Comparative Chloroplast Genomics of Fritillaria (Liliaceae), Inferences for Phylogenetic Relationships between Fritillaria and Lilium and Plastome Evolution


Abstract
:1. Introduction
2. Results
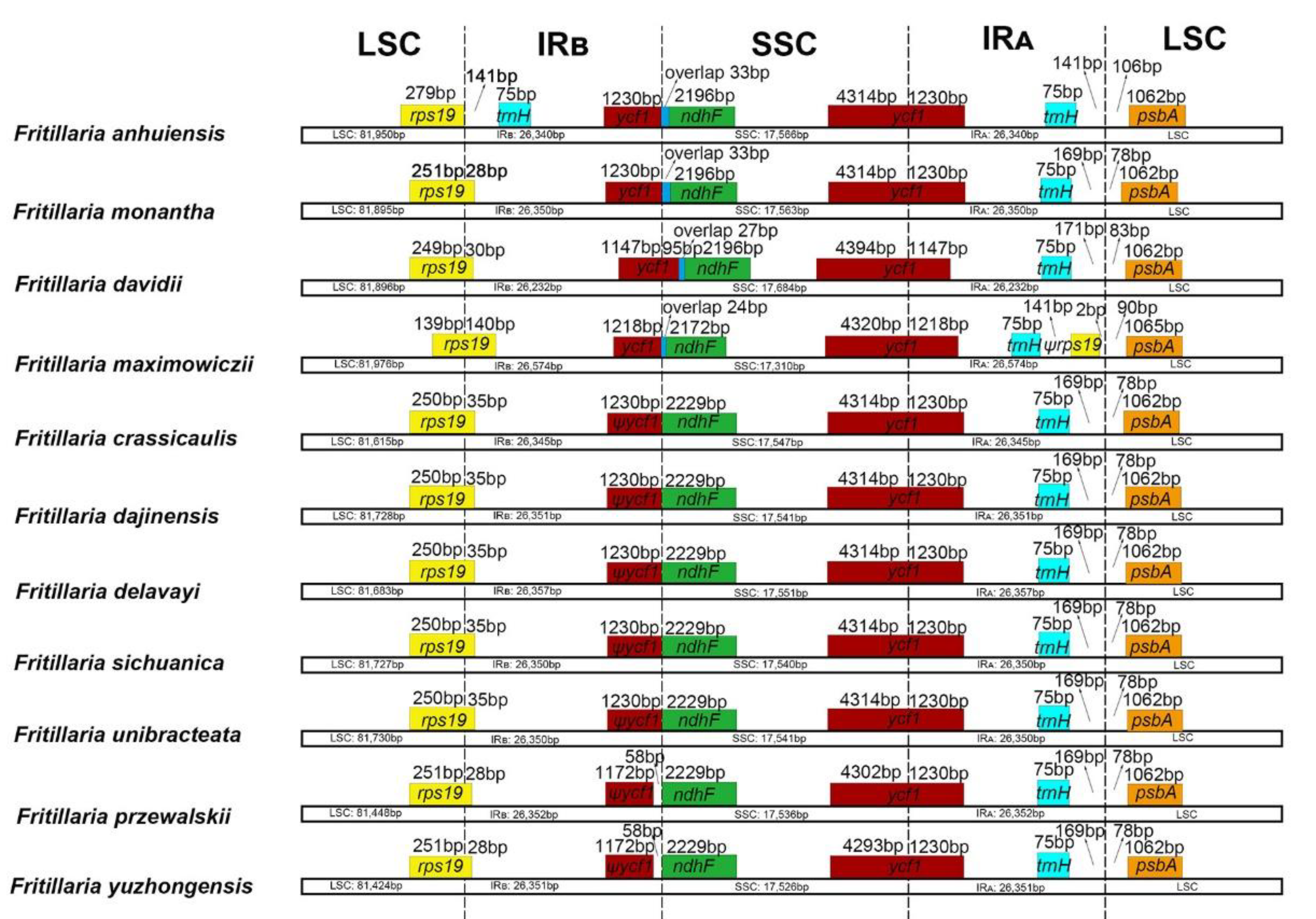
2.1. Plastome Features of Fritillaria and Comparison with Other Liliales Taxa
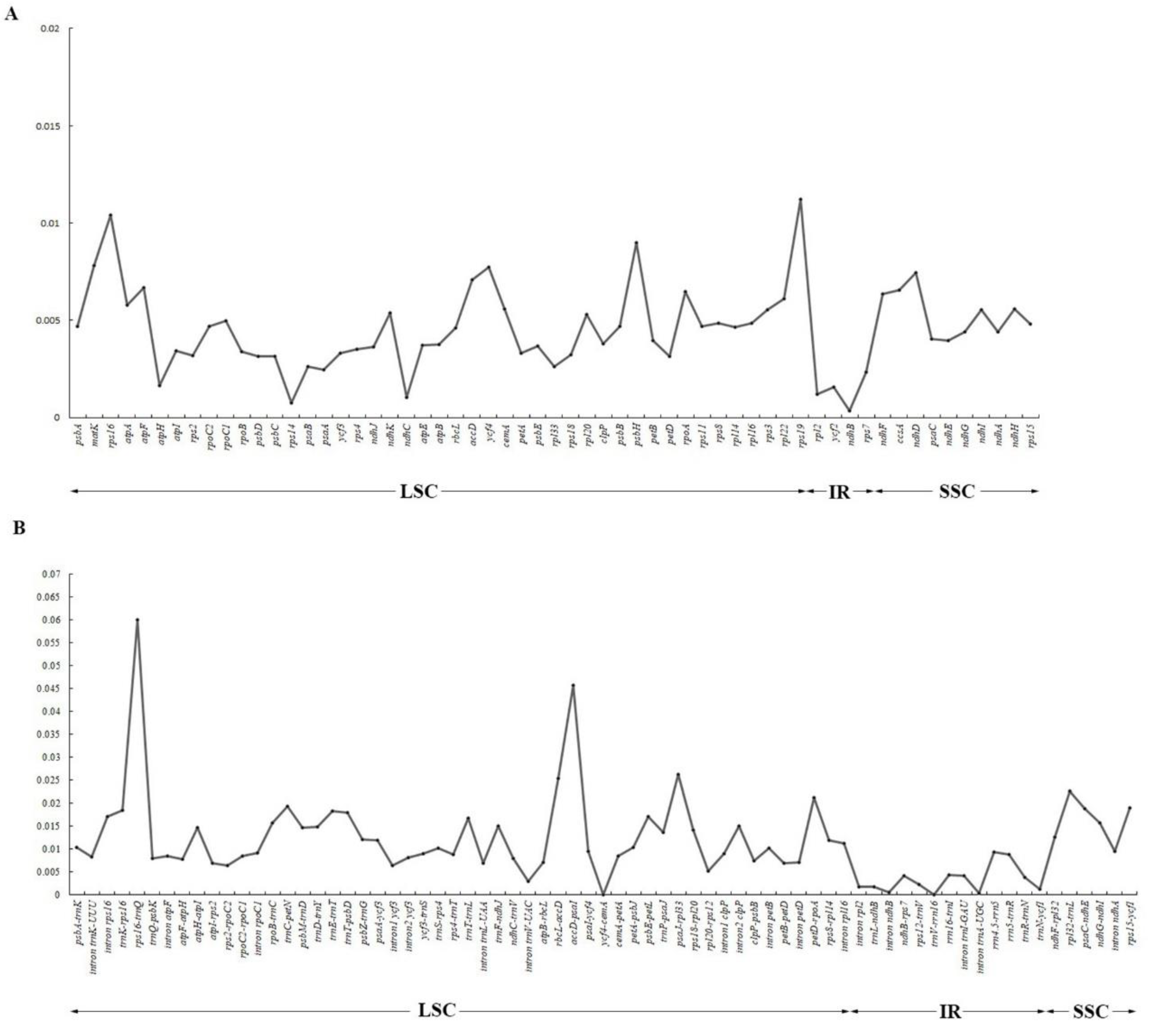
2.2. Comparative Genomic Analysis and Divergence Hotspot Regions
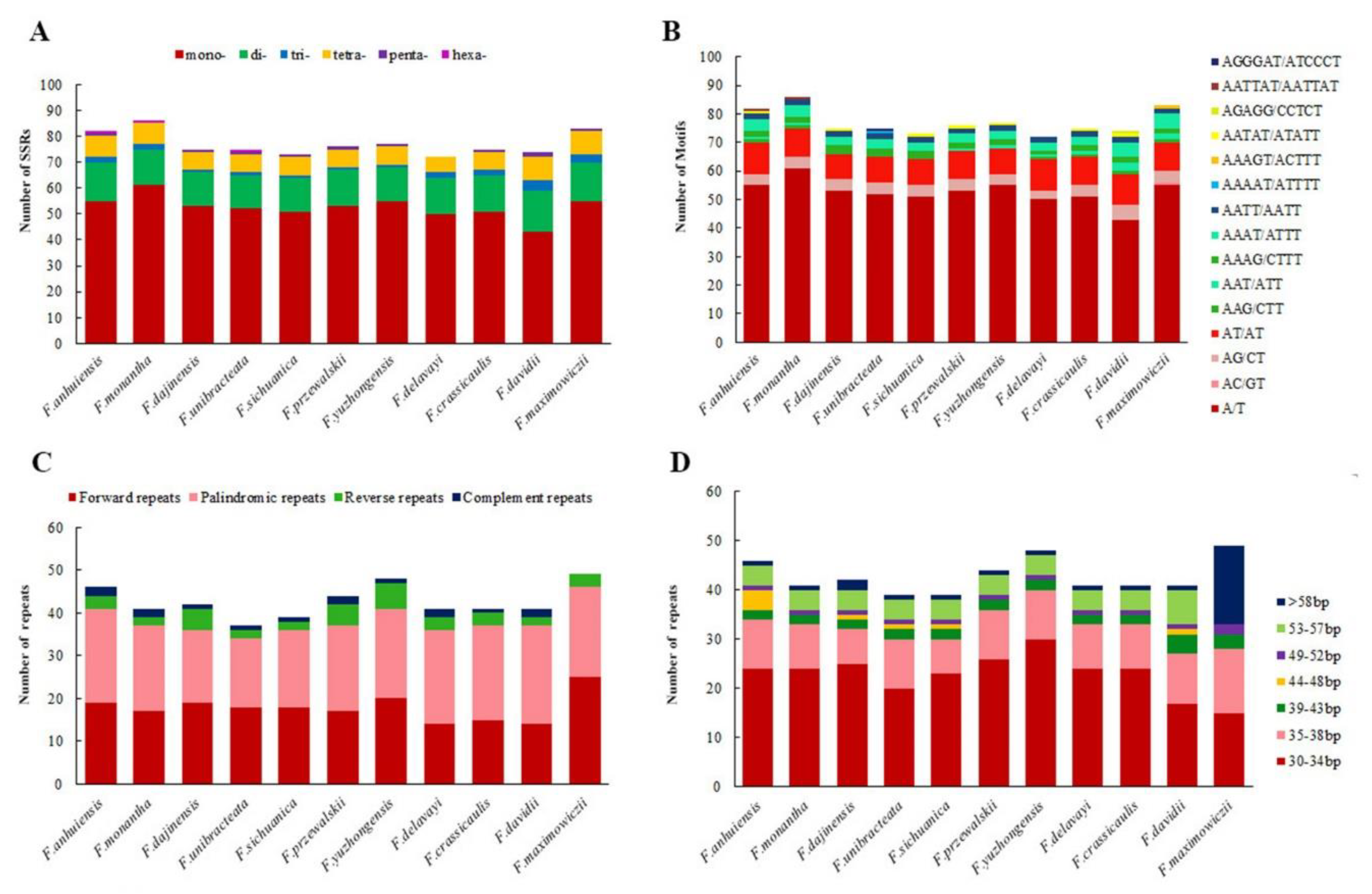
2.3. SSRs Analysis and Repeat Sequences
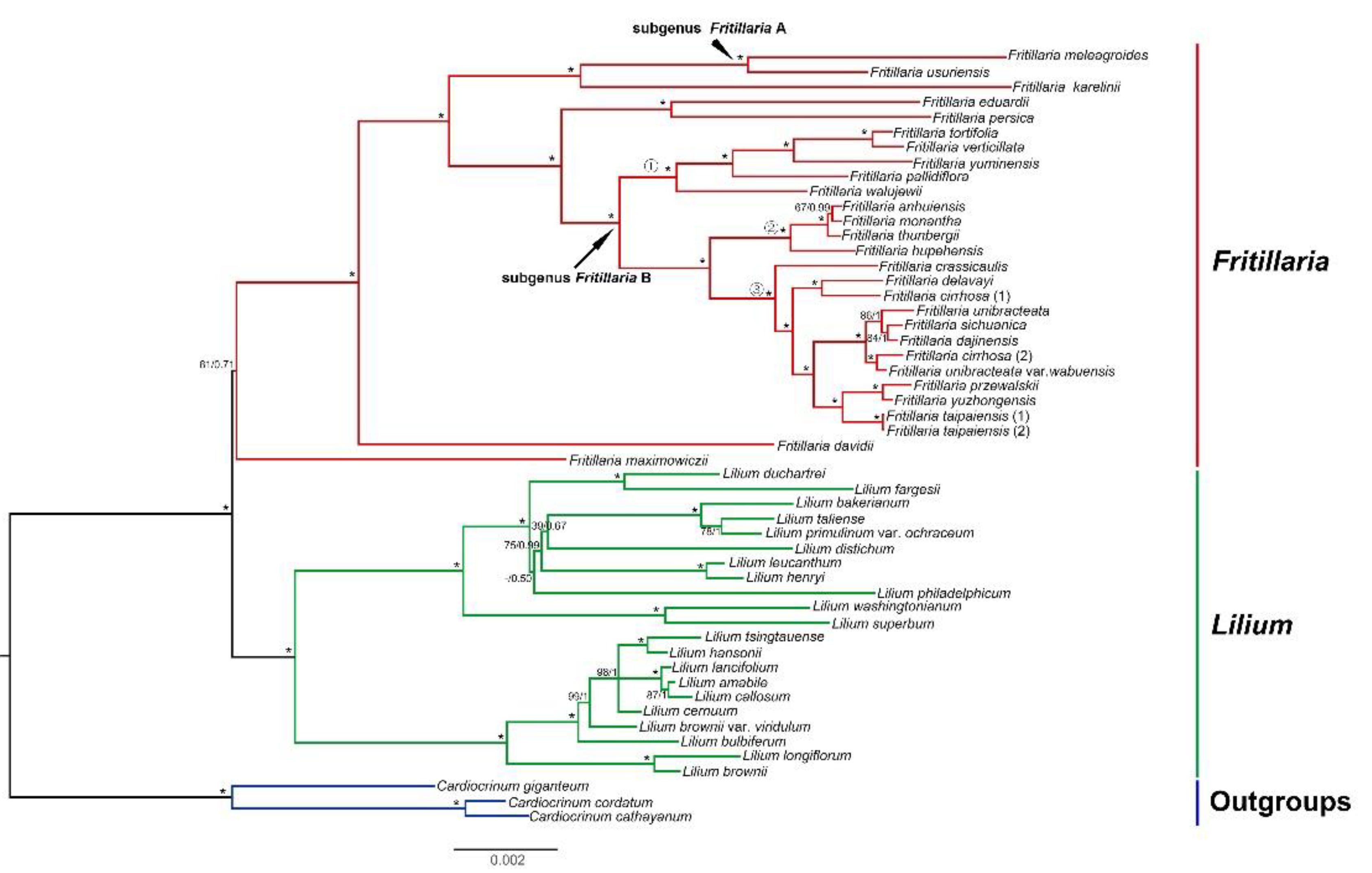
2.4. Phylogenetic Analyses
3. Discussion
3.1. Comparison of Fritillaria Plastomes and Phylogenetically Informative Markers
3.2. Phylogenetic Relationships and Implications
4. Materials and Methods
4.1. Taxon Sampling, DNA Extraction, and Sequencing
4.2. Chloroplast Genome Assembly, Annotation, and Structural Analyses
4.3. Genome Comparative Analysis and Identification of Hypervariable Regions
4.4. Characterization of SSRs and Repeat Sequences
4.5. Phylogenetic Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zhu, A.; Guo, W.; Gupta, S.; Fan, W.; Mower, J.P. Evolutionary dynamics of the plastid inverted repeat: The effects of expansion, contraction, and loss on substitution rates. New Phytol. 2016, 209, 1747–1756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, W.Q.; Yap, Z.Y.; Li, P.; Peter, H.C.; Qiu, Y.X. Plastome organization, genome-based phylogeny andevolution of plastid genes in Podophylloideae (Berberidaceae). Mol. Phylogenet. Evol. 2018, 127, 978–987. [Google Scholar] [CrossRef] [PubMed]
- Fu, C.N.; Li, H.T.; Milne, R.; Zhang, T.; Ma, P.F.; Yang, J.; Li, D.Z.; Gao, L.M. Comparative analyses of plastid genomes from fourteen Cornales species: Inferences for phylogenetic relationships and genome evolution. BMC Genom. 2017, 18, 956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Downie, S.R.; Jansen, R.K. A Comparative Analysis of Whole Plastid Genomes from the Apiales: Expansion and Contraction of the Inverted Repeat, Mitochondrial to Plastid Transfer of DNA, and Identification of Highly Divergent Noncoding Regions. Syst. Bot. 2015, 40, 336–351. [Google Scholar] [CrossRef]
- Hsu, C.Y.; Wu, C.S.; Chaw, S.M. Birth of Four Chimeric Plastid Gene Clusters in Japanese Umbrella Pine. Genome Biol. Evol. 2016, 8, 1776–1784. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Moore, M.J.; Lin, N.; Adelalu, K.F.; Meng, A.; Jian, S.; Yang, L.; Li, J.; Wang, H. Complete plastome sequencing of both living species of Circaeasteraceae (Ranunculales) reveals unusual rearrangements and the loss of the ndh gene family. BMC Genom. 2017, 18, 592. [Google Scholar] [CrossRef]
- Zhou, T.; Wang, J.; Jia, Y.; Li, W.; Xu, F.; Wang, X. Comparative Chloroplast Genome Analyses of Species in Gentiana section Cruciata (Gentianaceae) and the Development of Authentication Markers. Int. J. Mol. Sci. 2018, 19, 1962. [Google Scholar] [CrossRef] [Green Version]
- Shaw, J.; Lickey, E.B.; Beck, J.T.; Farmer, S.B.; Wusheng, L. The tortoise and the hare II: Relative utility of 21 noncoding chloroplast DNA sequences for phylogenetic analysis. Am. J. Bot. 2005, 92, 142–166. [Google Scholar] [CrossRef] [Green Version]
- Lu, R.S.; Li, P.; Qiu, Y.X. The complete chloroplast genomes of three Cardiocrinum (Liliaceae) species: Comparative genomic and phylogenetic analyses. Front. Plant Sci. 2017, 7, 2054–2066. [Google Scholar] [CrossRef]
- Group, C.P.W. A DNA barcode for land plants. Proc. Natl. Acad. Sci. USA 2009, 106, 12794–12797. [Google Scholar] [CrossRef] [Green Version]
- Hu, H.; Hu, Q.J.; Al-Shehbaz, I.A.; Luo, X.; Zeng, T.T.; Guo, X.Y.; Liu, J.Q. Species Delimitation and interspecific relationships of the genus Orychophragmus (Brassicaceae) inferred from whole chloroplast genomes. Front. Plant Sci. 2016, 7, 1826–1836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.D.; Jin, J.J.; Chen, S.Y.; Chase, M.W.; Soltis, D.E.; Li, H.T.; Yang, J.B.; Li, D.Z.; Yi, T.S. Diversification of Rosaceae since the Late Cretaceous based on plastid phylogenomics. New Phytol. 2017, 214, 1355–1368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, W.Q.; Losh, J.; Chen, C.; Pan, L.; Wang, R.H.; Zhao, Y.P.; Qiu, Y.X.; Fu, C.X. Comparative genomics of figworts (Scrophularia, Scrophulariaceae), with implications for the evolution of Scrophularia and Lamiales. J. Syst. Evol. 2018, 57, 55–65. [Google Scholar] [CrossRef]
- Xi, Z.X.; Ruhfel, B.R.; Schaefer, H.; Amorim, A.M.; Sugumara, M. Phylogenomics and a posteriori data partitioning resolve the Cretaceous angiosperm radiation Malpighiales. Proc. Natl. Acad. Sci. USA 2012, 109, 17519–17524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, N.; Wen, J.; Zimmer, E.A. Another look at the phylogenetic position of the grape order Vitales: Chloroplast phylogenomics with an expanded sampling of key lineages. Mol. Phylogenet. Evol. 2016, 101, 216–223. [Google Scholar] [CrossRef] [Green Version]
- Day, P.D.; Berger, M.; Hill, L.; Fay, M.F.; Leitch, A.R.; Leitch, I.J.; Kelly, L.J. Evolutionary relationships in the medicinally important genus Fritillaria L. (Liliaceae). Mol. Phylogenet. Evol. 2014, 80, 11–19. [Google Scholar] [CrossRef]
- Huang, J.; Yang, L.Q.; Yu, Y.; Liu, Y.M.; Xie, D.F. Molecular phylogenetics and historical biogeography of the tribe Lilieae (Liliaceae): Bi-directional dispersal between biodiversity hotspots in Eurasia. Ann. Bot. 2018, 122, 1245–1262. [Google Scholar] [CrossRef]
- Hao, D.C.; Xiao-Jie, G.U.; Xiao, P.G. Phytochemical and biological research of Fritillaria medicinal resources. Chin. J. Nat. Med. 2013, 11, 330–344. [Google Scholar] [CrossRef]
- Wang, D.; Li, Z.; Zhang, L.; Atanasov, A.G.; Wang, S. Characterization of the Isosteroidal Alkaloid Chuanbeinone from Bulbus of Fritillaria pallidiflora as Novel Antitumor Agent In Vitro and In Vivo. Planta Med. 2016, 82, 195–204. [Google Scholar] [CrossRef]
- Altinordu, F.; Peruzzi, L.; Yu, Y.; He, X.J. A tool for the analysis of chromosomes: KaryoType. Taxon 2016, 65, 586–592. [Google Scholar] [CrossRef]
- Rix, E.M. Fritillaria: A Revised Classification Together with an Updated List of Species; The Fritillaria Group of the Alpine Garden Society Press: Pershore, UK, 2001. [Google Scholar]
- Li, Q.S.; Li, Y.; Song, J.Y.; Xu, H.B.; Xu, J. High-accuracy de novo assembly and SNP detection of chloroplast genomes using a SMRT circular consensus sequencing strategy. New Phytol. 2014, 204, 1041–1049. [Google Scholar] [CrossRef] [PubMed]
- Park, I.; Kim, W.J.; Yeo, S.M.; Choi, G.; Kang, Y.M. The complete chloroplast genome sequences of Fritillaria ussuriensis Maxim. and Fritillaria cirrhosa D. Don, and comparative analysis with other Fritillaria species. Molecules 2017, 22, 982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bi, Y.; Zhang, M.F.; Xue, J.; Dong, R.; Du, Y.P.; Zhang, X.H. Chloroplast genomic resources for phylogeny and DNA barcoding: A case study on Fritillaria. Sci. Rep. 2018, 8, 1184–1196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Zhang, Z.; Yang, J.; Lv, G.H. Complete chloroplast genome of seven Fritillaria species, variable DNA markers identification and phylogenetic relationships within the genus. PLoS ONE 2018, 13, e0194613. [Google Scholar] [CrossRef] [Green Version]
- Li, P.; Lu, R.S.; Xu, W.Q.; Ohi-Toma, T.; Cai, M.Q.; Qiu, Y.X.; Cameron, K.M.; Fu, C.X. Comparative Genomics and Phylogenomics of East Asian Tulips (Amana, Liliaceae). Front. Plant Sci. 2017, 8, 451–463. [Google Scholar] [CrossRef] [Green Version]
- Raman, G.; Park, S. Analysis of the complete chloroplast genome of a medicinal plant, Dianthus superbus var longicalyncinus, from a comparative genomics perspective. PLoS ONE 2015, 10, e0141329. [Google Scholar] [CrossRef] [Green Version]
- Yan, L.; Lai, X.; Li, X.; Wei, C.; Tan, X.; Zhang, Y. Analyses of the complete genome and gene expression of chloroplast of sweet potato [Ipomoea batata]. PLoS ONE 2015, 10, e0124083. [Google Scholar] [CrossRef]
- Millen, R.S.; Olmstead, R.G.; Adams, K.L.; Palmer, J.D.; Lao, N.T.; Heggie, L.; Kavanagh, T.A.; Hibberd, J.M.; Gray, J.C.; Morden, C.W. Many parallel losses of infA from chloroplast DNA during angiosperm evolution with multiple independent transfers to the nucleus. Plant Cell 2001, 13, 645–658. [Google Scholar] [CrossRef] [Green Version]
- Wicke, S.; Schneeweiss, G.M.; Depamphilis, C.W.; Kai, F.M.; Quandt, D. The evolution of the plastid chromosome in land plants: Gene content, gene order, gene function. Plant Mol. Biol. 2011, 76, 273–297. [Google Scholar] [CrossRef] [Green Version]
- Ren, T.; Yang, Y.; Zhou, T.; Liu, Z.L. Comparative plastid genomes of Primula species: Sequence divergence and phylogenetic relationships. Int. J. Mol. Sci. 2018, 19, 1050. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Qi, Z.C.; Zhao, Y.P.; Fu, C.X.; Xiang, Q.Y. Complete cpDNA genome sequence of Smilax china and phylogenetic placement of Liliales–influences of gene partitions and taxon sampling. Mol. Phylogenet. Evol. 2012, 64, 545–562. [Google Scholar] [CrossRef] [PubMed]
- Do, H.D.; Kim, J.S.; Kim, J.H. Comparative genomics of four Liliales families inferred from the complete chloroplast genome sequence of Veratrum patulum O. Loes. (Melanthiaceae). Gene 2013, 530, 229–235. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.S.; Kim, J.H. Comparative genome analysis and phylogenetic relationship of order Liliales insight from the complete plastid genome sequences of two Lilies (Lilium longiflorum and Alstroemeria aurea). PLoS ONE 2013, 8, e68180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mennes, C.B.; Lam, V.K.Y.; Rudall, P.J.; Lyon, S.P.; Graham, S.W.; Smets, E.F.; Merckx, V.S.F.T. Ancient Gondwana break-up explains the distribution of the mycoheterotrophic family Corsiaceae (Liliales). J. Biogeogr. 2015, 42, 1123–1136. [Google Scholar] [CrossRef]
- Huang, Y.; Li, X.; Yang, Z.; Yang, C.; Yang, J.; Ji, Y. Analysis of complete chloroplast genome sequences improves phylogenetic resolution in Paris (Melanthiaceae). Front. Plant Sci. 2016, 7, 1797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, P.A.T.; Kim, J.S.; Kim, J.H. The complete chloroplast genome of colchicine plants (Colchicum autumnale L. and Gloriosa superba L.) and its application for identifying the genus. Planta 2015, 242, 223–237. [Google Scholar] [CrossRef]
- Palmer, J.D.; Thompson, W.F. Chloroplast DNA rearrangements are more frequent when a large inverted repeat sequence is lost. Cell 1982, 29, 537–550. [Google Scholar] [CrossRef]
- Xiao, H.; Jiang, N.; Schaffner, E.; Stockinger, E.J.; Knaap, E.V.D. A retrotransposon-mediated gene duplication underlies morphological variation of tomato fruit. Science 2008, 319, 1527–1530. [Google Scholar] [CrossRef]
- Yang, M.; Zhang, X.W.; Liu, G.M.; Yin, Y.X.; Chen, K.F. The complete chloroplast genome sequence of date palm (Phoenix dactylifera L.). PLoS ONE 2010, 5, e12762. [Google Scholar] [CrossRef] [Green Version]
- Do, H.D.K.; Kim, J.H. A dynamic tandem repeat in monocotyledons inferred from a comparative analysis of chloroplast genomes in Melanthiaceae. Front. Plant Sci. 2017, 8, 693. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.J.; Cheng, C.L.; Chang, C.C.; Wu, C.L.; Su, T.M.; Chaw, S.M. Dynamics and evolution of the inverted repeat-large single copy junctions in the chloroplast genomes of monocots. BMC Evol. Biol. 2008, 8, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plunkett, G.M.; Downie, S.R. Expansion and contraction of the chloroplast inverted repeat in Apiaceae subfamily Apioideae. Syst. Bot. 2000, 25, 648–667. [Google Scholar] [CrossRef]
- Kim, K.J.; Lee, H.L. Complete chloroplast genome sequences from Korean Ginseng (Panax schinseng Nees) and comparative analysis of sequence evolution among 17 vascular plants. DNA Res. 2005, 11, 247–261. [Google Scholar] [CrossRef] [PubMed]
- Dong, W.L.; Wang, R.N.; Zhang, N.Y.; Fan, W.B.; Fang, M.F.; Li, Z.H. Molecular evolution of chloroplast genomes of Orchid species: Insights into phylogenetic relationship and adaptive evolution. Int. J. Mol. Sci. 2018, 19, 716. [Google Scholar] [CrossRef] [Green Version]
- Rønsted, N.; Law, S.; Thornton, H.; Fay, M.F.; Chase, M.W. Molecular phylogenetic evidence for the monophyly of Fritillaria and Lilium (Liliaceae; Liliales) and the infrageneric classification of Fritillaria. Mol. Phylogenet. Evol. 2005, 35, 509–527. [Google Scholar] [CrossRef]
- Turktas, M.; Aslay, M.; Kaya, E.; Ertugrul, F. Molecular characterization of phylogenetic relationships in Fritillaria species inferred from chloroplast trnL-trnF sequences. Turk. J. Biol. 2012, 36, 552–560. [Google Scholar]
- Kim, J.S.; Hong, J.K.; Chase, M.W.; Fay, M.F.; Kim, J.H. Familial relationships of the monocot order Liliales based on a molecular phylogenetic analysis using four plastid loci: matK, rbcL, atpB and atpF-H. Bot. J. Linn. Soc. 2013, 172, 5–21. [Google Scholar] [CrossRef] [Green Version]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data 2015. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 6 October 2019).
- Luo, R.; Liu, B.; Xie, Y.; Li, Z.; Huang, W.; Yuan, J.; He, G.; Chen, Y.; Pan, Q.; Liu, Y. SOAPdenovo2: An empirically improved memory-efficient short-read de novo assembler. Gigascience 2012, 1, 18. [Google Scholar] [CrossRef]
- Kearse, M.; Moir, R.; Wilson, A.; Havas, S.S.; Cheung, M. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Lohse, M.; Drechsel, O.; Kahlau, S.; Bock, R. Organellar Genome DRAW—A suite of tools for generating physical maps of plastid and mitochondrial genomes and visualizing expression data sets. Nucleic Acids Res. 2013, 41, W575. [Google Scholar] [CrossRef]
- Frazer, K.A.; Lior, P.; Alexander, P.; Rubin, E.M.; Inna, D. VISTA: Computational tools for comparative genomics. Nucleic Acids Res. 2004, 32 (Suppl. S2), W273–W279. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef] [Green Version]
- Thiel, T.; Michalek, W.; Varshney, R.K.; Graner, A. Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.). Theor. Appl. Genet. 2003, 106, 411–422. [Google Scholar] [CrossRef] [PubMed]
- Kurtz, S.; Choudhuri, J.V.; Ohlebusch, E.; Schleiermacher, C.; Stoye, J.; Giegerich, R. REPuter: Themanifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001, 29, 4633–4642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2: More models, new heuristics and parallel computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.A.; Pfeiffer, W.; Schwartz, T. Creating the CIPRES Science Gateway for inference of large phylogenetic trees. In Proceedings of the 2010 Gateway Computing Environments Workshop (GCE), New Orleans, LA, USA, 14 November 2010; pp. 1–8. [Google Scholar]
- Ronquist, F.; Huelsenbeck, J.P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 2003, 19, 1572–1574. [Google Scholar] [CrossRef] [Green Version]
- Xie, D.F.; Yu, H.X.; Price, M.; Xie, C.; Deng, Y.Q.; Chen, J.P.; Yu, Y.; Zhou, S.D.; He, X.J. Phylogeny of Chinese Allium species in section Daghestanica and adaptive evolution of Allium (Amaryllidaceae, Allioideae) species revealed by the chloroplast complete genome. Front. Plant Sci. 2019, 10, 460. [Google Scholar] [CrossRef] [Green Version]
- Xie, D.F.; Yu, Y.; Deng, Y.Q.; Li, J.; Liu, H.Y.; Zhou, S.D.; He, X.J. Comparative analysis of the chloroplast genomes of the chinese endemic genus Urophysa and their contribution to chloroplast phylogeny and adaptive evolution. Int. J. Mol. Sci. 2018, 19, 1847. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Genome Size | LSC Length | IR Length | SSC Length | G-C | Number of Genes | Number of CDS | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| (bp) | (bp) | (bp) | (bp) | (%) | Total | CDS | rRNAs | tRNAs | LSC | IRA | SSC | IRB | |
| F. anhuiensis | 152,196 | 81,950 | 26,340 | 17,566 | 36.9 | 131[18] | 85[6] | 8[4] | 38[8] | 60 | 7 | 11 | 7 |
| F. crassicaulis | 151,852 | 81,615 | 26,345 | 17,547 | 37.0 | 136[20] | 90[8] | 8[4] | 38[8] | 60(1) | 9(3) | 11 | 9(2) |
| F. dajinensis | 151,971 | 81,728 | 26,351 | 17,541 | 37.0 | 136[20] | 90[8] | 8[4] | 38[8] | 60(1) | 9(3) | 11 | 9(2) |
| F. davidii | 152,044 | 81,896 | 26,232 | 17,684 | 37.0 | 131[18] | 85[6] | 8[4] | 38[8] | 60 | 7 | 11 | 7 |
| F. delavayi | 151,948 | 81,683 | 26,357 | 17,551 | 36.9 | 136[20] | 90[8] | 8[4] | 38[8] | 60(1) | 9(3) | 11 | 9(2) |
| F. maximowiczii | 152,434 | 81,976 | 26,574 | 17,310 | 37.1 | 132[18] | 86[6] | 8[4] | 38[8] | 60(1) | 7 | 11 | 7 |
| F. monantha | 152,158 | 81,895 | 26,350 | 17,563 | 37.0 | 131[18] | 85[6] | 8[4] | 38[8] | 60 | 7 | 11 | 7 |
| F. przewalskii | 151,688 | 81,448 | 26,352 | 17,536 | 37.0 | 132[18] | 86[6] | 8[4] | 38[8] | 60(1) | 7(1) | 11 | 7 |
| F. sichuanica | 151,967 | 81,727 | 26,350 | 17,540 | 37.0 | 136[20] | 90[8] | 8[4] | 38[8] | 60(1) | 9(3) | 11 | 9(2) |
| F. unibracteata | 151,971 | 81,730 | 26,350 | 17,541 | 36.9 | 136[20] | 90[8] | 8[4] | 38[8] | 60(1) | 9(3) | 11 | 9(2) |
| F. yuzhongensis | 151,652 | 81,424 | 26,351 | 17,526 | 37.0 | 132[18] | 86[6] | 8[4] | 38[8] | 60(1) | 7(1) | 11 | 7 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, J.; Yu, Y.; Liu, Y.-M.; Xie, D.-F.; He, X.-J.; Zhou, S.-D. Comparative Chloroplast Genomics of Fritillaria (Liliaceae), Inferences for Phylogenetic Relationships between Fritillaria and Lilium and Plastome Evolution. Plants 2020, 9, 133. https://doi.org/10.3390/plants9020133
Huang J, Yu Y, Liu Y-M, Xie D-F, He X-J, Zhou S-D. Comparative Chloroplast Genomics of Fritillaria (Liliaceae), Inferences for Phylogenetic Relationships between Fritillaria and Lilium and Plastome Evolution. Plants. 2020; 9(2):133. https://doi.org/10.3390/plants9020133
Chicago/Turabian StyleHuang, Jiao, Yan Yu, Yan-Mei Liu, Deng-Feng Xie, Xing-Jin He, and Song-Dong Zhou. 2020. "Comparative Chloroplast Genomics of Fritillaria (Liliaceae), Inferences for Phylogenetic Relationships between Fritillaria and Lilium and Plastome Evolution" Plants 9, no. 2: 133. https://doi.org/10.3390/plants9020133
APA StyleHuang, J., Yu, Y., Liu, Y. -M., Xie, D. -F., He, X. -J., & Zhou, S. -D. (2020). Comparative Chloroplast Genomics of Fritillaria (Liliaceae), Inferences for Phylogenetic Relationships between Fritillaria and Lilium and Plastome Evolution. Plants, 9(2), 133. https://doi.org/10.3390/plants9020133