

A Multi-Scale Model for Cholera Outbreaks

Abstract
:1. Introduction
2. Model
3. Pathogen Distribution for Constant Environmental Pathogen Load
3.1. Semigroup
3.2. Stationary Solution and Spectral Gap
3.2.1. Eigenvalues and Fixed Point Operator
3.2.2. A Priori Estimates
3.2.3. Regularized Operator
- , is a simple eigenvalue with an eigenvector in the non-empty interior and no other eigenvalue has a positive eigenvector;
- ∀ eigenvalues, .
Regularized Operator
Eigenvalues
3.2.4. De-Regularization and Spectral Gap
Uniqueness of Fixed Point for
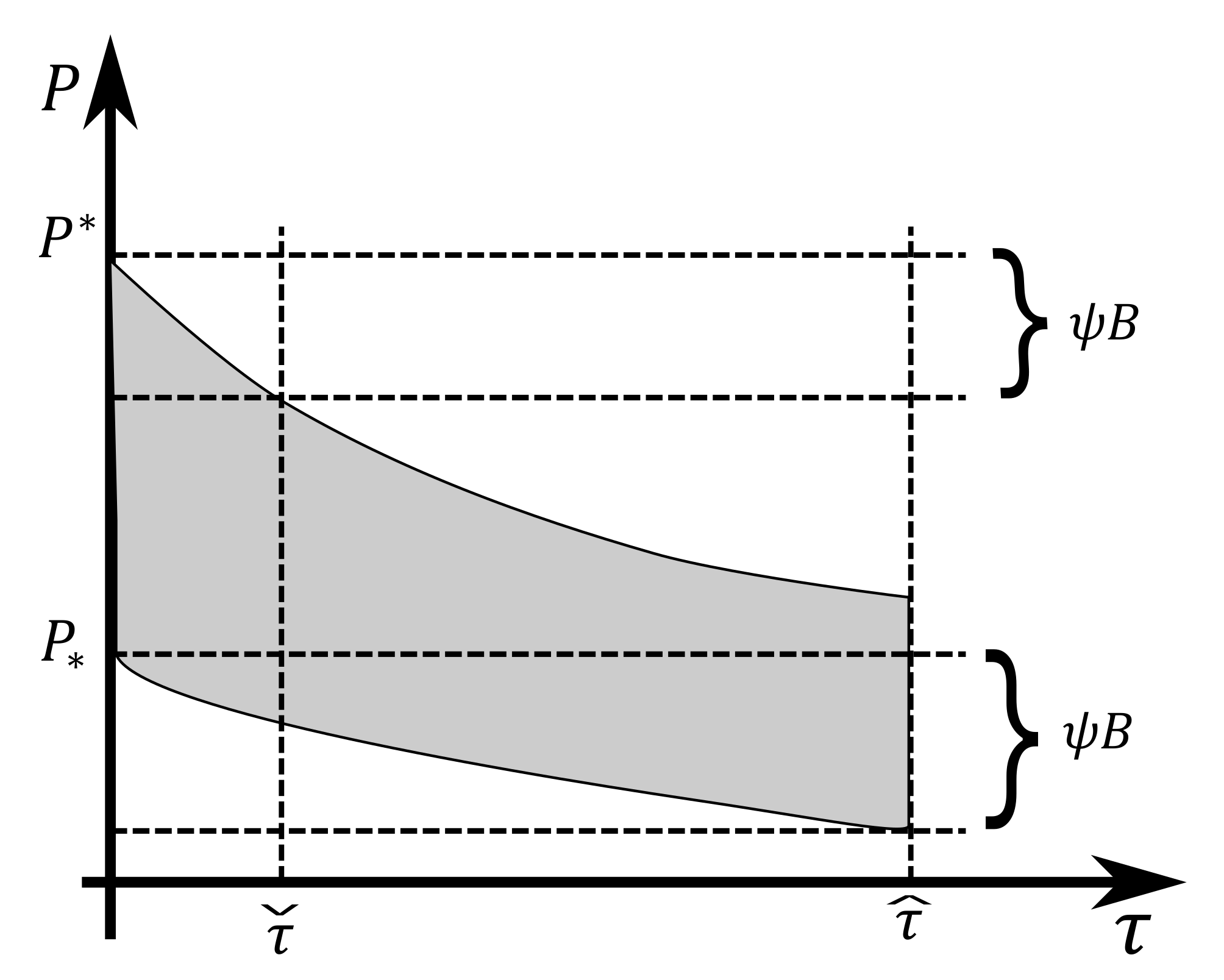
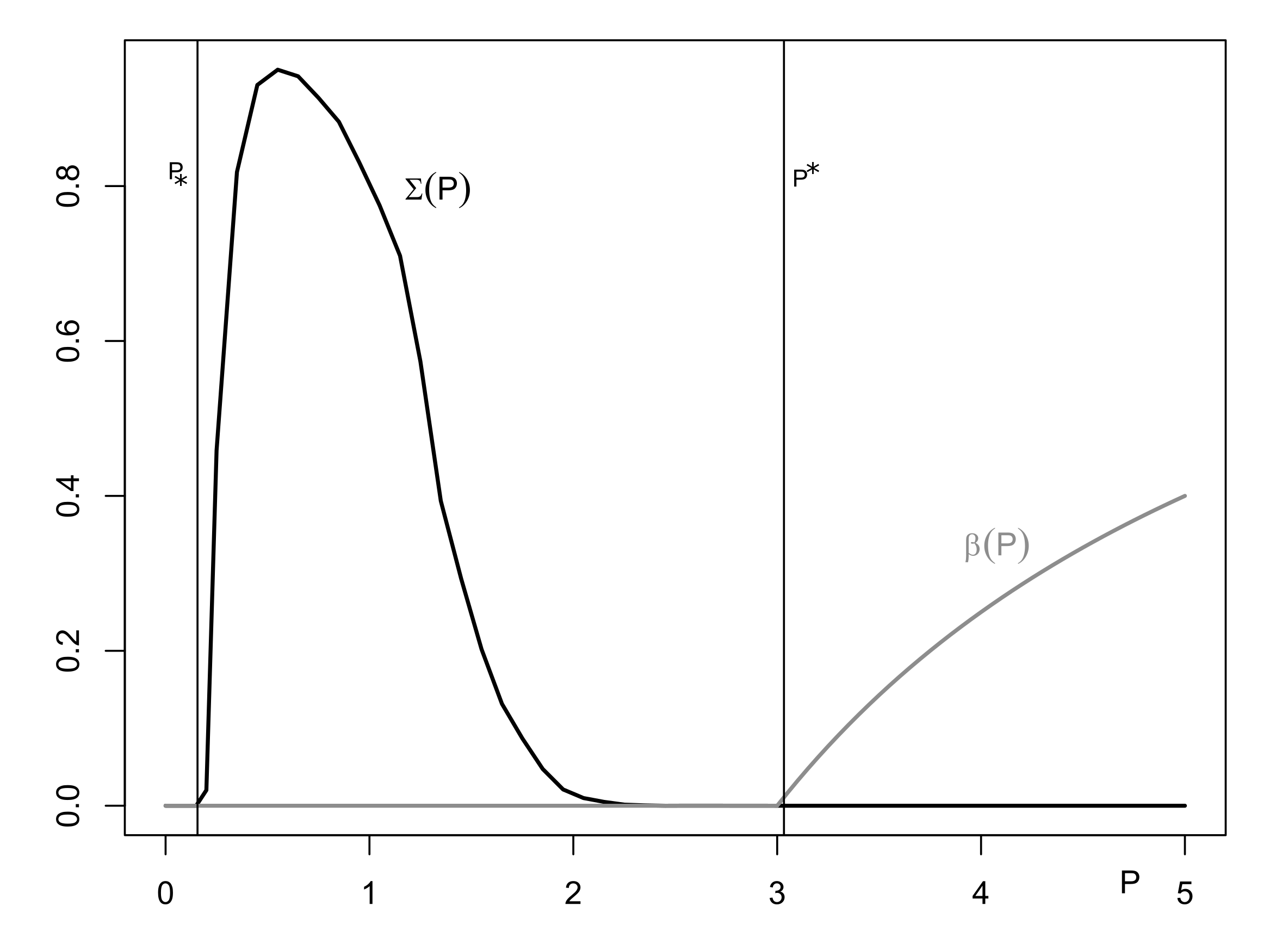
Spectral Gap
4. Reduced Model
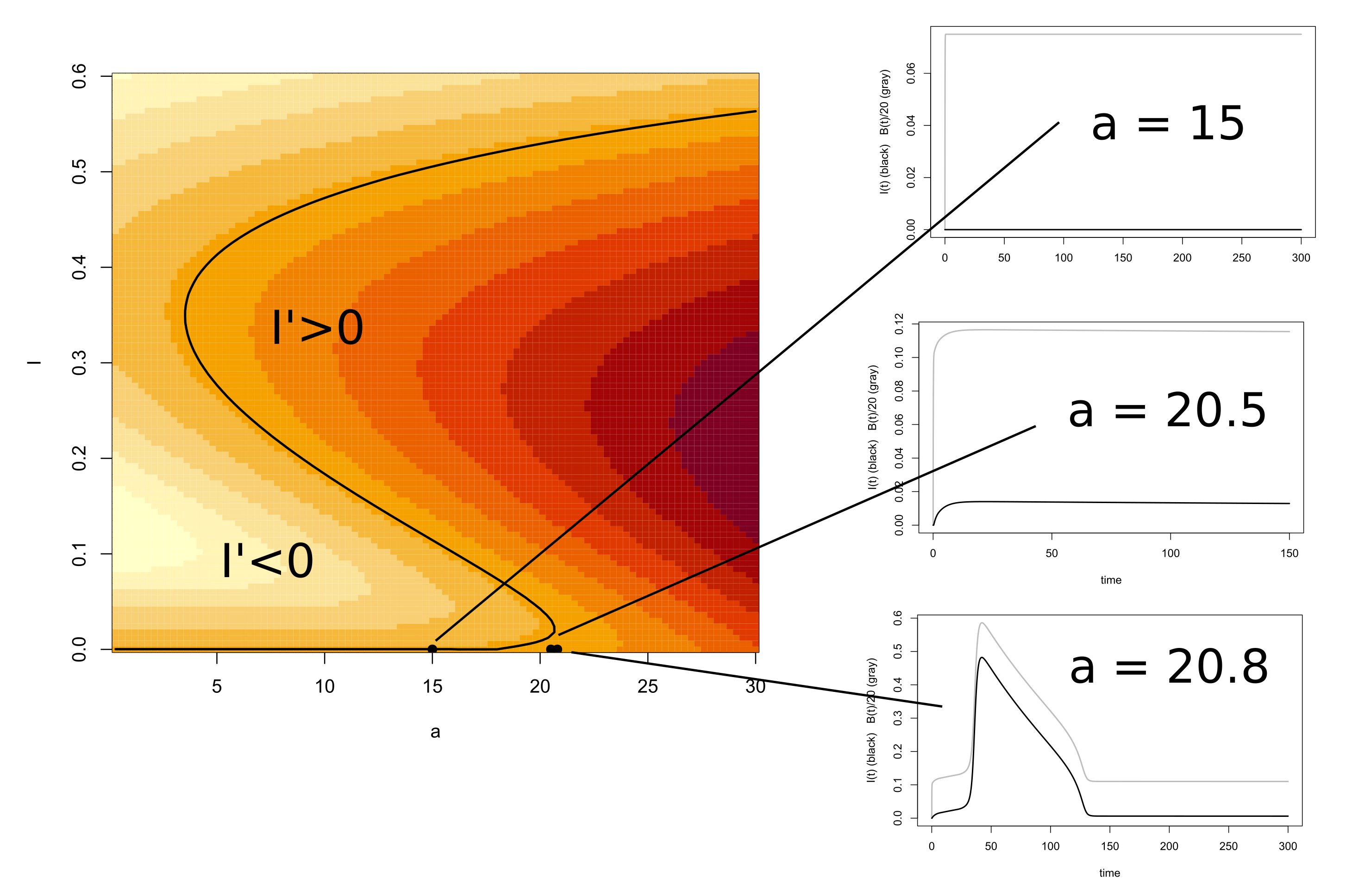
4.1. Fast-Slow Analysis
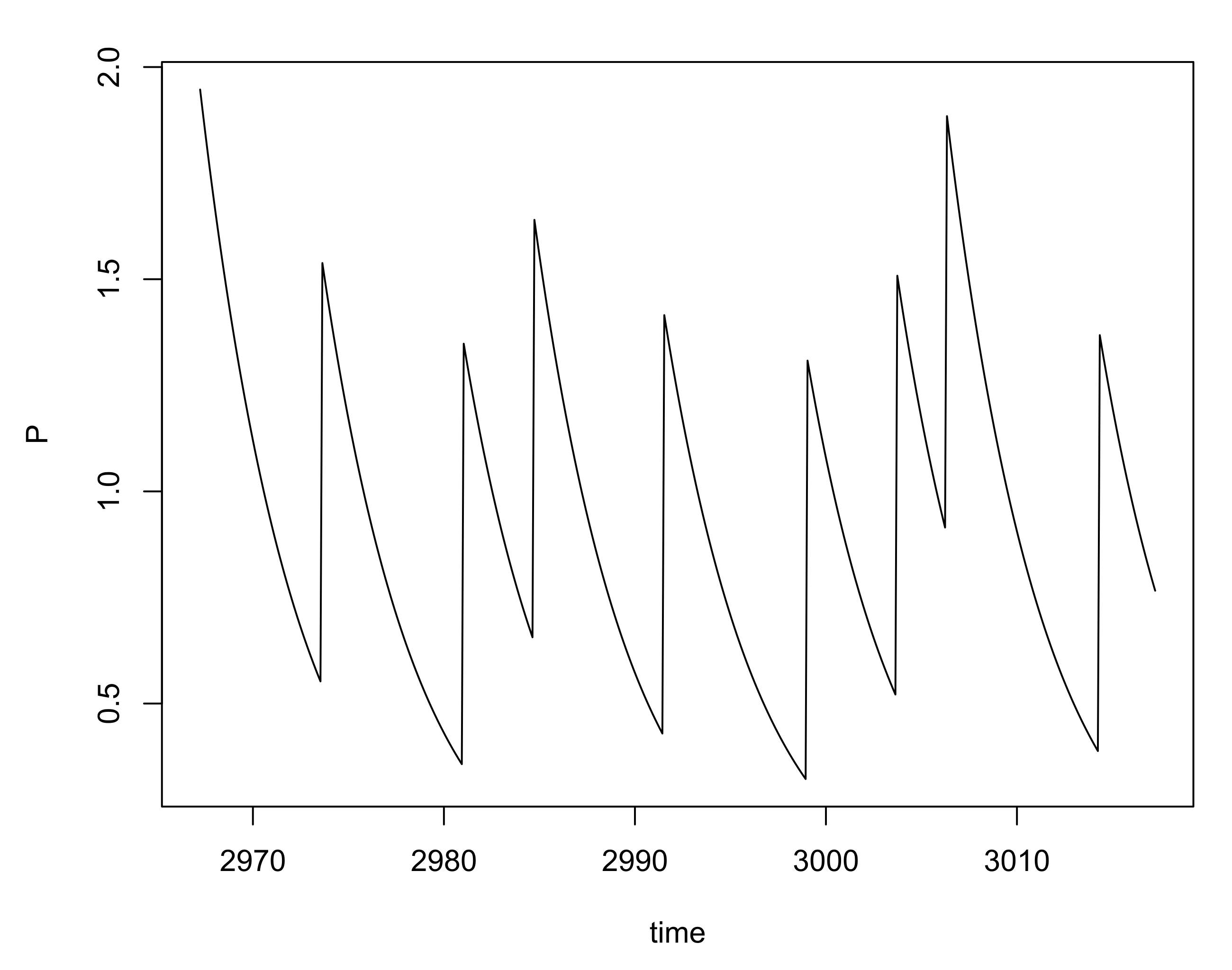
Behavior of the Reduced Model: A Simulation Study
5. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Value |
|---|---|
| 2 | |
| 10 | |
| 0.2 | |
| 12 | |
| 2000 | |
| 10 | |
| 0.01 | |
| 3 | |
| s(0) | 100 |
| I(0), B(0) | 0 |
Appendix B
Appendix B.1
Appendix B.2
Appendix B.3
References
- Codeço, C.T. Endemic and epidemic dynamics of cholera: The role of the aquatic reservoir. BMC Infect. Dis. 2001, 1, 1. [Google Scholar]
- Kermack, W.O.; McKendrick, A.G. A contribution to the mathematical theory of epidemics. Proc. R. Soc. Lond. Ser. Contain. Pap. Math. Phys. Character 1927, 115, 700–721. [Google Scholar]
- Kermack, W.O.; McKendrick, A.G. Contributions to the mathematical theory of epidemics. II.—The problem of endemicity. Proc. R. Soc. Lond. Ser. Contain. Pap. Math. Phys. Character 1932, 138, 55–83. [Google Scholar]
- Garira, W. A complete categorization of multiscale models of infectious disease systems. J. Biol. Dyn. 2017, 11, 378–435. [Google Scholar] [CrossRef]
- Gog, J.R.; Pellis, L.; Wood, J.L.; McLean, A.R.; Arinaminpathy, N.; Lloyd-Smith, J.O. Seven challenges in modeling pathogen dynamics within-host and across scales. Epidemics 2015, 10, 45–48. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, J. Modeling the within-host dynamics of cholera: Bacterial–viral interaction. J. Biol. Dyn. 2017, 11, 484–501. [Google Scholar] [CrossRef]
- Anderson, R.M.; May, R.M. Infectious Diseases of Humans: Dynamics and Control; Oxford University Press: Oxford, UK, 1992. [Google Scholar]
- Martcheva, M. An Introduction to Mathematical Epidemiology; Springer: Berlin/Heidelberg, Germany, 2015; Volume 61. [Google Scholar]
- Hu, Z.; Ma, W.; Ruan, S. Analysis of SIR epidemic models with nonlinear incidence rate and treatment. Math. Biosci. 2012, 238, 12–20. [Google Scholar] [CrossRef]
- Feng, Z.; Velasco-Hernandez, J.; Tapia-Santos, B. A mathematical model for coupling within-host and between-host dynamics in an environmentally-driven infectious disease. Math. Biosci. 2013, 241, 49–55. [Google Scholar] [CrossRef]
- Feng, Z.; Velasco-Hernandez, J.; Tapia-Santos, B.; Leite, M.C.A. A model for coupling within-host and between-host dynamics in an infectious disease. Nonlinear Dyn. 2012, 68, 401–411. [Google Scholar] [CrossRef]
- Angulo, O.; Milner, F.; Sega, L. A SIR epidemic model structured by immunological variables. J. Biol. Syst. 2013, 21, 1340013. [Google Scholar] [CrossRef]
- Martcheva, M.; Tuncer, N.; St Mary, C. Coupling within-host and between-host infectious diseases models. Biomath 2015, 4, 1510091. [Google Scholar] [CrossRef]
- Gandolfi, A.; Pugliese, A.; Sinisgalli, C. Epidemic dynamics and host immune response: A nested approach. J. Math. Biol. 2015, 70, 399–435. [Google Scholar] [CrossRef] [PubMed]
- Metz, J.A.; Diekmann, O. The Dynamics of Physiologically Structured Populations; Springer: Berlin/Heidelberg, Germany, 1986; Volume 68. [Google Scholar]
- Thieme, H.R. Mathematics in population biology. In Mathematics in Population Biology; Princeton University Press: Princeton, NJ, USA, 2018. [Google Scholar]
- Brauer, F.; Shuai, Z.; Van Den Driessche, P. Dynamics of an age-of-infection cholera model. Math. Biosci. Eng. 2013, 10, 1335–1349. [Google Scholar]
- Hartley, D.M.; Morris, J.G., Jr.; Smith, D.L. Hyperinfectivity: A critical element in the ability of V. cholerae to cause epidemics? PLoS Med. 2005, 3, e7. [Google Scholar] [CrossRef]
- Mukandavire, Z.; Liao, S.; Wang, J.; Gaff, H.; Smith, D.L.; Morris, J.G. Estimating the reproductive numbers for the 2008–2009 cholera outbreaks in Zimbabwe. Proc. Natl. Acad. Sci. USA 2011, 108, 8767–8772. [Google Scholar] [CrossRef]
- Cai, L.M.; Modnak, C.; Wang, J. An age-structured model for cholera control with vaccination. Appl. Math. Comput. 2017, 299, 127–140. [Google Scholar] [CrossRef]
- Jiang, X. Global Dynamics for an Age-Structured Cholera Infection Model with General Infection Rates. Mathematics 2021, 9, 2993. [Google Scholar] [CrossRef]
- Wang, X.; Wang, J. Disease dynamics in a coupled cholera model linking within-host and between-host interactions. J. Biol. Dyn. 2017, 11, 238–262. [Google Scholar] [CrossRef]
- Ratchford, C.; Wang, J. Modeling cholera dynamics at multiple scales: Environmental evolution, between-host transmission, and within-host interaction. Math. Biosci. Eng. 2019, 16, 782–812. [Google Scholar] [CrossRef]
- Musundi, B. An Immuno-epidemiological Model Linking Between-host and Within-host Dynamics of Cholera. arXiv 2021, arXiv:2105.12675. [Google Scholar]
- Reidl, J.; Klose, K.E. Vibrio cholerae and cholera: Out of the water and into the host. FEMS Microbiol. Rev. 2002, 26, 125–139. [Google Scholar] [CrossRef] [PubMed]
- Nelson, E.J.; Harris, J.B.; Morris, J.G.; Calderwood, S.B.; Camilli, A. Cholera transmission: The host, pathogen and bacteriophage dynamic. Nat. Rev. Microbiol. 2009, 7, 693–702. [Google Scholar]
- Schmid-Hempel, P.; Frank, S.A. Pathogenesis, virulence, and infective dose. PLoS Pathog. 2007, 3, e147. [Google Scholar] [CrossRef]
- Alavi, S.; Mitchell, J.D.; Cho, J.Y.; Liu, R.; Macbeth, J.C.; Hsiao, A. Interpersonal gut microbiome variation drives susceptibility and resistance to cholera infection. Cell 2020, 181, 1533–1546. [Google Scholar] [CrossRef]
- Cho, J.Y.; Liu, R.; Macbeth, J.C.; Hsiao, A. The interface of Vibrio cholerae and the gut microbiome. Gut Microbes 2021, 13, 1937015. [Google Scholar]
- Gillman, A.N.; Mahmutovic, A.; Abel zur Wiesch, P.; Abel, S. The Infectious Dose Shapes Vibrio cholerae Within-Host Dynamics. Msystems 2021, 6, e00659-21. [Google Scholar] [CrossRef]
- Heijmans, H.J.A.M. An eigenvalue problem related to cell growth. J. Math. Anal. Appl. 1985, 111, 253–280. [Google Scholar] [CrossRef] [Green Version]
- Heijmans, H.J. The dynamical behaviour of the age-size-distribution of a cell population. In The Dynamics of Physiologically Structured Populations; Springer: Berlin/Heidelberg, Germany, 1986; pp. 185–202. [Google Scholar]
- Jauffret, M.D.; Gabriel, P. Eigenelements of a general aggregation-fragmentation model. Math. Model. Methods Appl. Sci. 2010, 20, 757–783. [Google Scholar] [CrossRef]
- Campillo, F.; Champagnat, N.; Fritsch, C. Links between deterministic and stochastic approaches for invasion in growth-fragmentation-death models. J. Math. Biol. 2016, 73, 1781–1821. [Google Scholar]
- Stadler, E. Eigensolutions and spectral analysis of a model for vertical gene transfer of plasmids. J. Math. Biol. 2019, 78, 1299–1330. [Google Scholar] [CrossRef]
- Stadler, E.; Müller, J. Analyzing plasmid segregation: Existence and stability of the eigensolution in a non-compact case. Discret. Contin. Dyn. Syst. B 2020, 25, 4127. [Google Scholar] [CrossRef]
- Schilling, K.A.; Cartwright, E.J.; Stamper, J.; Locke, M.; Esposito, D.H.; Balaban, V.; Mintz, E. Diarrheal illness among US residents providing medical services in Haiti during the cholera epidemic, 2010 to 2011. J. Travel Med. 2014, 21, 55–57. [Google Scholar] [CrossRef] [PubMed]
- Morris, J.G., Jr. Cholera—Modern pandemic disease of ancient lineage. Emerg. Infect. Dis. 2011, 17, 2099. [Google Scholar] [CrossRef]
- Gyllenberg, M.; Heijmans, H.J. An abstract delay-differential equation modelling size dependent cell growth and division. SIAM J. Math. Anal. 1987, 18, 74–88. [Google Scholar] [CrossRef]
- Yosida, K. Functional Analysis; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2012. [Google Scholar]
- Banasiak, J.; Miekisz, J. Multiscale Problems in the Life Sciences: From Microscopic to Macroscopic; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2008. [Google Scholar]
- Magal, P.; Ruan, S. Structured Population Models in Biology and Epidemiology; Springer: Berlin/Heidelberg, Germany, 2008; Volume 1936. [Google Scholar]
- Webb, G.F. Theory of Nonlinear Age-Dependent Population Dynamics; CRC Press: Boca Raton, FL, USA, 1985. [Google Scholar]
- Inaba, H. Age-Structured Population Dynamics in Demography and Epidemiology; Springer: Berlin/Heidelberg, Germany, 2017. [Google Scholar]
- Drnovšek, R. Spectral inequalities for compact integral operators on Banach function spaces. Math. Proc. Camb. Philos. Soc. 1992, 112, 589–598. [Google Scholar] [CrossRef]
- Mokhtar-Kharroubi, M. On spectral gaps of growth-fragmentation semigroups with mass loss or death. Commun. Pure Appl. Anal. 2022, 21, 1293. [Google Scholar] [CrossRef]
- Webb, G.F. An operator-theoretic formulation of asynchronous exponential growth. Trans. Am. Math. Soc. 1987, 303, 751–763. [Google Scholar] [CrossRef]
- Müller, J.; Kuttler, C. Methods and models in mathematical biology. In Lecture Notes on Mathematical Modelling in Life Sciences; Springer: Berlin, Germany, 2015. [Google Scholar]
- Kuehn, C. Multiple Time Scale Dynamics; Springer: Berlin/Heidelberg, Germany, 2015. [Google Scholar]
- Umoh, J.; Adesiyun, A.; Adekeye, J.; Nadarajah, M. Epidemiological features of an outbreak of gastroenteritis/cholera in Katsina, Northern Nigeria. Epidemiol. Infect. 1983, 91, 101–111. [Google Scholar] [CrossRef] [Green Version]
- Tuckwell, H.C. Stochastic Processes in the Neurosciences; Chapter IV; SIAM: Philadelphia, PA, USA, 1989. [Google Scholar]
- Lymperopoulos, I.N. #Stayhome to contain COVID-19: Neuro-SIR–Neurodynamical epidemic modeling of infection patterns in social networks. Expert Syst. Appl. 2021, 165, 113970. [Google Scholar]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Musundi, B.; Müller, J.; Feng, Z. A Multi-Scale Model for Cholera Outbreaks. Mathematics 2022, 10, 3114. https://doi.org/10.3390/math10173114
Musundi B, Müller J, Feng Z. A Multi-Scale Model for Cholera Outbreaks. Mathematics. 2022; 10(17):3114. https://doi.org/10.3390/math10173114
Chicago/Turabian StyleMusundi, Beryl, Johannes Müller, and Zhilan Feng. 2022. "A Multi-Scale Model for Cholera Outbreaks" Mathematics 10, no. 17: 3114. https://doi.org/10.3390/math10173114
APA StyleMusundi, B., Müller, J., & Feng, Z. (2022). A Multi-Scale Model for Cholera Outbreaks. Mathematics, 10(17), 3114. https://doi.org/10.3390/math10173114