

DExMA: An R Package for Performing Gene Expression Meta-Analysis with Missing Genes
 , , , ,
, , , ,  and
and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Meta-Analysis Methods
2.1.1. Effect Size Combination Methods
- There is independence between the experimental and the control group.
- Both the experimental and control groups are distributed according to a normal distribution with means μE and μC, respectively, and with the same σ2 variance.
- , is a factor that corrects the positive bias. nE and nC are the sample sizes of the experimental and control groups, respectively.
- and are the gene expression means of the experimental and control group, respectively.
- is the standard deviation between studies. and are the variances in the experimental and control groups, respectively.
- Ti is the effect size of the i-th study.
- ωi is the weight assigned to the i-th study. In the case of a meta-analysis, the inverse of the variance is used as weights, .
- represents the total variance, where:
- ωi is the calculated weight for the Fixed Effects Model.
- T. is the combined effect size for the Fixed Effects Model (Equation (2)).
- is a scaling-related factor related to the fact that Q is a weighted sum of squares.
- df = k − 1 are the degrees of freedom for the meta-analysis.
2.1.2. p-Values Combination Methods
- p1, …, pk are the p-values from the k independent studies.
- The t1, …, tk test statistics have absolute continuous probability distributions under their corresponding null hypotheses.
2.2. Control of Missing Genes
3. Results
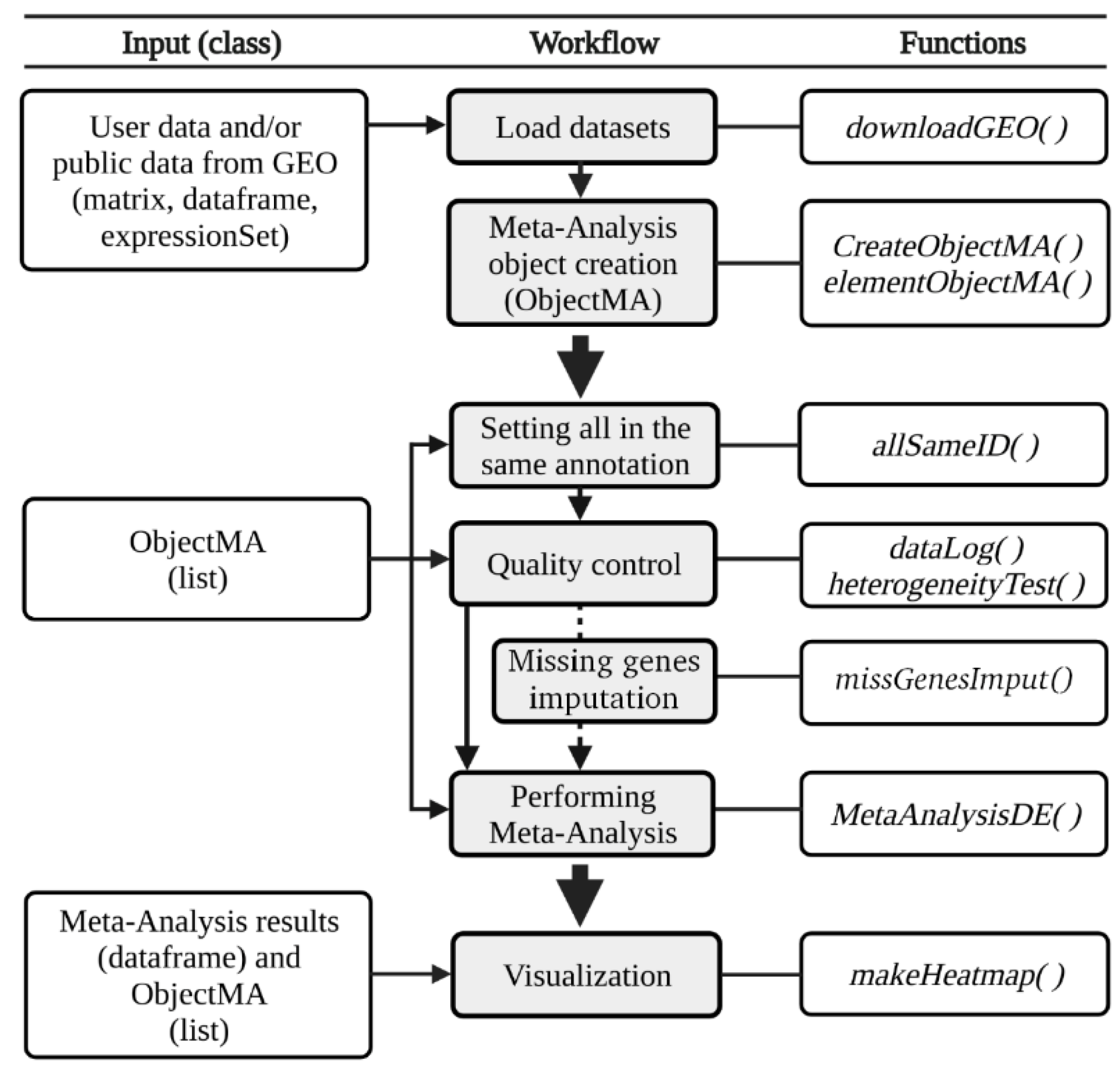
3.1. The DExMA Package
- “listMatrixEX”: a list of four expression matrices.
- “listPhenodatas”: a list of the four phenodata dataframes corresponding to four expression matrices.
- “listExpressionSets”: a list of four ExpressionSet objects. It contains the same information as listMatrixEX and listPhenodatas.
- “ExpressionSetStudy5”: an ExpressionSet object similar to the ExpressionSets objects of listExpressionSets.
- “maObjectDif”: the meta-analysis object (objectMA) created from the listMatrixEx and listPhenodatas objects.
- “maObject”: the meta-analysis object (objectMA) after setting all the studies in Official Gene Symbol annotation.
3.1.1. Meta-Analysis Object Creation
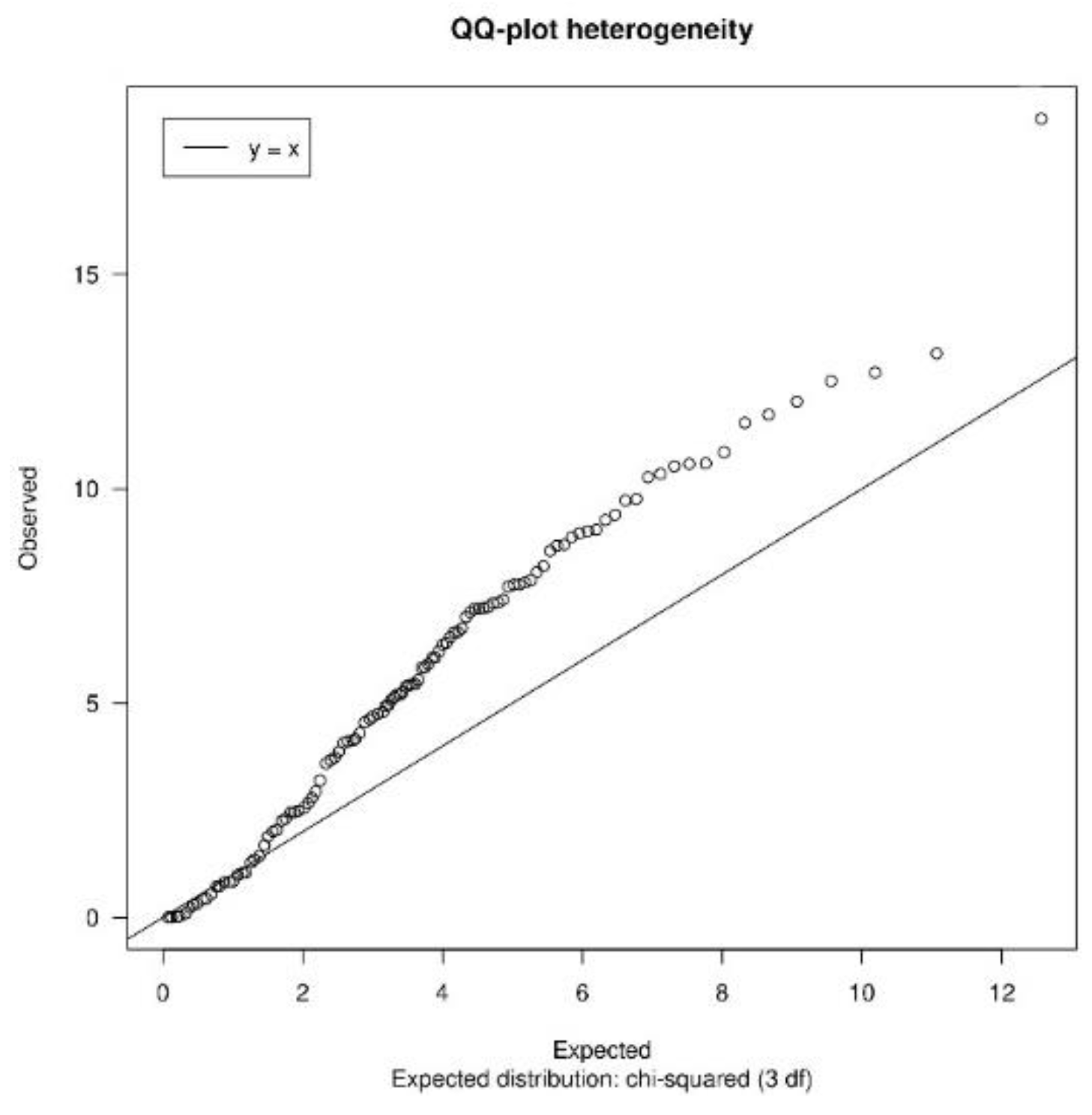
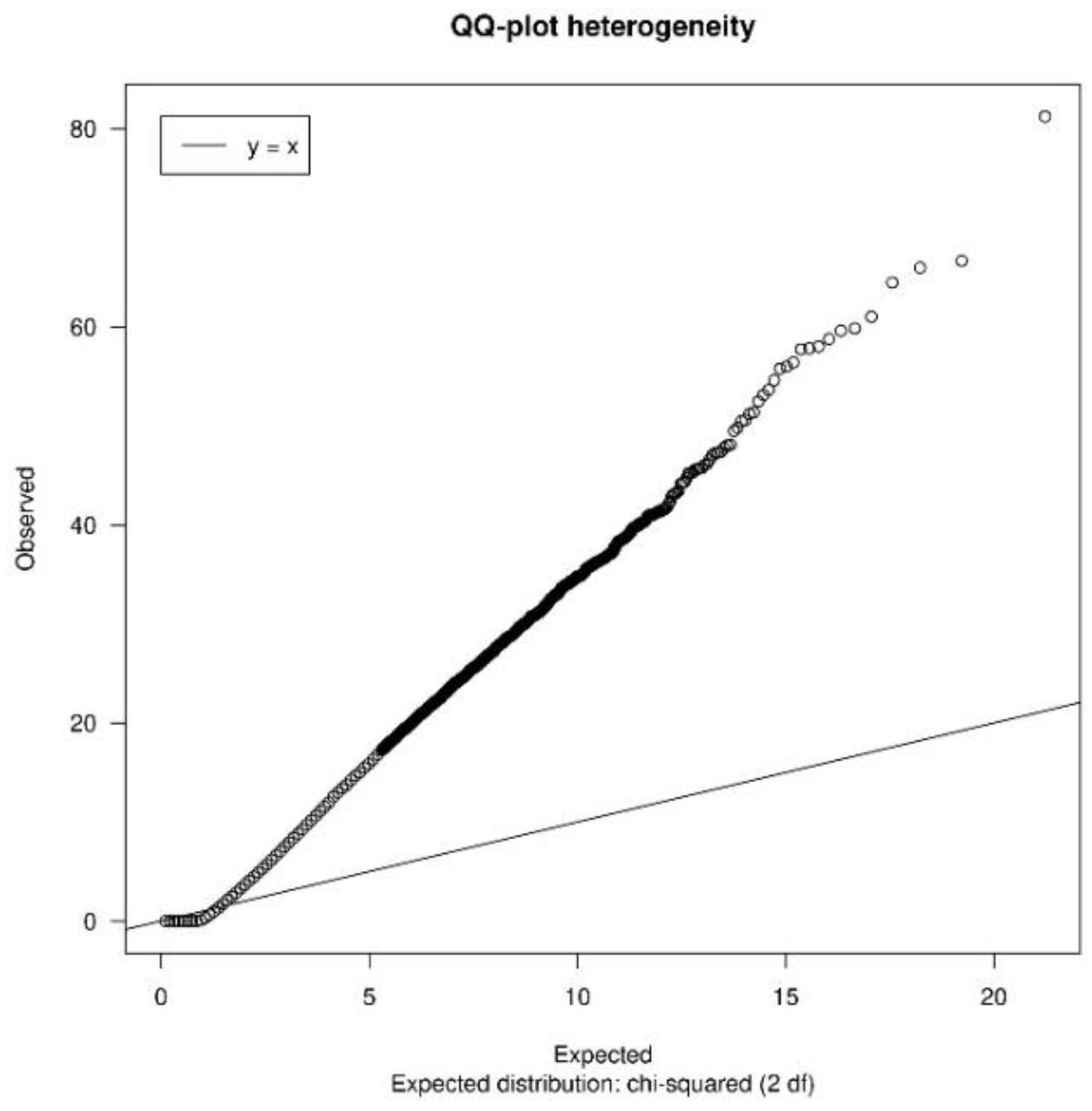
3.1.2. Quality Control
3.1.3. Missing Gene Imputation
- imputValuesSample: the number of missing values imputed per sample.
- imputPercentageSample: the percentage of missing values imputed per sample.
- imputValuesGene: the number of missing values imputed per gene.
- imputPercentageGene: the percentage of missing values imputed per gene.
3.1.4. Performing Gene Expression Meta-Analysis
3.1.5. Other Useful Functions
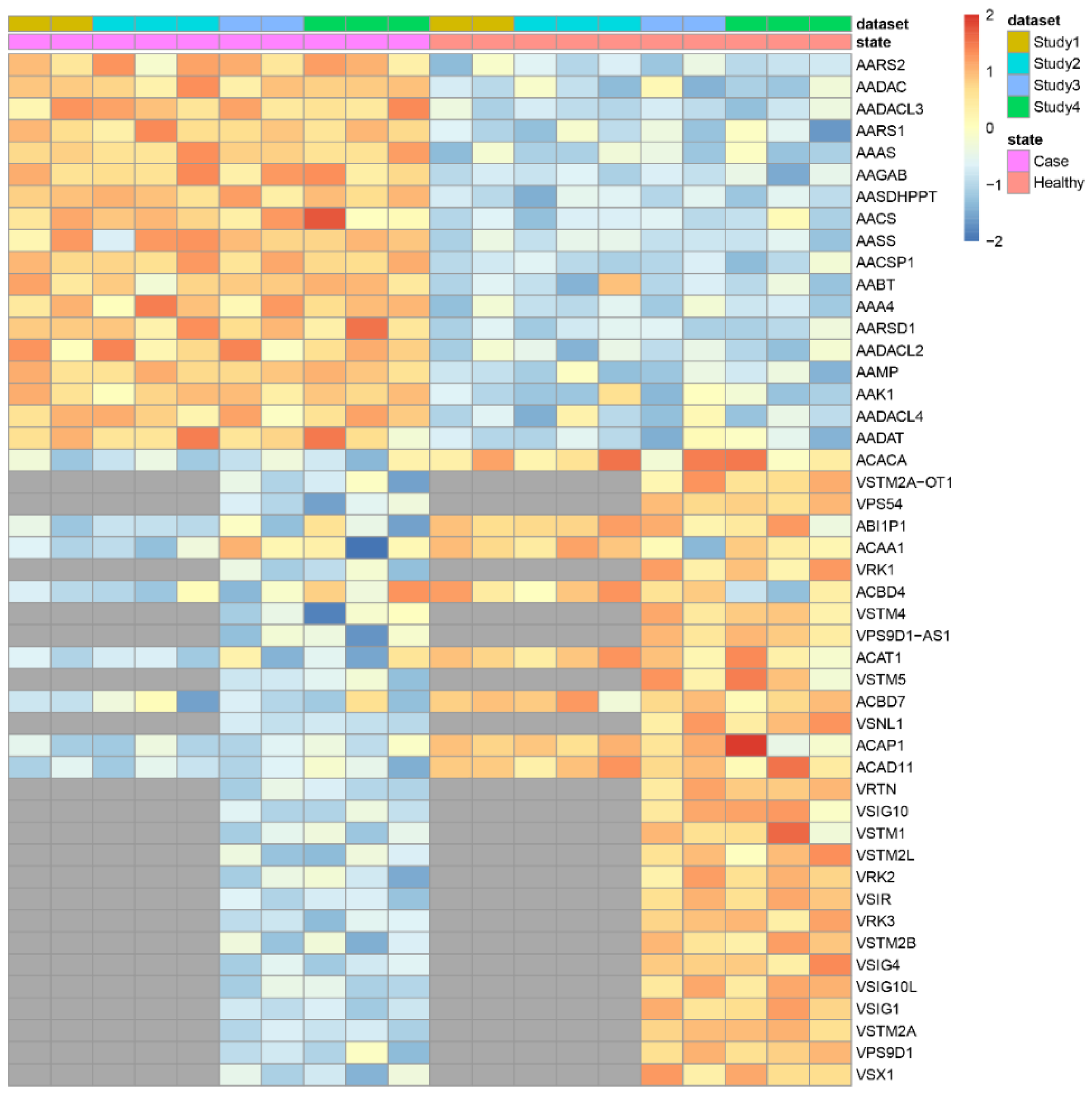
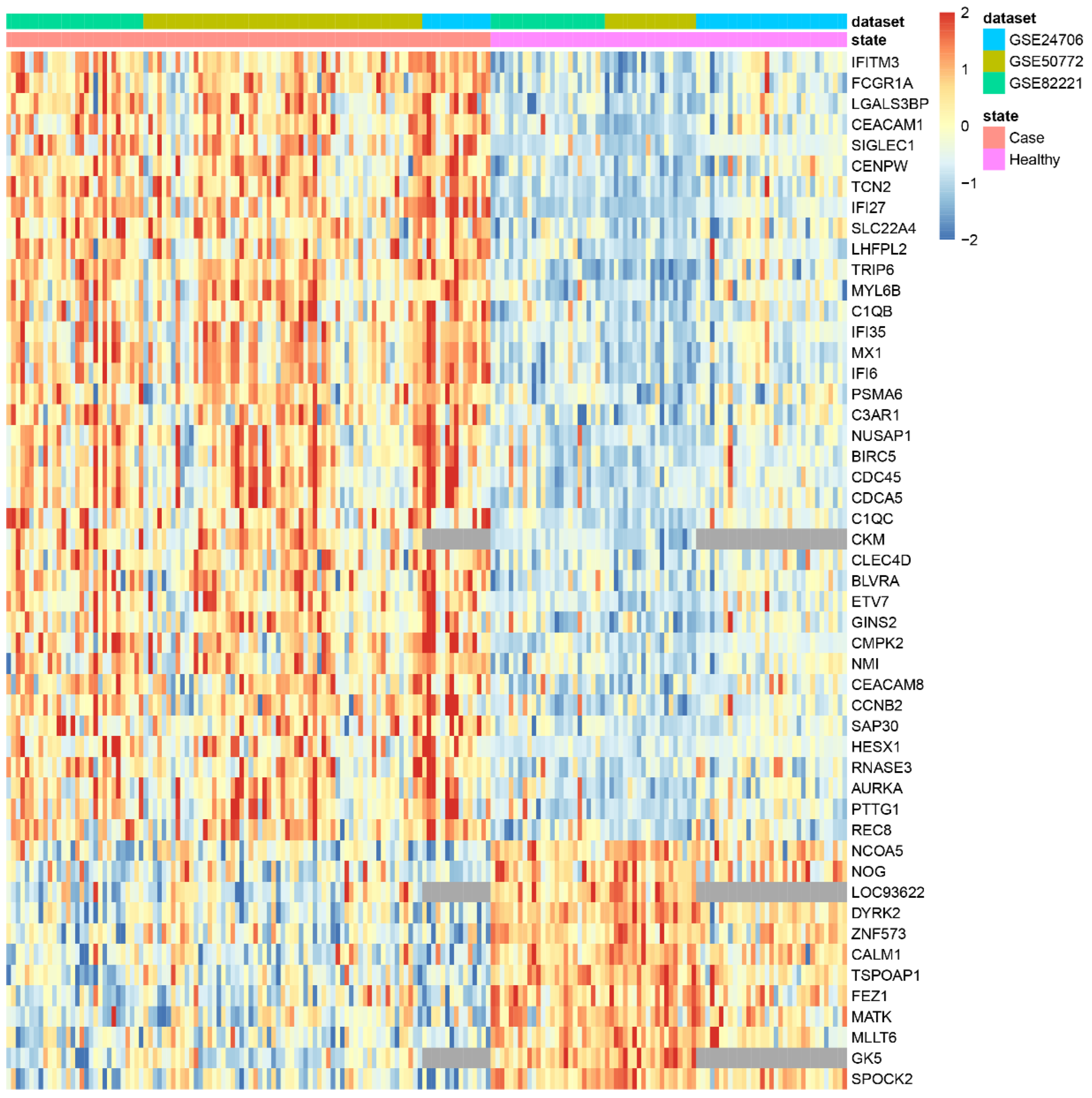
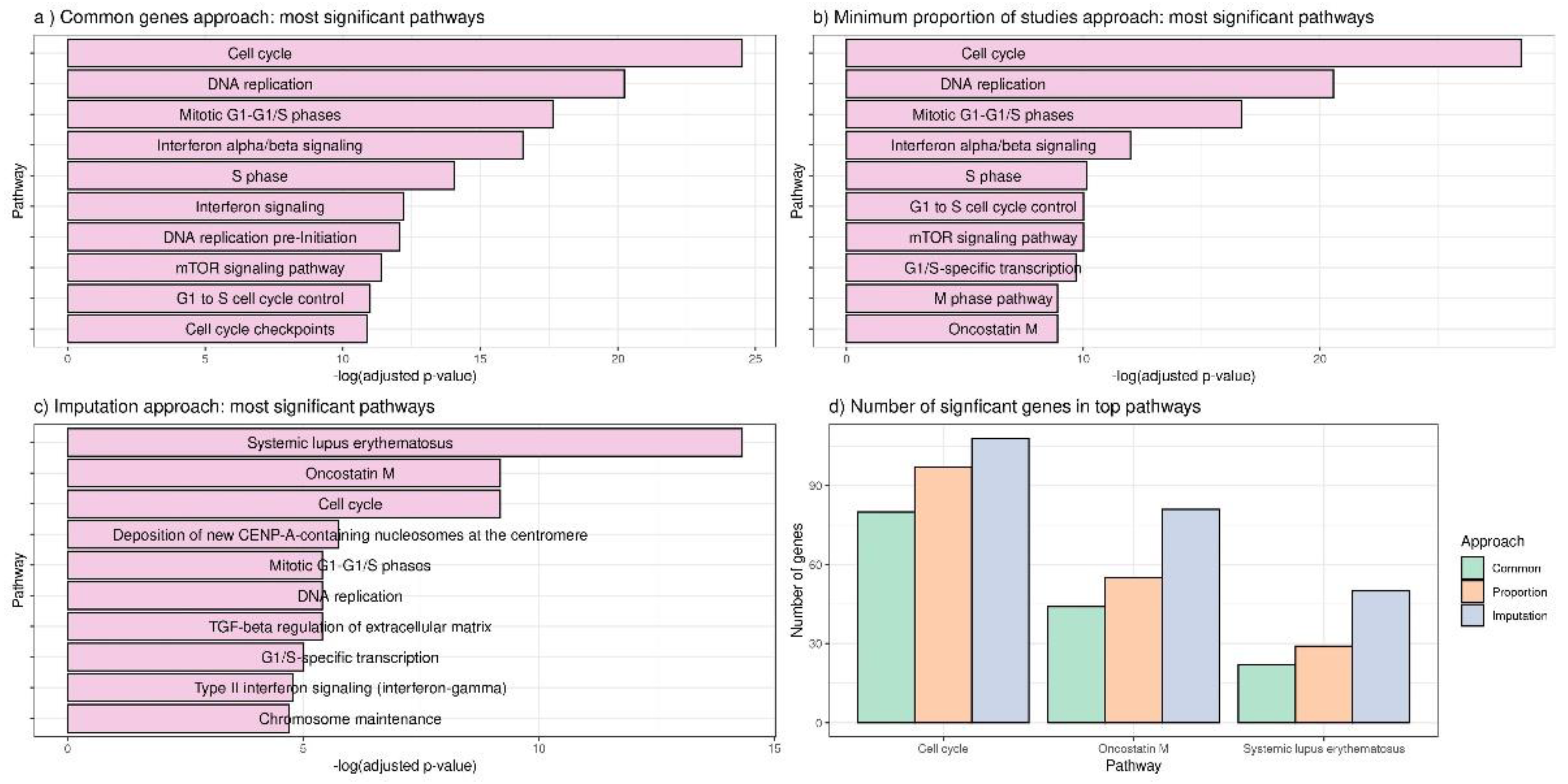
3.2. Applying DExMA to Real Data
- Using only common genes (common genes approach).
- Considering the genes that are present in at least two of the studies (66%) (minimum proportion approach).
- Performing a previous imputation of missing genes before accomplishing the meta-analysis (called the imputing missing genes approach).
3.3. Comparison to Other Available R Packages
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| KNN K | nearest neighbours |
| FEM | Fixed Effects Model |
| REM | Random Effects Model |
| ID | Identifier |
| SLE | systemic lupus erythematosus |
Appendix A
Loading and Preparing the Case Study Data Directly from the ADEX Database
- GSE24706.tsv: gene expression matrix of the study GSE24706.
- GSE50772.tsv: gene expression matrix of the study GSE50772.
- GSE82221_GPL10558.tsv: gene expression matrix of the study GSE82221.
- metadata.tsv: dataframe with the information from the different samples of the studies (phenodata).
References
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for Functional Genomics Data Sets—Update. Nucleic Acids Res. 2013, 41, D991–D995. [Google Scholar] [CrossRef] [PubMed]
- Perez-Riverol, Y.; Zorin, A.; Dass, G.; Vu, M.-T.; Xu, P.; Glont, M.; Vizcaíno, J.A.; Jarnuczak, A.F.; Petryszak, R.; Ping, P.; et al. Quantifying the Impact of Public Omics Data. Nat. Commun. 2019, 10, 3512. [Google Scholar] [CrossRef] [PubMed]
- Song, G.G.; Kim, J.-H.; Seo, Y.H.; Choi, S.J.; Ji, J.D.; Lee, Y.H. Meta-Analysis of Differentially Expressed Genes in Primary Sjogren’s Syndrome by Using Microarray. Hum. Immunol. 2014, 75, 98–104. [Google Scholar] [CrossRef] [PubMed]
- Afroz, S.; Giddaluru, J.; Vishwakarma, S.; Naz, S.; Khan, A.A.; Khan, N. A Comprehensive Gene Expression Meta-Analysis Identifies Novel Immune Signatures in Rheumatoid Arthritis Patients. Front. Immunol. 2017, 8, 74. [Google Scholar] [CrossRef]
- Badr, M.T.; Häcker, G. Gene Expression Profiling Meta-Analysis Reveals Novel Gene Signatures and Pathways Shared between Tuberculosis and Rheumatoid Arthritis. PLoS ONE 2019, 14, e0213470. [Google Scholar] [CrossRef]
- Kelly, J.; Moyeed, R.; Carroll, C.; Albani, D.; Li, X. Gene Expression Meta-Analysis of Parkinson’s Disease and Its Relationship with Alzheimer’s Disease. Mol. Brain 2019, 12, 16. [Google Scholar] [CrossRef]
- Ibáñez, K.; Boullosa, C.; Tabarés-Seisdedos, R.; Baudot, A.; Valencia, A. Molecular Evidence for the Inverse Comorbidity between Central Nervous System Disorders and Cancers Detected by Transcriptomic Meta-Analyses. PLoS Genet. 2014, 10, e1004173. [Google Scholar] [CrossRef]
- Ioannidis, J.P.A. The Mass Production of Redundant, Misleading, and Conflicted Systematic Reviews and Meta-Analyses. Milbank Q. 2016, 94, 485–514. [Google Scholar] [CrossRef]
- Park, J.H.; Eisenhut, M.; van der Vliet, H.J.; Shin, J.I. Statistical Controversies in Clinical Research: Overlap and Errors in the Meta-Analyses of MicroRNA Genetic Association Studies in Cancers. Ann. Oncol. 2017, 28, 1169–1182. [Google Scholar] [CrossRef]
- Haynes, W.A.; Vallania, F.; Liu, C.; Bongen, E.; Tomczak, A.; Andres-Terrè, M.; Lofgren, S.; Tam, A.; Deisseroth, C.A.; Li, M.D.; et al. Empowering Multi-Cohort Gene Expression Analysis to Increase Reproducibility. Pac. Symp. Biocomput. 2016, 22, 144–153. [Google Scholar]
- Prada, C.; Lima, D.; Nakaya, H. MetaVolcanoR: Gene Expression Meta-Analysis Visualization Tool. 2022. Available online: https://www.bioconductor.org/packages/release/bioc/html/MetaVolcanoR.html (accessed on 1 July 2022).
- Bobak, C.A.; McDonnell, L.; Nemesure, M.D.; Lin, J.; Hill, J.E. Assessment of Imputation Methods for Missing Gene Expression Data in Meta-Analysis of Distinct Cohorts of Tuberculosis Patients. Pac. Symp. Biocomput. 2020, 25, 307–318. [Google Scholar] [PubMed]
- Mancuso, C.A.; Canfield, J.L.; Singla, D.; Krishnan, A. A Flexible, Interpretable, and Accurate Approach for Imputing the Expression of Unmeasured Genes. Nucleic Acids Res. 2020, 48, e125. [Google Scholar] [CrossRef] [PubMed]
- Toro-Domínguez, D.; Villatoro-García, J.A.; Martorell-Marugán, J.; Román-Montoya, Y.; Alarcón-Riquelme, M.E.; Carmona-Sáez, P. A Survey of Gene Expression Meta-Analysis: Methods and Applications. Brief. Bioinform. 2021, 22, 1694–1705. [Google Scholar] [CrossRef] [PubMed]
- Borenstein, M.; Hedges, L.V.; Higgins, J.P.T.; Rothstein, H.R. Introduction to Meta-Analysis; John Wiley & Sons: Hoboken, NJ, USA, 2021; ISBN 978-1-119-55838-5. [Google Scholar]
- Heard, N.A.; Rubin-Delanchy, P. Choosing between Methods of Combining p-Values. Biometrika 2018, 105, 239–246. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma Powers Differential Expression Analyses for RNA-Sequencing and Microarray Studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Tseng, G.C. An Adaptively Weighted Statistic for Detecting Differential Gene Expression When Combining Multiple Transcriptomic Studies. Ann. Appl. Stat. 2011, 5, 994–1019. [Google Scholar] [CrossRef]
- Zaykin, D.V. Optimally Weighted Z-Test Is a Powerful Method for Combining Probabilities in Meta-Analysis. J. Evol. Biol. 2011, 24, 1836–1841. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, S.; Li, Z.; Morrison, A.C.; Boerwinkle, E.; Lin, X. ACAT: A Fast and Powerful p Value Combination Method for Rare-Variant Analysis in Sequencing Studies. Am. J. Hum. Genet. 2019, 104, 410–421. [Google Scholar] [CrossRef]
- Liu, Y.; Xie, J. Cauchy Combination Test: A Powerful Test with Analytic p-Value Calculation under Arbitrary Dependency Structures. J. Am. Stat. Assoc. 2020, 115, 393–402. [Google Scholar] [CrossRef]
- Higgins, J.P.T.; Thompson, S.G. Quantifying Heterogeneity in a Meta-Analysis. Stat. Med. 2002, 21, 1539–1558. [Google Scholar] [CrossRef]
- Higgins, J.P.T.; Thompson, S.G.; Deeks, J.J.; Altman, D.G. Measuring Inconsistency in Meta-Analyses. BMJ 2003, 327, 557–560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wickham, H.; Seidel, D. Scales: Scale Functions for Visualization. 2020. Available online: https://cran.r-project.org/web/packages/scales/index.html (accessed on 30 June 2022).
- Lazar, C.; Meganck, S.; Taminau, J.; Steenhoff, D.; Coletta, A.; Molter, C.; Weiss-Solís, D.Y.; Duque, R.; Bersini, H.; Nowé, A. Batch Effect Removal Methods for Microarray Gene Expression Data Integration: A Survey. Brief. Bioinform. 2013, 14, 469–490. [Google Scholar] [CrossRef] [PubMed]
- Martorell-Marugán, J.; López-Domínguez, R.; García-Moreno, A.; Toro-Domínguez, D.; Villatoro-García, J.A.; Barturen, G.; Martín-Gómez, A.; Troule, K.; Gómez-López, G.; Al-Shahrour, F.; et al. A Comprehensive Database for Integrated Analysis of Omics Data in Autoimmune Diseases. BMC Bioinform. 2021, 22, 343. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.-Z.; Karp, D.R.; Quan, J.; Branch, V.K.; Zhou, J.; Lian, Y.; Chong, B.F.; Wakeland, E.K.; Olsen, N.J. Risk Factors for ANA Positivity in Healthy Persons. Arthritis Res. Ther. 2011, 13, R38. [Google Scholar] [CrossRef]
- Kennedy, W.P.; Maciuca, R.; Wolslegel, K.; Tew, W.; Abbas, A.R.; Chaivorapol, C.; Morimoto, A.; McBride, J.M.; Brunetta, P.; Richardson, B.C.; et al. Association of the Interferon Signature Metric with Serological Disease Manifestations but Not Global Activity Scores in Multiple Cohorts of Patients with SLE. Lupus Sci. Med. 2015, 2, e000080. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Mi, W.; Luo, H.; Chen, T.; Liu, S.; Raman, I.; Zuo, X.; Li, Q.-Z. Whole-Genome Transcription and DNA Methylation Analysis of Peripheral Blood Mononuclear Cells Identified Aberrant Gene Regulation Pathways in Systemic Lupus Erythematosus. Arthritis Res. Ther. 2016, 18, 162. [Google Scholar] [CrossRef]
- Carmona-Saez, P.; Chagoyen, M.; Tirado, F.; Carazo, J.M.; Pascual-Montano, A. GENECODIS: A Web-Based Tool for Finding Significant Concurrent Annotations in Gene Lists. Genome Biol. 2007, 8, R3. [Google Scholar] [CrossRef]
- Garcia-Moreno, A.; López-Domínguez, R.; Villatoro-García, J.A.; Ramirez-Mena, A.; Aparicio-Puerta, E.; Hackenberg, M.; Pascual-Montano, A.; Carmona-Saez, P. Functional Enrichment Analysis of Regulatory Elements. Biomedicines 2022, 10, 590. [Google Scholar] [CrossRef]
- Huang, R.; Grishagin, I.; Wang, Y.; Zhao, T.; Greene, J.; Obenauer, J.C.; Ngan, D.; Nguyen, D.-T.; Guha, R.; Jadhav, A.; et al. The NCATS BioPlanet—An Integrated Platform for Exploring the Universe of Cellular Signaling Pathways for Toxicology, Systems Biology, and Chemical Genomics. Front. Pharmacol. 2019, 10, 445. [Google Scholar] [CrossRef] [Green Version]
- Stevens, J.R.; Nicholas, G. Metahdep: Hierarchical Dependence in Meta-Analysis. 2022. Available online: https://www.bioconductor.org/packages/release/bioc/html/metahdep.html (accessed on 25 June 2022).
- Lusa, L.; Gentleman, R.; Ruschhaupt, M. GeneMeta: MetaAnalysis for High Throughput Experiments 2021. Available online: https://www.bioconductor.org/packages/release/bioc/html/GeneMeta.html (accessed on 25 June 2022).
- Marot, G.; Rau, A.; Jaffrezic, F.; Blanck, S. MetaRNASeq: Meta-Analysis of RNA-Seq Data 2021. Available online: https://cran.r-project.org/web/packages/metaRNASeq/index.html (accessed on 27 June 2022).
- Tsuyuzaki, K.; Nikaido, I. MetaSeq: Meta-Analysis of RNA-Seq Count Data in Multiple Studies 2022. Available online: https://www.bioconductor.org/packages/release/bioc/html/metaSeq.html (accessed on 27 June 2022).
- Marot, G. MetaMA: Meta-Analysis for MicroArrays 2022. Available online: https://cran.r-project.org/web/packages/metaMA/index.html (accessed on 27 June 2022).
- Pickering, A. Crossmeta: Cross Platform Meta-Analysis of Microarray Data 2022. Available online: https://www.bioconductor.org/packages/release/bioc/html/crossmeta.html (accessed on 27 June 2022).







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Function | Description |
|---|---|
| allsameID | Sets all datasets of objectMA in the same annotation (Official Gene Symbol, Entrez, or Ensembl) |
| batchRemove | Reduces the effects of batch or bias through the use of covariates |
| calculateES | Calculates the effects sizes and their variances for each gene and each dataset using Hedges’ g estimator |
| createObjectMA | Creates the meta-analysis object (objectMA) |
| dataLog | Checks if data are log transformed and transforms them if they are not |
| downloadGEOData | Downloads ExpressionSets objects from GEO database |
| elementObjectMA | Creates an object that can be added to a meta-analysis object (objectMA) |
| heterogeneityTest | Shows a QQ-plot of Cochran’s test and the quantiles of I2 statistic values to measure heterogeneity |
| makeHeatmap | Shows a heatmap with the expression of significant genes along samples |
| metaAnalysisDE | Performs a meta-analysis using the selected method |
| pvalueIndAnalysis | Performs a differential expression analysis in each of the studies to obtain the p-values |
| missGenesImput | Imputes missing genes using the sampleKNN method |
| Package | Input | QC | ES | PV | Considers Missing Genes | Imputes Missing Genes | Visualization |
|---|---|---|---|---|---|---|---|
| DExMA | GEO/User data | Yes | Yes | Yes | Yes | Yes | Yes |
| MetaIntegrator [10] | User data | Yes | Yes | Yes | Yes | No | Yes |
| GeneMeta [34] | User data | No | Yes | No | No | No | Yes |
| Metahdep [33] | User data | No | Yes | No | No | No | No |
| Crossmeta [38] | User data | No | Yes | No | Yes | No | No |
| metaMA [37] | User data | No | Yes | Yes | No | No | No |
| metaRNASeq [35] | User data | No | No | No | No | No | Yes |
| metaSeq [36] | User data | No | No | No | No | No | No |
| MetaVolcanoR [11] | User data | No | Yes | Yes | Yes | No | Yes |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Villatoro-García, J.A.; Martorell-Marugán, J.; Toro-Domínguez, D.; Román-Montoya, Y.; Femia, P.; Carmona-Sáez, P. DExMA: An R Package for Performing Gene Expression Meta-Analysis with Missing Genes. Mathematics 2022, 10, 3376. https://doi.org/10.3390/math10183376
Villatoro-García JA, Martorell-Marugán J, Toro-Domínguez D, Román-Montoya Y, Femia P, Carmona-Sáez P. DExMA: An R Package for Performing Gene Expression Meta-Analysis with Missing Genes. Mathematics. 2022; 10(18):3376. https://doi.org/10.3390/math10183376
Chicago/Turabian StyleVillatoro-García, Juan Antonio, Jordi Martorell-Marugán, Daniel Toro-Domínguez, Yolanda Román-Montoya, Pedro Femia, and Pedro Carmona-Sáez. 2022. "DExMA: An R Package for Performing Gene Expression Meta-Analysis with Missing Genes" Mathematics 10, no. 18: 3376. https://doi.org/10.3390/math10183376
APA StyleVillatoro-García, J. A., Martorell-Marugán, J., Toro-Domínguez, D., Román-Montoya, Y., Femia, P., & Carmona-Sáez, P. (2022). DExMA: An R Package for Performing Gene Expression Meta-Analysis with Missing Genes. Mathematics, 10(18), 3376. https://doi.org/10.3390/math10183376