In Silico Analyses of Autophagy-Related Genes in Rapeseed (Brassica napus L.) under Different Abiotic Stresses and in Various Tissues

 ,
,  and
and 
Abstract
:1. Introduction
2. Results
2.1. The Identification of B. napus ATG Genes
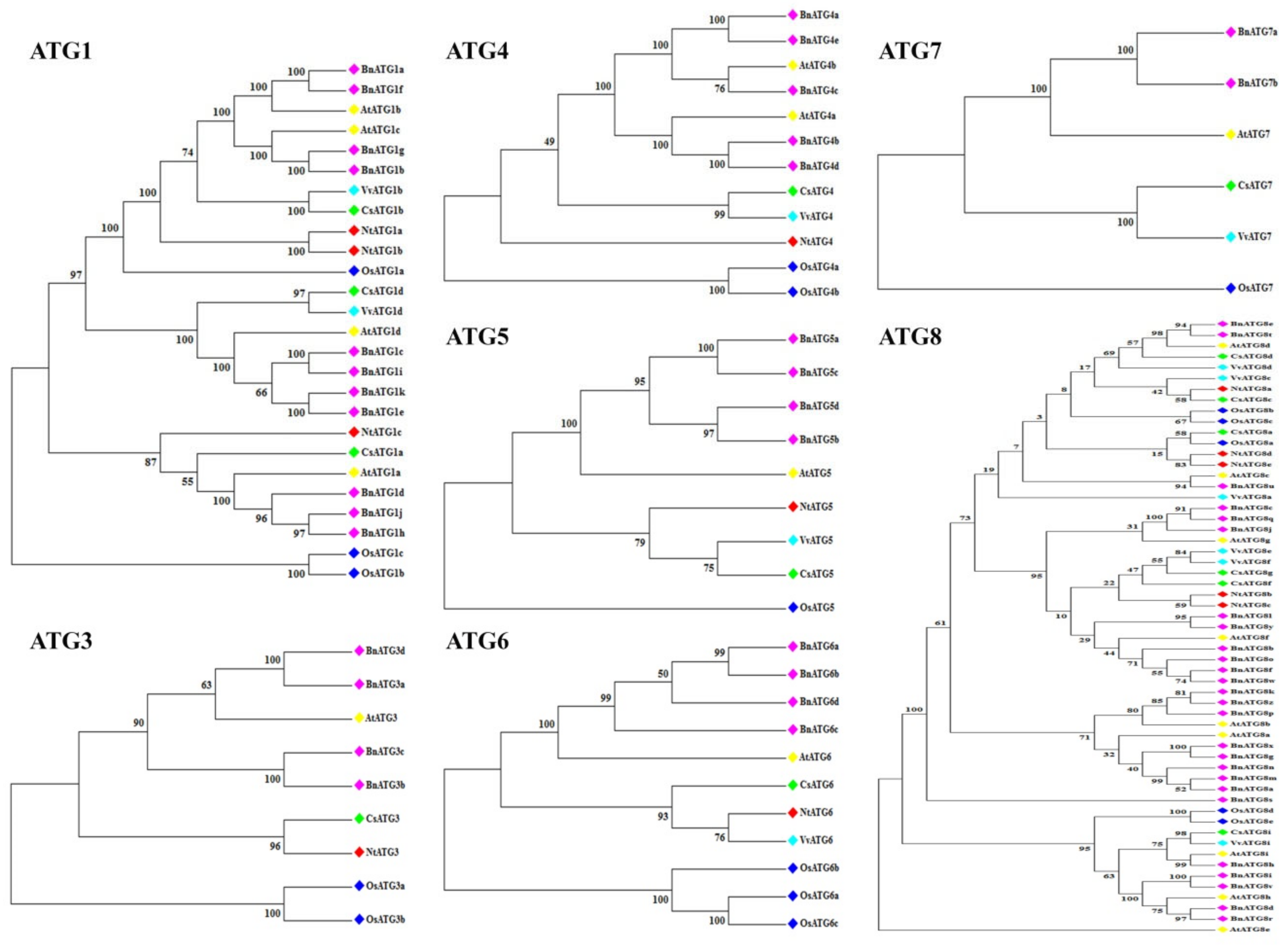
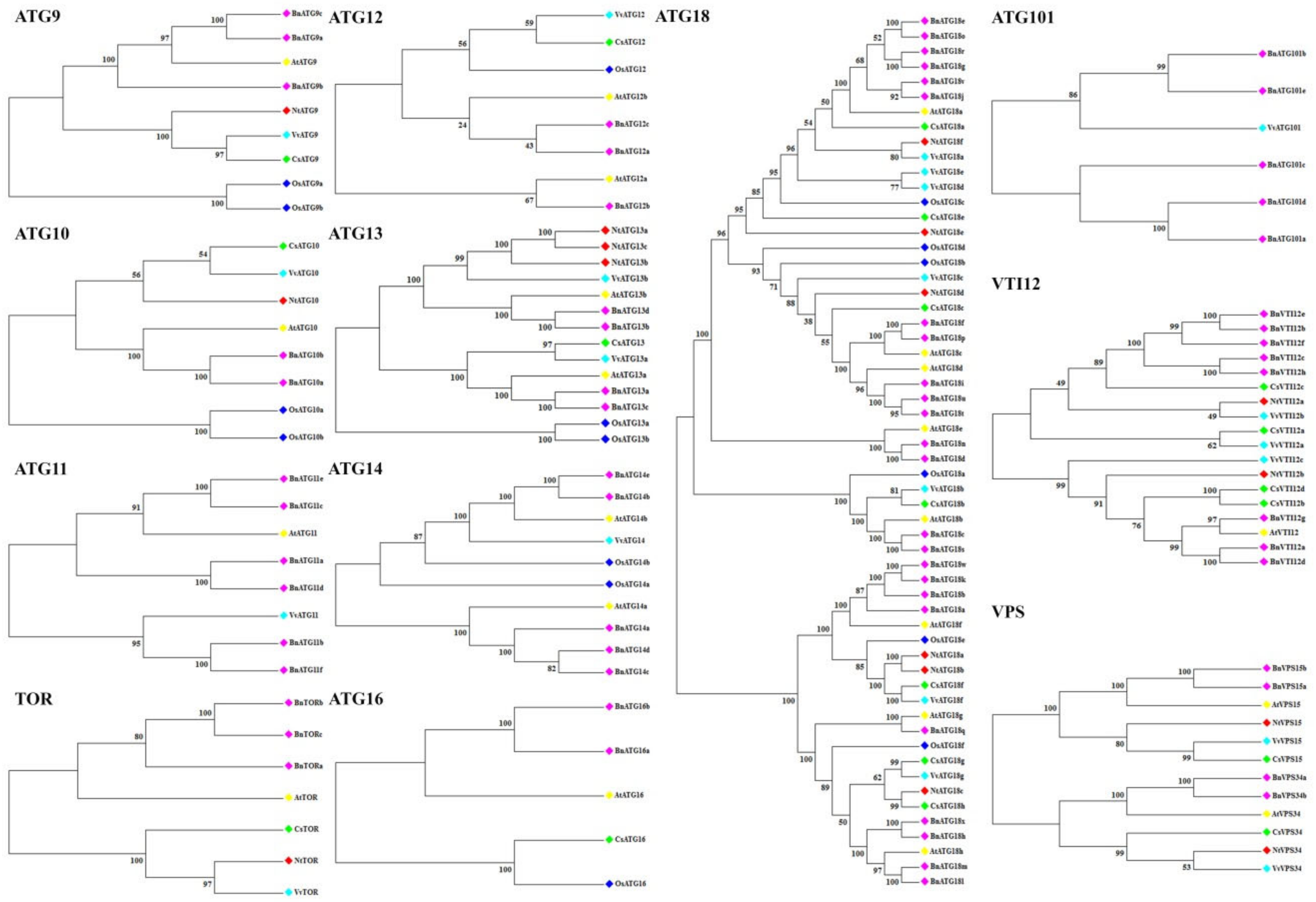
2.2. The Phylogenetic Analysis of B. napus ATG Gene Family
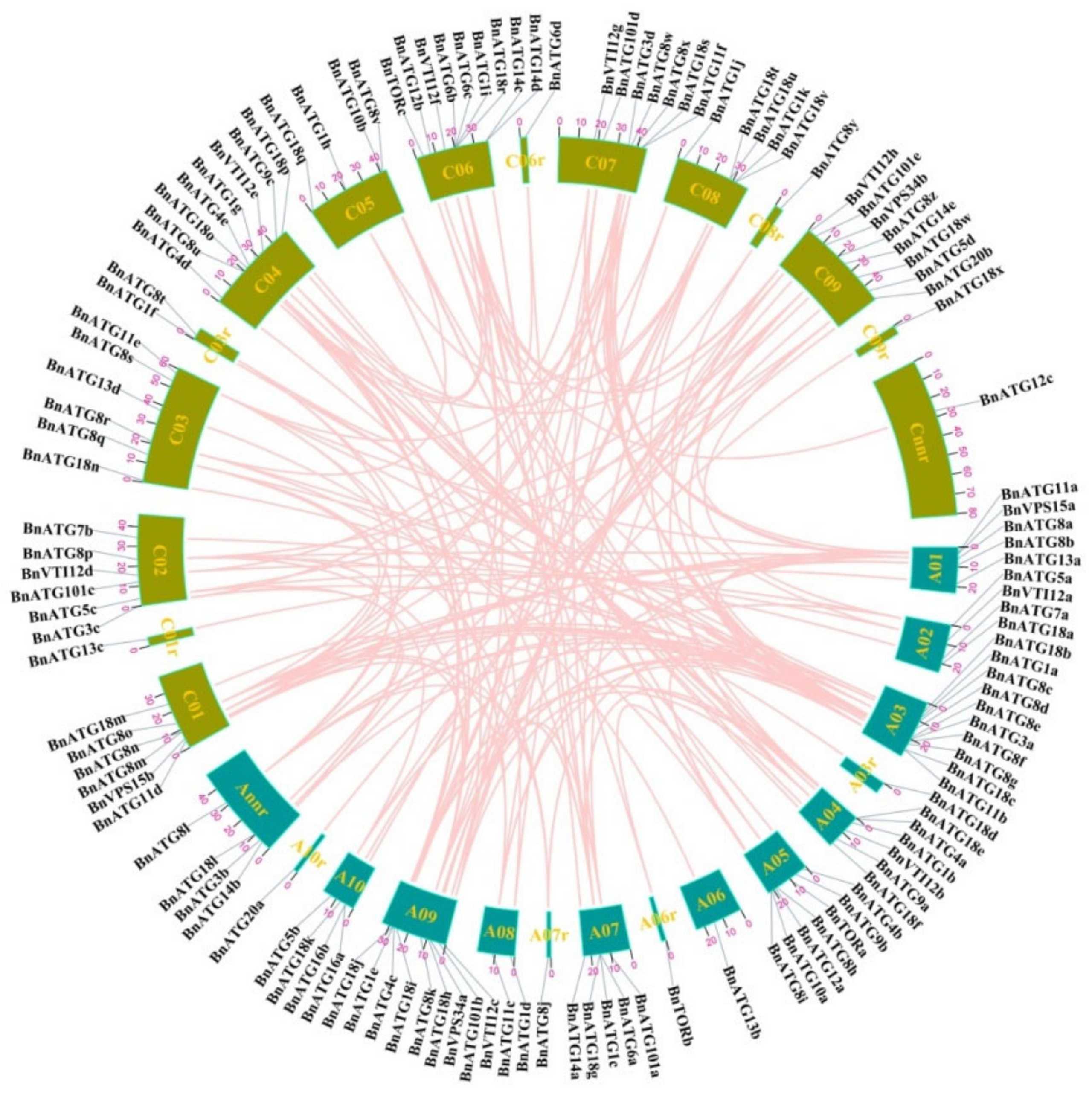
2.3. Gene Location, Duplication, and Selection Pressure of BnATGs
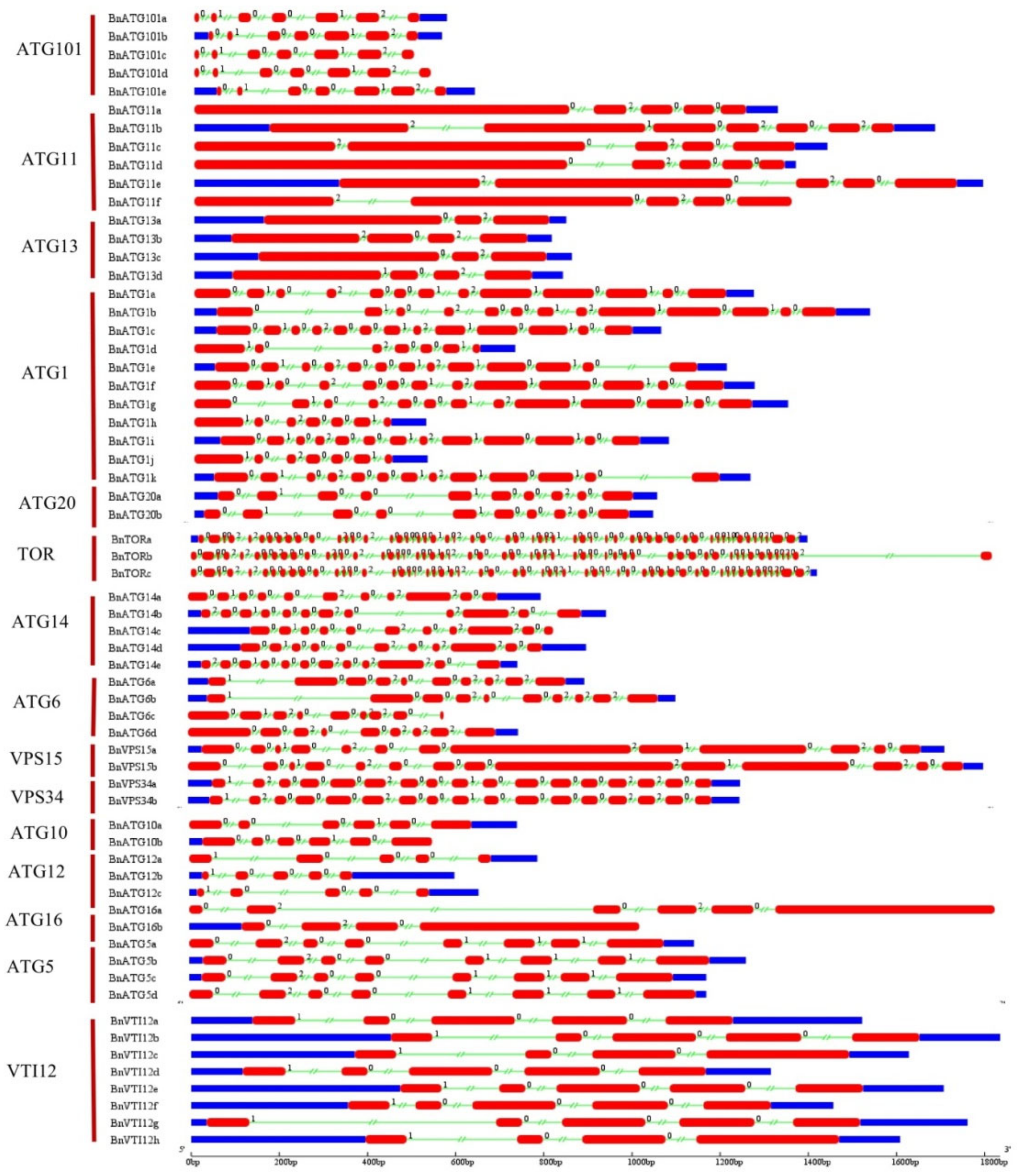
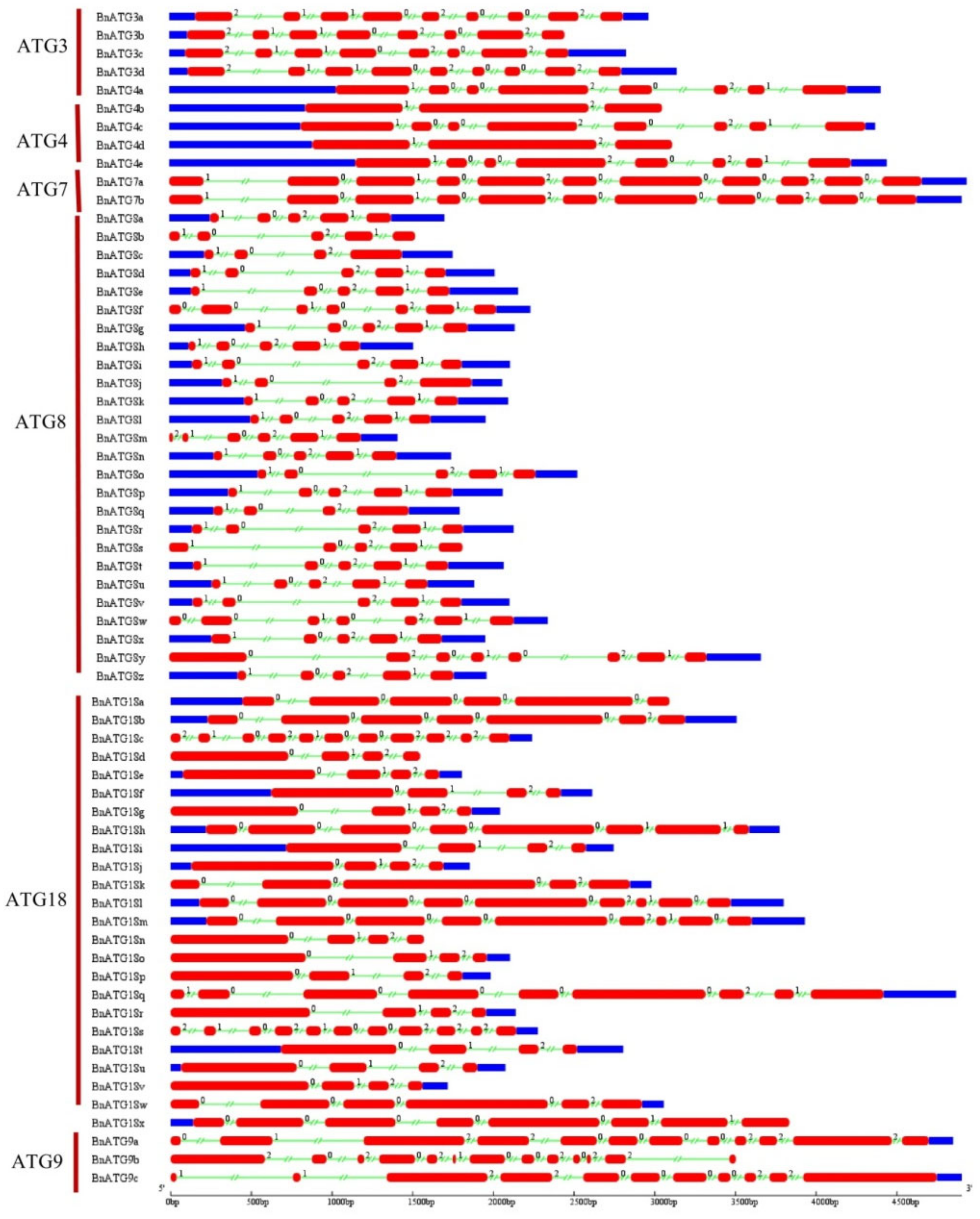
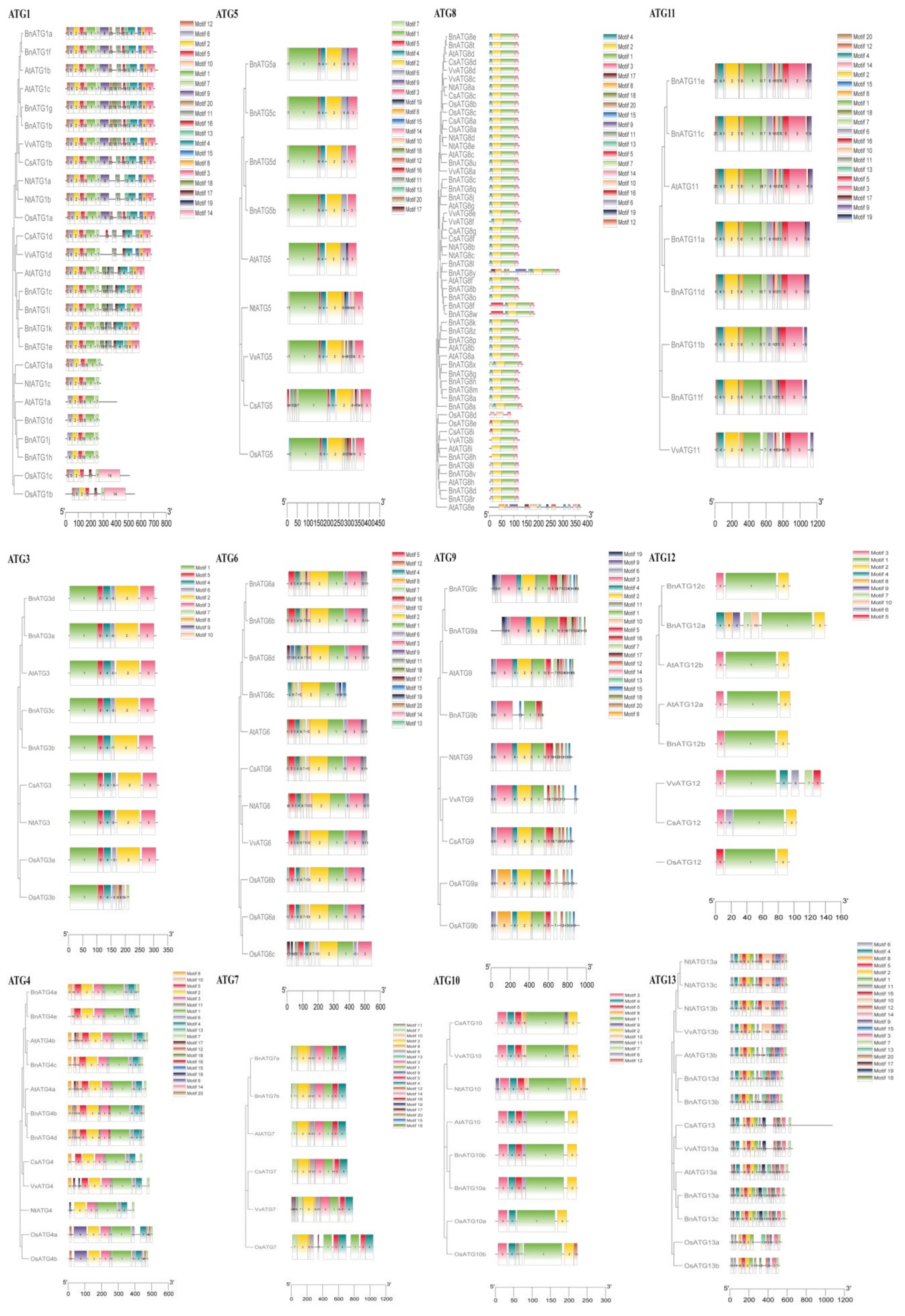
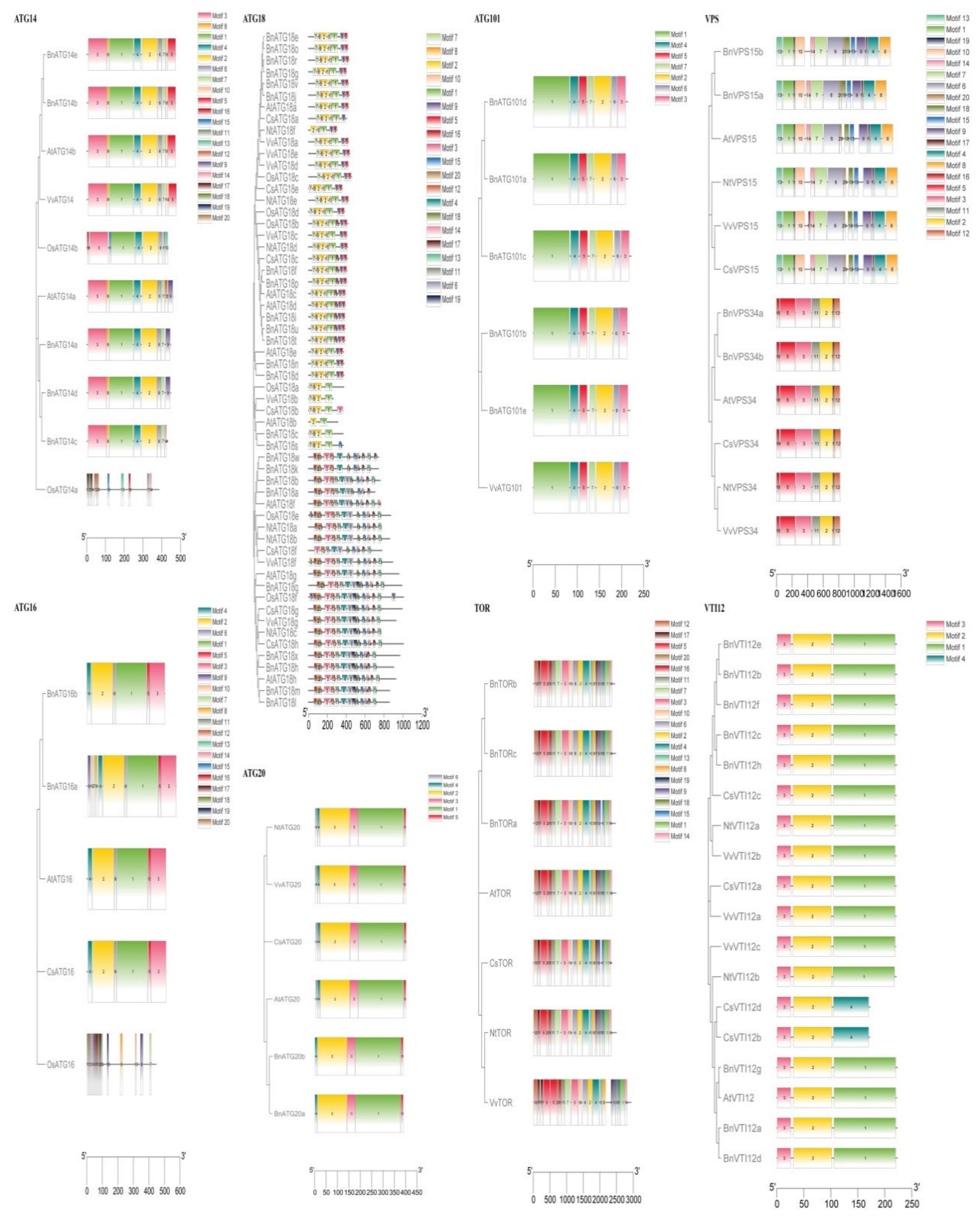
2.4. The Exon–Intron Structure and Conserved Motifs of B. napus ATGs
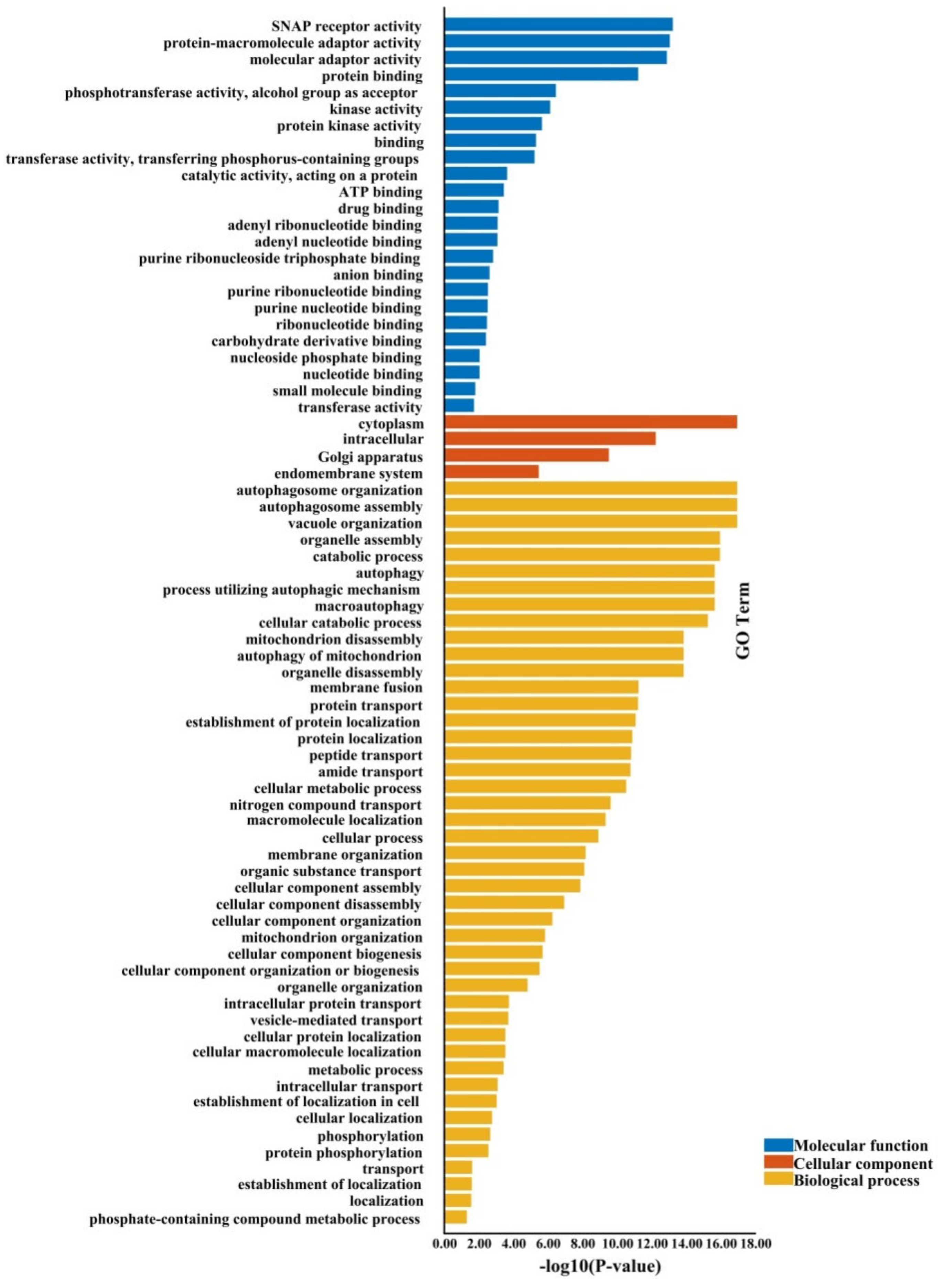
2.5. The Gene Ontology Annotations and Cis-Regulatory Elements of BnATGs
2.6. The Prediction of Simple Sequence Repeats (SSRs) in BnATGs
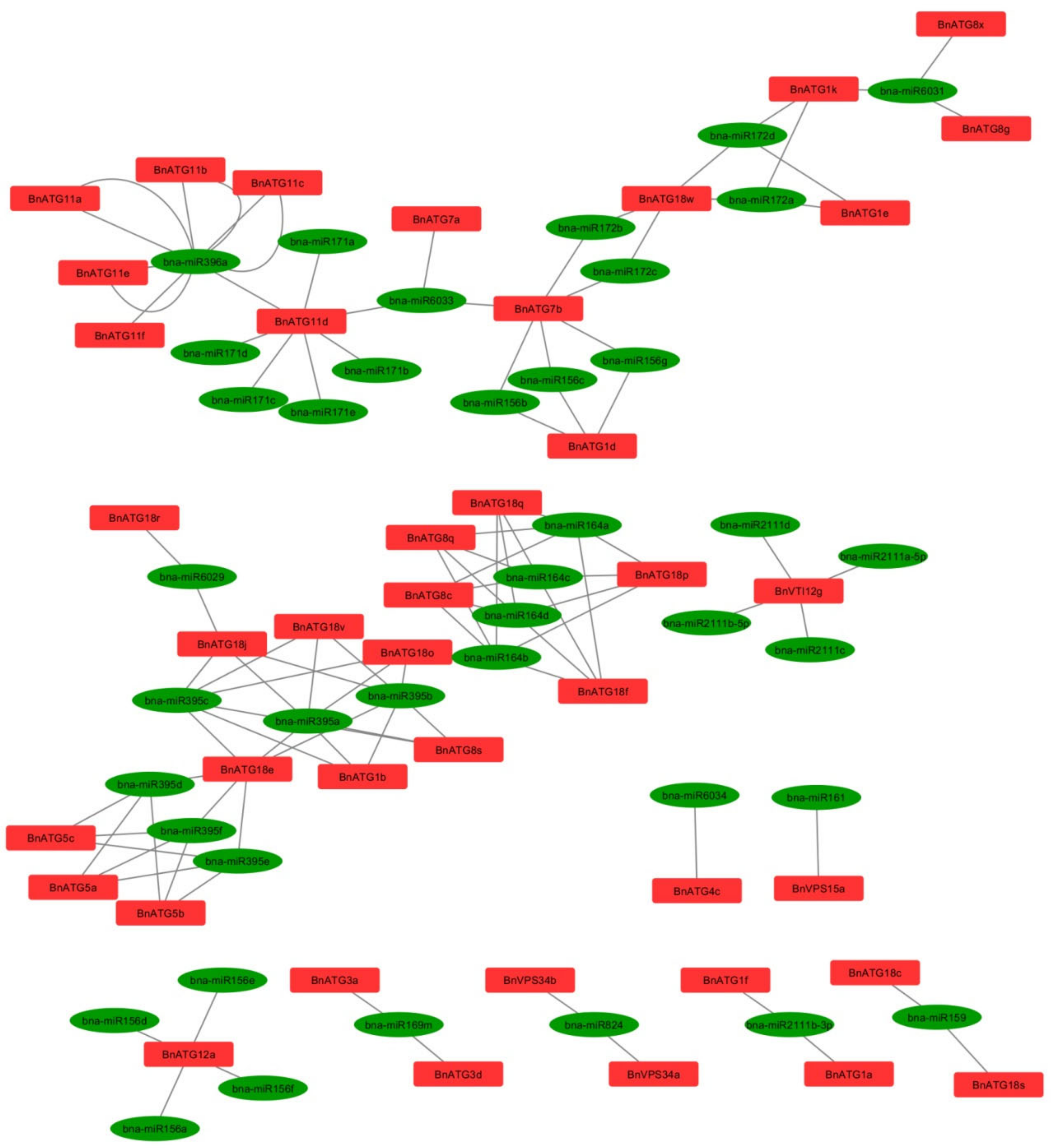
2.7. BnATG-Targeted miRNAs Prediction
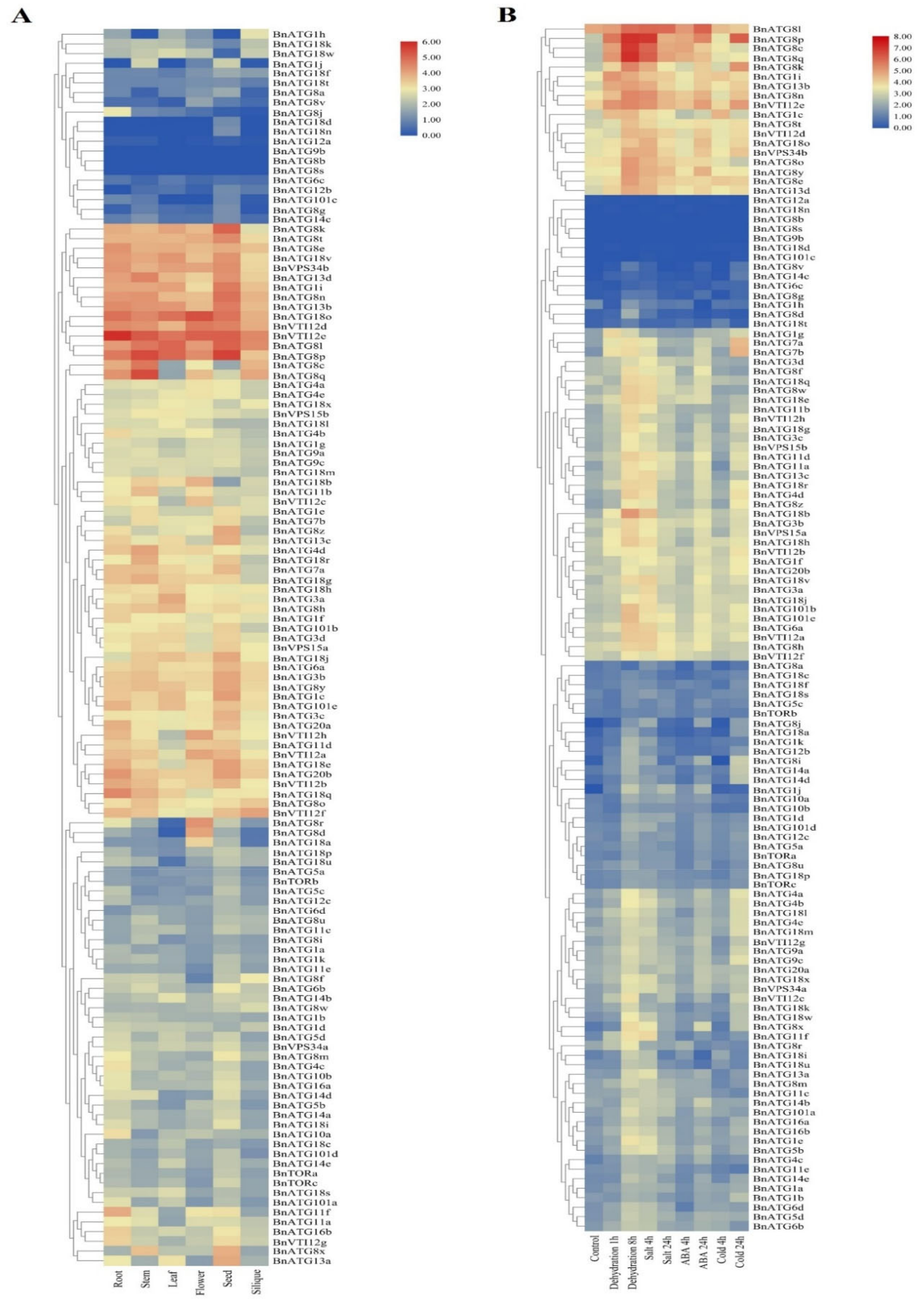
2.8. The Expression Analysis of BnATGs at Various Developmental Stages
2.9. The Expression Profile of BnATGs under Abiotic Stresses
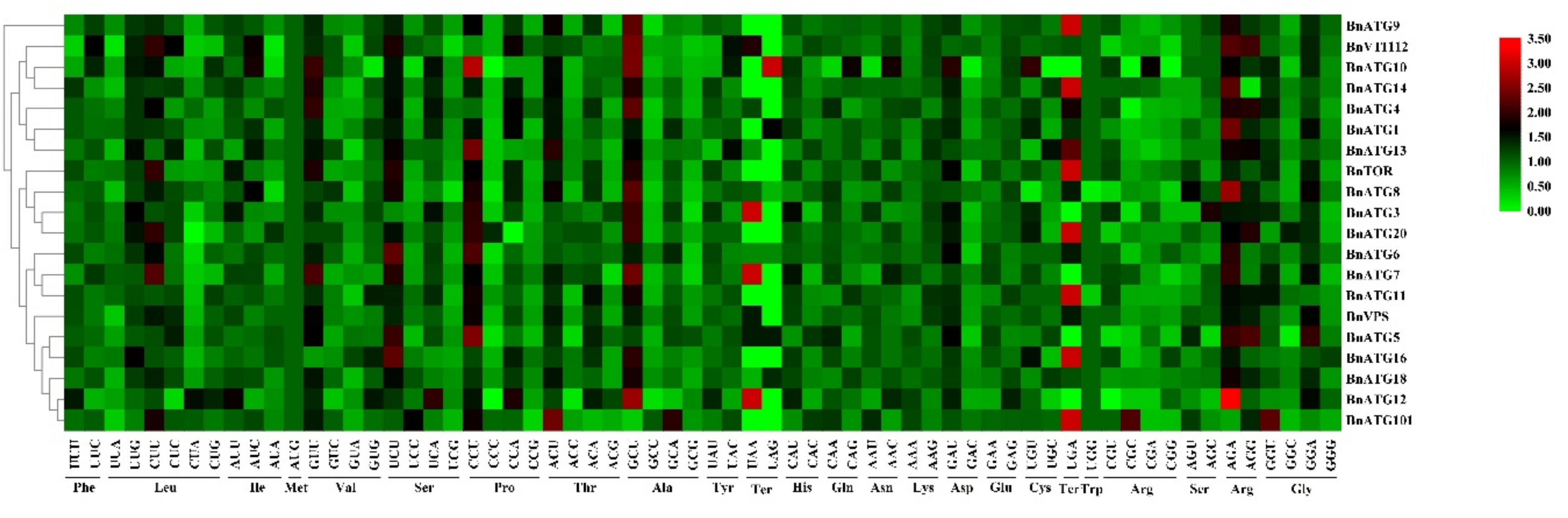
2.10. The Codon Usage Bias Analysis of BnATGs
3. Discussion
4. Materials and Methods
4.1. In Silico Identification of BnATG Genes
4.2. Phylogenetic Analysis of B. napus ATG Gene Family
4.3. Chromosome Localization, Gene Duplication, and Selection Pressure
4.4. Exon–Intron Structure and Conserved Motifs
4.5. Gene Ontology Annotations and Cis-Regulatory Element Identification
4.6. The Prediction of Simple Sequence Repeats (SSR) Markers and BnATG-Targeted miRNAs
4.7. Analysis of Previously Published B. napus RNA-Seq Data
4.8. Codon Usage Bias Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Li, F.; Vierstra, R.D. Autophagy: A multifaceted intracellular system for bulk and selective recycling. Trend. Plant Sci. 2012, 17, 526–537. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Meijer, A.J.; Codogno, P.; Neufeld, T.P.; Scott, R.C. Autophagy and p70S6 Kinase. Autophagy 2005, 1, 59–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, X.; Klionsky, D.J. An overview of macroautophagy in yeast. J. Mol. Biol. 2016, 428, 1681–1699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.-W.; Li, J.; Bao, J. Microautophagy: Lesser-known self-eating. Cell. Mol. Life Sci. 2011, 69, 1125–1136. [Google Scholar] [CrossRef] [PubMed]
- Avin-Wittenberg, T.; Baluška, F.; Bozhkov, P.V.; Elander, P.H.; Fernie, A.R.; Galili, G.; Hassan, A.; Hofius, D.; Isono, E.; Le Bars, R.; et al. Autophagy-related approaches for improving nutrient use efficiency and crop yield protection. J. Exp. Bot. 2018, 69, 1335–1353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Liao, B.; Qin-Fang, C.; Xie, L.-J.; Huang, L.; Tan, W.-J.; Zhai, N.; Yuan, L.-B.; Zhou, Y.; Yu, L.-J.; et al. Autophagy contributes to regulation of the hypoxia response during submergence in Arabidopsis thaliana. Autophagy 2015, 11, 2233–2246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guiboileau, A.; Yoshimoto, K.; Soulay, F.; Bataillé, M.-P.; Avice, J.-C.; Masclaux-Daubresse, C. Autophagy machinery controls nitrogen remobilization at the whole-plant level under both limiting and ample nitrate conditions in Arabidopsis. New Phytol. 2012, 194, 732–740. [Google Scholar] [CrossRef] [PubMed]
- Li, W.-W.; Chen, M.; Zhong, L.; Liu, J.-M.; Xu, Z.-S.; Li, L.-C.; Zhou, Y.-B.; Guo, C.-H.; Ma, Y.-Z. Overexpression of the autophagy-related gene SiATG8a from foxtail millet (Setaria italica L.) confers tolerance to both nitrogen starvation and drought stress in Arabidopsis. Biochem. Biophys. Res. Commun. 2015, 468, 800–806. [Google Scholar] [CrossRef] [PubMed]
- Luo, L.; Zhang, P.; Zhu, R.; Fu, J.; Su, J.; Zheng, J.; Wang, Z.; Wang, D.; Gong, Q. Autophagy Is Rapidly Induced by Salt Stress and Is Required for Salt Tolerance in Arabidopsis. Front. Plant Sci. 2017, 8, 1459. [Google Scholar] [CrossRef] [Green Version]
- Slobodkin, M.R.; Elazar, Z. The Atg8 family: Multifunctional ubiquitin-like key regulators of autophagy. Essays Biochem. 2013, 55, 51–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshimoto, K.; Hanaoka, H.; Sato, S.; Kato, T.; Tabata, S.; Noda, T.; Ohsumi, Y. Processing of ATG8s, Ubiquitin-Like Proteins, and Their Deconjugation by ATG4s Are Essential for Plant Autophagy. Plant Cell 2004, 16, 2967–2983. [Google Scholar] [CrossRef] [PubMed]
- Shangguan, L.; Fang, X.; Chen, L.; Cui, L.; Fang, J. Genome-wide analysis of autophagy-related genes (ARGs) in grapevine and plant tolerance to copper stress. Planta 2018, 247, 1449–1463. [Google Scholar] [CrossRef] [PubMed]
- Kuzuoğlu-Öztürk, D.; Yalcinkaya, O.C.; Akpınar, B.A.; Mitou, G.; Korkmaz, G.; Gozuacik, D.; Budak, H.; Akpinar, B.A. Autophagy-related gene, TdAtg8, in wild emmer wheat plays a role in drought and osmotic stress response. Planta 2012, 236, 1081–1092. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Xiong, Y.; Bassham, D.C. Autophagy is required for tolerance of drought and salt stress in plants. Autophagy 2009, 5, 954–963. [Google Scholar] [CrossRef] [Green Version]
- Shin, J.-H.; Yoshimoto, K.; Ohsumi, Y.; Jeon, J.-S.; An, G. OsATG10b, an autophagosome component, is needed for cell survival against oxidative stresses in rice. Mol. Cells 2009, 27, 67–74. [Google Scholar] [CrossRef]
- Minina, E.A.; Moschou, P.N.; Vetukuri, R.R.; Sanchez-Vera, V.; Cardoso, C.; Liu, Q.; Elander, P.H.; Dalman, K.; Beganovic, M.; Yilmaz, J.L.; et al. Transcriptional stimulation of rate-limiting components of the autophagic pathway improves plant fitness. J. Exp. Bot. 2018, 69, 1415–1432. [Google Scholar] [CrossRef]
- Wang, P.; Sun, X.; Jia, X.; Ma, F. Apple autophagy-related protein MdATG3s afford tolerance to multiple abiotic stresses. Plant Sci. 2017, 256, 53–64. [Google Scholar] [CrossRef]
- Hanaoka, H.; Noda, T.; Shirano, Y.; Kato, T.; Hayashi, H.; Shibata, D.; Tabata, S.; Ohsumi, Y. Leaf Senescence and Starvation-Induced Chlorosis Are Accelerated by the Disruption of an Arabidopsis Autophagy Gene. Plant Physiol. 2002, 129, 1181–1193. [Google Scholar] [CrossRef] [Green Version]
- Islam, S.; Proshad, R.; Kormoker, T.; Tusher, T.R. Autophagy-mediated Nutrient Recycling and Regulation in Plants: A Molecular View. J. Plant Biol. 2019, 62, 307–319. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, J.; Abeliovich, H.; Arozena, A.A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy 3rd ed. Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef] [Green Version]
- Tang, J.; Bassham, D.C. Autophagy in crop plants: What’s new beyond Arabidopsis? Open Biol. 2018, 8, 180162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, T.; Li, X.; Yang, M.; Shao, Q.; Zhao, Y.; Ma, C.; Wang, P. Autophagy: An Intracellular Degradation Pathway Regulating Plant Survival and Stress Response. Front. Plant Sci. 2020, 11, 164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Yan, H.; Mei, W.; Tse, Y.C.; Wang, H. Boosting autophagy in sexual reproduction: A plant perspective. New Phytol. 2020, 226, 679–689. [Google Scholar] [CrossRef] [PubMed]
- Chung, T.; Suttangkakul, A.; Vierstra, R.D. The ATG Autophagic Conjugation System in Maize: ATG Transcripts and Abundance of the ATG8-Lipid Adduct Are Regulated by Development and Nutrient Availability. Plant Physiol. 2008, 149, 220–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breeze, E.; Harrison, E.; McHattie, S.; Hughes, L.; Hickman, R.; Hill, C.; Kiddle, S.; Kim, Y.-S.; Penfold, C.A.; Jenkins, D.; et al. High-Resolution Temporal Profiling of Transcripts during Arabidopsis Leaf Senescence Reveals a Distinct Chronology of Processes and Regulation. Plant Cell 2011, 23, 873–894. [Google Scholar] [CrossRef] [Green Version]
- Di Berardino, J.; Marmagne, A.; Berger, A.; Yoshimoto, K.; Cueff, G.; Chardon, F.; Masclaux-Daubresse, C.; Reisdorf-Cren, M. Autophagy controls resource allocation and protein storage accumulation in Arabidopsis seeds. J. Exp. Bot. 2018, 69, 1403–1414. [Google Scholar] [CrossRef] [Green Version]
- Kurusu, T.; Koyano, T.; Hanamata, S.; Kubo, T.; Noguchi, Y.; Yagi, C.; Nagata, N.; Yamamoto, T.; Ohnishi, T.; Okazaki, Y.; et al. OsATG7 is required for autophagy-dependent lipid metabolism in rice postmeiotic anther development. Autophagy 2014, 10, 878–888. [Google Scholar] [CrossRef]
- Wada, S.; Hayashida, Y.; Izumi, M.; Kurusu, T.; Hanamata, S.; Kanno, K.; Kojima, S.; Yamaya, T.; Kuchitsu, K.; Makino, A.; et al. Autophagy Supports Biomass Production and Nitrogen Use Efficiency at the Vegetative Stage in Rice. Plant Physiol. 2015, 168, 60–73. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Chung, T.; Pennington, J.G.; Federico, M.L.; Kaeppler, H.F.; Kaeppler, S.M.; Otegui, M.S.; Vierstra, R.D. Autophagic Recycling Plays a Central Role in Maize Nitrogen Remobilization. Plant Cell 2015, 27, 1389–1408. [Google Scholar] [CrossRef] [Green Version]
- Zomorodian, A.; Kavoosi, Z.; Momenzadeh, L. Determination of EMC isotherms and appropriate mathematical models for canola. Food Bioprod. Process. 2011, 89, 407–413. [Google Scholar] [CrossRef]
- Selvam, A.; Wong, J.W. Cadmium uptake potential of Brassica napus cocropped with Brassica parachinensis and Zea mays. J. Hazard. Mater. 2009, 167, 170–178. [Google Scholar] [CrossRef]
- Wang, B.; Liu, L.; Gao, Y.; Chen, J. Improved phytoremediation of oilseed rape (Brassica napus) by Trichoderma mutant constructed by restriction enzyme-mediated integration (REMI) in cadmium polluted soil. Chemosphere 2009, 74, 1400–1403. [Google Scholar] [CrossRef] [PubMed]
- Fiebelkorn, D.; Rahman, M. Development of a protocol for frost-tolerance evaluation in rapeseed/canola (Brassica napus L.). Crop. J. 2016, 4, 147–152. [Google Scholar] [CrossRef] [Green Version]
- Bassham, D.C.; Laporte, M.; Marty, F.; Moriyasu, Y.; Ohsumi, Y.; Olsen, L.J.; Yoshimoto, K. Autophagy in Development and Stress Responses of Plants. Autophagy 2006, 2, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Xia, K.; Liu, T.; Ouyang, J.; Wang, R.; Fan, T.; Zhang, M. Genome-Wide Identification, Classification, and Expression Analysis of Autophagy-Associated Gene Homologues in Rice (Oryza sativa L.). DNA Res. 2011, 18, 363–377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, X.-M.; Zhao, P.; Wang, W.; Zou, J.; Cheng, T.-H.; Peng, X.-B.; Sun, M.-X. A comprehensive, genome-wide analysis of autophagy-related genes identified in tobacco suggests a central role of autophagy in plant response to various environmental cues. DNA Res. 2015, 22, 245–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ezhai, Y.; Eguo, M.; Ewang, H.; Elu, J.; Eliu, J.; Ezhang, C.; Egong, Z.-H.; Lu, M.-H. Autophagy, a Conserved Mechanism for Protein Degradation, Responds to Heat, and Other Abiotic Stresses in Capsicum annuum L. Front. Plant Sci. 2016, 7, 131. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Chen, M.; Wang, E.; Hu, L.; Hawkesford, M.J.; Zhong, L.; Chen, Z.; Xu, Z.; Li, L.; Zhou, Y.; et al. Genome-wide analysis of autophagy-associated genes in foxtail millet (Setaria italica L.) and characterization of the function of SiATG8a in conferring tolerance to nitrogen starvation in rice. BMC Genom. 2016, 17, 797. [Google Scholar] [CrossRef] [Green Version]
- Wei, Y.; Liu, W.; Hu, W.; Liu, G.; Wu, C.; Liu, W.; Zeng, H.; He, C.; Shi, H. Genome-wide analysis of autophagy-related genes in banana highlights MaATG8s in cell death and autophagy in immune response to Fusarium wilt. Plant Cell Rep. 2017, 36, 1237–1250. [Google Scholar] [CrossRef]
- Fu, X.-Z.; Zhou, X.; Xu, Y.-Y.; Hui, Q.-L.; Chang-Pin, C.; Li-Li, L.; Peng, L.-Z. Comprehensive Analysis of Autophagy-Related Genes in Sweet Orange (Citrus sinensis) Highlights Their Roles in Response to Abiotic Stresses. Int. J. Mol. Sci. 2020, 21, 2699. [Google Scholar] [CrossRef] [Green Version]
- Han, B.; Xu, H.; Feng, Y.; Xu, W.; Cui, Q.; Liu, A. Genomic Characterization and Expressional Profiles of Autophagy-Related Genes (ATGs) in Oilseed Crop Castor Bean (Ricinus communis L.). Int. J. Mol. Sci. 2020, 21, 562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, S.; Yu, B.; Wang, Y.; Liu, Y. Role of plant autophagy in stress response. Protein Cell 2011, 2, 784–791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, F.; Liu, S.; Wu, J.; Fang, L.; Sun, S.; Liu, B.; Li, P.; Hua, W.; Wang, X. BRAD, the genetics and genomics database for Brassica plants. BMC Plant Biol. 2011, 11, 136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharp, P.A. Speculations on RNA splicing. Cell 1981, 23, 643–646. [Google Scholar] [CrossRef]
- Altenhoff, A.M.; Studer, R.A.; Robinson-Rechavi, M.; Dessimoz, C. Resolving the Ortholog Conjecture: Orthologs Tend to Be Weakly, but Significantly, More Similar in Function than Paralogs. PLoS Comput. Biol. 2012, 8, e1002514. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Chen, H.; He, Y.; Xia, R. TBtools, a toolkit for biologists integrating various biological data handling tools with a user-friendly interface. BioRxiv 2018, 289660. [Google Scholar] [CrossRef]
- Zhang, Y.; Ali, U.; Zhang, G.; Yu, L.; Fang, S.; Iqbal, S.; Li, H.; Lu, S.; Guo, L. Transcriptome analysis reveals genes commonly responding to multiple abiotic stresses in rapeseed. Mol. Breed. 2019, 39, 158. [Google Scholar] [CrossRef]
- Sueoka, N.; Kawanishi, Y. DNA G+C content of the third codon position and codon usage biases of human genes. Gene 2000, 261, 53–62. [Google Scholar] [CrossRef]
- Sharp, P.M.; Tuohy, T.M.; Mosurski, K.R. Codon usage in yeast: Cluster analysis clearly differentiates highly and lowly expressed genes. Nucl. Acids Res. 1986, 14, 5125–5143. [Google Scholar] [CrossRef]
- Yue, W.; Nie, X.; Cui, L.; Zhi, Y.; Zhang, T.; Du, X.; Song, W. Genome-wide sequence and expressional analysis of autophagy Gene family in bread wheat (Triticum aestivum L.). J. Plant Physiol. 2018, 229, 7–21. [Google Scholar] [CrossRef]
- Xu, G.; Guo, C.; Shan, H.; Kong, H. Divergence of duplicate genes in exon-intron structure. Proc. Natl. Acad. Sci. USA 2012, 109, 1187–1192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, G.-M.; Yin, H.; Qiao, X.; Tan, X.; Gu, C.; Wang, B.-H.; Cheng, R.; Wang, Y.-Z.; Zhang, S.-L. F-box genes: Genome-wide expansion, evolution and their contribution to pollen growth in pear (Pyrus bretschneideri). Plant Sci. 2016, 253, 164–175. [Google Scholar] [CrossRef] [PubMed]
- Bai, C.; Sen, P.; Hofmann, K.; Ma, L.; Goebl, M.; Harper, J.; Elledge, S.J. SKP1 Connects Cell Cycle Regulators to the Ubiquitin Proteolysis Machinery through a Novel Motif, the F-Box. Cell 1996, 86, 263–274. [Google Scholar] [CrossRef] [Green Version]
- Dinant, S.; Clark, A.M.; Zhu, Y.; Vilaine, F.; Palauqui, J.-C.; Kusiak, C.; Thompson, G.A. Diversity of the Superfamily of Phloem Lectins (Phloem Protein 2) in Angiosperms. Plant Physiol. 2003, 131, 114–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haasl, R.J.; Payseur, B.A. Microsatellites as targets of natural selection. Mol. Biol. Evol. 2012, 30, 285–298. [Google Scholar] [CrossRef] [Green Version]
- Qin, Z.; Wang, Y.; Wang, Q.; Li, A.; Hou, F.; Zhang, L. Evolution Analysis of Simple Sequence Repeats in Plant Genome. PLoS ONE 2015, 10, e0144108. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Qiao, Y.; Zhang, J.; Shi, W.; Zhang, J. Genome wide identification of microRNAs involved in fatty acid and lipid metabolism of Brassica napus by small RNA and degradome sequencing. Gene 2017, 619, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Park, M.Y.; Conway, S.R.; Wang, J.-W.; Weigel, D.; Poethig, R.S. The Sequential Action of miR156 and miR172 Regulates Developmental Timing in Arabidopsis. Cell 2009, 138, 750–759. [Google Scholar] [CrossRef] [Green Version]
- Chuck, G.; Cigan, A.M.; Saeteurn, K.; Hake, S. The heterochronic maize mutant Corngrass1 results from overexpression of a tandem microRNA. Nat. Genet. 2007, 39, 544–549. [Google Scholar] [CrossRef]
- Zhao, L.; Kim, Y.; Dinh, T.T.; Chen, X. miR172 regulates stem cell fate and defines the inner boundary of APETALA3 and PISTILLATA expression domain in Arabidopsis floral meristems. Plant J. 2007, 51, 840–849. [Google Scholar] [CrossRef] [Green Version]
- Jones-Rhoades, M.W.; Bartel, D.P. Computational Identification of Plant MicroRNAs and Their Targets, Including a Stress-Induced miRNA. Mol. Cell 2004, 14, 787–799. [Google Scholar] [CrossRef] [PubMed]
- Nonogaki, H. MicroRNA Gene Regulation Cascades During Early Stages of Plant Development. Plant Cell Physiol. 2010, 51, 1840–1846. [Google Scholar] [CrossRef] [Green Version]
- Zhao, B.; Ge, L.; Liang, R.-Q.; Li, W.; Ruan, K.; Lin, H.; Jin, Y. Members of miR-169 family are induced by high salinity and transiently inhibit the NF-YA transcription factor. BMC Mol. Biol. 2009, 10, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jian, H.; Wang, J.; Wang, T.; Wei, L.; Li, J.; Liu, L. Identification of Rapeseed MicroRNAs Involved in Early Stage Seed Germination under Salt and Drought Stresses. Front. Plant Sci. 2016, 7, 658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.-H.; Tian, X.; Li, Y.-J.; Wu, C.-A.; Zheng, C.-C. Microarray-based analysis of stress-regulated microRNAs in Arabidopsis thaliana. RNA 2008, 14, 836–843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakashima, K.; Takasaki, H.; Mizoi, J.; Shinozaki, K.; Yamaguchi-Shinozaki, K. NAC transcription factors in plant abiotic stress responses. Biochim. Biophys. Acta (BBA) Bioenerg. 2012, 1819, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhang, S.; Han, S.; Li, X.; Tong, Z.; Qi, L. Deciphering Small Noncoding RNAs during the Transition from Dormant Embryo to Germinated Embryo in Larches (Larix leptolepis). PLoS ONE 2013, 8, e81452. [Google Scholar] [CrossRef]
- Szaker, H.M.; Darkó, É.; Medzihradszky, A.; Janda, T.; Liu, H.-C.; Charng, Y.-Y.; Csorba, T. miR824/AGAMOUS-LIKE16 Module Integrates Recurring Environmental Heat Stress Changes to Fine-Tune Poststress Development. Front. Plant Sci. 2019, 10, 10. [Google Scholar] [CrossRef]
- Pant, B.D.; Musialak-Lange, M.; Nuc, P.; May, P.; Buhtz, A.; Kehr, J.; Walther, D.; Scheible, W.-R. Identification of Nutrient-Responsive Arabidopsis and Rapeseed MicroRNAs by Comprehensive Real-Time Polymerase Chain Reaction Profiling and Small RNA Sequencing. Plant Physiol. 2009, 150, 1541–1555. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Jian, H.; Wang, T.; Wei, L.; Li, J.; Li, C.; Liu, L. Identification of microRNAs Actively Involved in Fatty Acid Biosynthesis in Developing Brassica napus Seeds Using High-Throughput Sequencing. Front. Plant Sci. 2016, 7, 1570. [Google Scholar] [CrossRef] [Green Version]
- Sláviková, S.; Shy, G.; Yao, Y.; Glozman, R.; Levanony, H.; Pietrokovski, S.; Elazar, Z.; Egalili, G. The autophagy-associated Atg8 gene family operates both under favourable growth conditions and under starvation stresses in Arabidopsis plants. J. Exp. Bot. 2005, 56, 2839–2849. [Google Scholar] [CrossRef] [PubMed]
- Bu, F.; Yang, M.; Guo, X.; Huang, W.; Chen, L. Multiple Functions of ATG8 Family Proteins in Plant Autophagy. Front. Cell Dev. Biol. 2020, 8, 466. [Google Scholar] [CrossRef] [PubMed]
- Nakatogawa, H.; Ichimura, Y.; Ohsumi, Y. Atg8, a Ubiquitin-like Protein Required for Autophagosome Formation, Mediates Membrane Tethering and Hemifusion. Cell 2007, 130, 165–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slavikova, S.; Ufaz, S.; Avin-Wittenberg, T.; Levanony, H.; Egalili, G. An autophagy-associated Atg8 protein is involved in the responses of Arabidopsis seedlings to hormonal controls and abiotic stresses. J. Exp. Bot. 2008, 59, 4029–4043. [Google Scholar] [CrossRef] [Green Version]
- Xia, T.; Xiao, D.; Liu, N.; Chai, W.; Gong, Q.; Wang, N.N. Heterologous Expression of ATG8c from Soybean Confers Tolerance to Nitrogen Deficiency and Increases Yield in Arabidopsis. PLoS ONE 2012, 7, e37217. [Google Scholar] [CrossRef]
- Avila-Ospina, L.; Marmagne, A.; Soulay, F.; Masclaux-Daubresse, C. Identification of Barley (Hordeum vulgare L.) Autophagy Genes and Their Expression Levels during Leaf Senescence, Chronic Nitrogen Limitation and in Response to Dark Exposure. Agronomy 2016, 6, 15. [Google Scholar] [CrossRef] [Green Version]
- He, H.; Liang, G.; Lu, S.; Wang, P.; Liu, T.; Ma, Z.; Zuo, C.; Sun, X.; Chen, B.; Mao, J. Genome-Wide Identification and Expression Analysis of GA2ox, GA3ox, and GA20ox Are Related to Gibberellin Oxidase Genes in Grape (Vitis Vinifera L.). Genes 2019, 10, 680. [Google Scholar] [CrossRef] [Green Version]
- Goodstein, D.M.; Shu, S.; Howson, R.; Neupane, R.; Hayes, R.D.; Fazo, J.; Mitros, T.; Dirks, W.; Hellsten, U.; Putnam, N.; et al. Phytozome: A comparative platform for green plant genomics. Nucleic Acids Res. 2011, 40, D1178–D1186. [Google Scholar] [CrossRef]
- Finn, R.D.; Bateman, A.; Clements, J.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Heger, A.; Hetherington, K.; Holm, L.; Mistry, J.; et al. Pfam: The protein families database. Nucleic Acids Res. 2013, 42, D222–D230. [Google Scholar] [CrossRef] [Green Version]
- Artimo, P.; Jonnalagedda, M.; Arnold, K.; Baratin, D.; Csardi, G.; De Castro, E.; Duvaud, S.; Flegel, V.; Fortier, A.; Gasteiger, E.; et al. ExPASy: SIB bioinformatics resource portal. Nucleic Acids Res. 2012, 40, W597–W603. [Google Scholar] [CrossRef]
- Yu, C.-S.; Chen, Y.-C.; Lu, C.-H.; Hwang, J.-K. Prediction of protein subcellular localization. Proteins Struct. Funct. Bioinform. 2006, 64, 643–651. [Google Scholar] [CrossRef]
- Felsenstein, J. Confidence Limits on Phylogenies: An Approach Using the Bootstrap. Evolution 1985, 39, 783. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, K.; Pan, S.; Li, Y. Functional Characterization of Maize C2H2 Zinc-Finger Gene Family. Plant Mol. Biol. Rep. 2015, 34, 761–776. [Google Scholar] [CrossRef]
- Wu, C.; Ding, X.; Ding, Z.; Tie, W.; Yan, Y.; Wang, Y.; Yang, H.; Hu, W. The Class III Peroxidase (POD) Gene Family in Cassava: Identification, Phylogeny, Duplication, and Expression. Int. J. Mol. Sci. 2019, 20, 2730. [Google Scholar] [CrossRef] [Green Version]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef] [Green Version]
- Hu, B.; Jin, J.; Guo, A.-Y.; Zhang, H.; Luo, J.; Gao, G. GSDS 2.0: An upgraded gene feature visualization server. Bioinformatics 2014, 31, 1296–1297. [Google Scholar] [CrossRef] [Green Version]
- Bailey, T.L.; Williams, N.; Misleh, C.; Li, W.W. MEME: Discovering and analyzing DNA and protein sequence motifs. Nucleic Acids Res. 2006, 34, W369–W373. [Google Scholar] [CrossRef]
- Howe, K.L.; Contreras-Moreira, B.; De Silva, N.; Maslen, G.; Akanni, W.; Allen, J.; Alvarez-Jarreta, J.; Barba, M.; Bolser, D.M.; Cambell, L.; et al. Ensembl Genomes 2020—enabling non-vertebrate genomic research. Nucleic Acids Res. 2019, 48, D689–D695. [Google Scholar] [CrossRef] [Green Version]
- Lescot, M. PlantCARE, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Res. 2002, 30, 325–327. [Google Scholar] [CrossRef]
- You, F.M.; Huo, N.; Gu, Y.Q.; Luo, M.-C.; Ma, Y.; Hane, D.; Lazo, G.R.; Dvorák, J.; Anderson, O.D. BatchPrimer3: A high throughput web application for PCR and sequencing primer design. BMC Bioinform. 2008, 9, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://digd.big.ac.cn/ (accessed on 18 October 2020).
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data; Babraham Bioinformatics, Babraham Institute: Cambridge, UK, 2010; Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 18 October 2020).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobin, A.; Gingeras, T.R. Mapping RNA-seq Reads with STAR. Curr. Protoc. Bioinform. 2015, 51, 11–14. [Google Scholar] [CrossRef] [Green Version]
- Peden, J.F. Analysis of Codon Usage. Ph.D. Thesis, Department of Genetics, University of Nottingham, Nottingham, UK, 1999. [Google Scholar]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene ID | Count | Motif | Gene ID | Count | Motif |
|---|---|---|---|---|---|
| BnATG1a | 2 | (GGTT)3, (ATC)4 | BnATG13a | 2 | (AAC)4, (TCC)4 |
| BnATG1b | 4 | (ATTT)3, (TC)7, (GGA)4, (GGC)4 | BnATG13b | 1 | (TCT)4 |
| BnATG1c | 1 | (GATG)3 | BnATG13d | 2 | (GAT)4, (TCT)5 |
| BnATG1f | 2 | (GGTT)3, (ATC)4 | BnATG14a | 1 | (AC)6 |
| BnATG1g | 1 | (ATTT)3 | BnATG14c | 1 | (AC)6 |
| BnATG1i | 2 | (GATG)3, (TTTG)3 | BnATG14e | 1 | (GGAAC)3 |
| BnATG3d | 1 | (GAG)5 | BnATG16a | 1 | (TGATT)3 |
| BnATG4c | 1 | (GAAGA)3 | BnATG16b | 1 | (TTTGA)6 |
| BnATG4d | 1 | (TCTA)3 | BnATG18a | 3 | (TTCC)4, (TCT)4, (GGA)5 |
| BnATG5a | 2 | (CTTT)3, (CCT)5 | BnATG18b | 1 | (GCA)4 |
| BnATG5b | 1 | (AGA)7 | BnATG18e | 1 | (CTC)4 |
| BnATG5c | 1 | (TTTC)3 | BnATG18g | 1 | (GGT)4 |
| BnATG6a | 3 | (GAA)4, (TG)10, (GT)7 | BnATG18h | 1 | (TTTTAT)3 |
| BnATG6b | 1 | (GAA)4 | BnATG18i | 1 | (CAG)5 |
| BnATG6c | 1 | (GAA)4 | BnATG18l | 4 | (GAT)4, (AGC)5, (ATG)4, (TTC)4 |
| BnATG6d | 1 | (GAA)4 | BnATG18m | 2 | (GAT)6, (TTC)4 |
| BnATG7a | 1 | (TTTC)3 | BnATG18r | 1 | (CTC)4 |
| BnATG8c | 2 | (TTTG)3, (TTC)4 | BnATG18x | 3 | (ATG)4, (CAT)4, (TTC)5 |
| BnATG8d | 2 | (TATTT)3, (TTG)5 | BnATG20a | 2 | (AAC)4, (TC)7 |
| BnATG8h | 2 | (ATTCA)3, (GTT)4 | BnATG20b | 3 | (ATA), (TC)7, (AAC)4 |
| BnATG8j | 2 | (TCT)5, (AT)12 | BnATG101b | 1 | (TCG)4 |
| BnATG8k | 1 | (TTTGA)3 | BnATG101c | 5 | (TGGCCT)3, (CTA)6, (TTTA)4, (GATG)3, (CCAT)3 |
| BnATG8l | 3 | (AAGC)3, (TTGA)3, (TTCT)4 | BnATG101d | 3 | (CCAT)3, (AT)7, (TTC)4 |
| BnATG8o | 1 | (TA)8 | BnATG101e | 2 | (TCT)4, (TC)6 |
| BnATG8p | 1 | (CTT)4 | BnTORa | 5 | (TTTA)3, (TC)7, (CT)8, (TC)8, (CTTT)3 |
| BnATG8q | 3 | (TTTG)3, (TC)7, (TTC)4 | BnTORb | 3 | (TCTT)3, (AT)9, (ATT)4 |
| BnATG8r | 1 | (TTG)6 | BnTORc | 2 | (TATTT)3, (TTA)5 |
| BnATG8y | 2 | (AAGC)4, (AT)7 | BnVPS15a | 2 | (TTTC)3, (TTTG)3 |
| BnATG9a | 7 | (TCAAT)5, (CT)10, (CTC)4, (GAG)5, (GAT)5, (AAGA)3, (GTTA)3 | BnVPS15b | 2 | (TTTC)3, (TTTG)3 |
| BnATG9c | 6 | (TCAAT)4, (CT)10, (GAT)4, (TATT)3, (AAGA)3, (GTTA)3 | BnVPS34a | 1 | (TA)7 |
| BnATG10a | 1 | (CGGCAG)3 | BnVPS34b | 1 | (TC)6 |
| BnATG11a | 2 | (TTCT)3, (TTTA)5 | BnVTI12a | 1 | (AAG)6 |
| BnATG11b | 3 | (TTAT)3, (AGA)6, (AAC)4 | BnVTI12b | 3 | (CCTT)3, (AAT)5, (TCA)4 |
| BnATG11c | 1 | (ATC)4 | BnVTI12d | 1 | (AAG)5 |
| BnATG11e | 1 | (AT)6 | BnVTI12e | 1 | (CCTT)4 |
| BnATG11f | 1 | (AAC)7 | BnVTI12f | 2 | (ATAC)3, (CTT)4 |
| BnATG12a | 1 | (TTTGT)3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eshkiki, E.M.; Hajiahmadi, Z.; Abedi, A.; Kordrostami, M.; Jacquard, C. In Silico Analyses of Autophagy-Related Genes in Rapeseed (Brassica napus L.) under Different Abiotic Stresses and in Various Tissues. Plants 2020, 9, 1393. https://doi.org/10.3390/plants9101393
Eshkiki EM, Hajiahmadi Z, Abedi A, Kordrostami M, Jacquard C. In Silico Analyses of Autophagy-Related Genes in Rapeseed (Brassica napus L.) under Different Abiotic Stresses and in Various Tissues. Plants. 2020; 9(10):1393. https://doi.org/10.3390/plants9101393
Chicago/Turabian StyleEshkiki, Elham Mehri, Zahra Hajiahmadi, Amin Abedi, Mojtaba Kordrostami, and Cédric Jacquard. 2020. "In Silico Analyses of Autophagy-Related Genes in Rapeseed (Brassica napus L.) under Different Abiotic Stresses and in Various Tissues" Plants 9, no. 10: 1393. https://doi.org/10.3390/plants9101393
APA StyleEshkiki, E. M., Hajiahmadi, Z., Abedi, A., Kordrostami, M., & Jacquard, C. (2020). In Silico Analyses of Autophagy-Related Genes in Rapeseed (Brassica napus L.) under Different Abiotic Stresses and in Various Tissues. Plants, 9(10), 1393. https://doi.org/10.3390/plants9101393