyma.12G043600 | 3157432–3162480 | Protein phosphorylation; protein tyrosine kinase; leucine-rich repeat |
| Scale | Damage Degree | Resistance Degree |
|---|---|---|
| 0 | No visible sign of necrotic lesions on root | Immune |
| 1 | Only tap root exhibits small necrotic lesions without obvious changes in its form and shape | Resistant |
| 2 | Necrotic lesions spread to the crown and root system and seedling mortality less than 10% | Moderately Resistant |
| 3 | Roots show serious necrotic lesions with less than 50% loss by rot and seedling mortality of 10–20% | Moderately Susceptible |
| 4 | Roots show serious necrotic lesions with more than 50% root loss by rot and seedling mortality of 21–50% | Susceptible |
| 5 | Over 50% of root loss by rot with seedling mortality of more than 50% | Highly Susceptible |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Antwi-Boasiako, A.; Jia, S.; Liu, J.; Guo, N.; Chen, C.; Karikari, B.; Feng, J.; Zhao, T. Identification and Genetic Dissection of Resistance to Red Crown Rot Disease in a Diverse Soybean Germplasm Population. Plants 2024, 13, 940. https://doi.org/10.3390/plants13070940
Antwi-Boasiako A, Jia S, Liu J, Guo N, Chen C, Karikari B, Feng J, Zhao T. Identification and Genetic Dissection of Resistance to Red Crown Rot Disease in a Diverse Soybean Germplasm Population. Plants. 2024; 13(7):940. https://doi.org/10.3390/plants13070940
Chicago/Turabian StyleAntwi-Boasiako, Augustine, Shihao Jia, Jiale Liu, Na Guo, Changjun Chen, Benjamin Karikari, Jianying Feng, and Tuanjie Zhao. 2024. "Identification and Genetic Dissection of Resistance to Red Crown Rot Disease in a Diverse Soybean Germplasm Population" Plants 13, no. 7: 940. https://doi.org/10.3390/plants13070940
APA StyleAntwi-Boasiako, A., Jia, S., Liu, J., Guo, N., Chen, C., Karikari, B., Feng, J., & Zhao, T. (2024). Identification and Genetic Dissection of Resistance to Red Crown Rot Disease in a Diverse Soybean Germplasm Population. Plants, 13(7), 940. https://doi.org/10.3390/plants13070940