


A Real Neural Network State for Quantum Chemistry

 ,
,
Abstract
:1. Introduction
2. Methods
2.1. Real Neural Network Ansatz
2.2. Variational Monte Carlo
3. Results
3.1. Training Details
3.2. Effect of Hidden Size
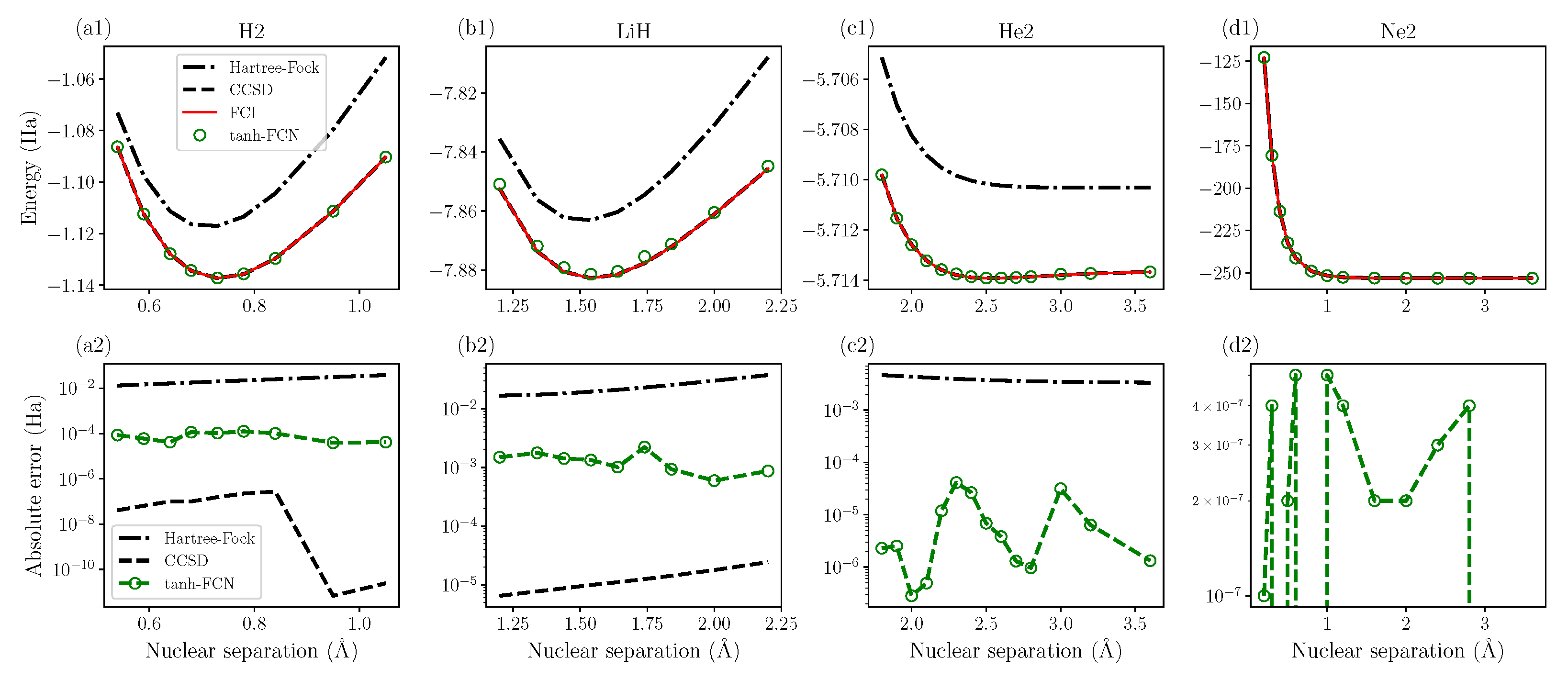
3.3. Potential Energy Surfaces
3.4. Final Energies for Several Molecular Systems
3.5. Effect of Hartree–Fock Re-Initialization
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Helgaker, T.; Jørgensen, P.; Olsen, J. Molecular Electronic-Structure Theory; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2000. [Google Scholar] [CrossRef]
- Vogiatzis, K.D.; Ma, D.; Olsen, J.; Gagliardi, L.; de Jong, W.A. Pushing configuration-interaction to the limit: Towards massively parallel MCSCF calculations. J. Chem. Phys. 2017, 147, 184111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, S.R. Density matrix formulation for quantum renormalization groups. Phys. Rev. Lett. 1992, 69, 2863–2866. [Google Scholar] [CrossRef]
- White, S.R. Density-matrix algorithms for quantum renormalization groups. Phys. Rev. B 1993, 48, 10345–10356. [Google Scholar] [CrossRef] [PubMed]
- Brabec, J.; Brandejs, J.; Kowalski, K.; Xantheas, S.; Legeza, Ö.; Veis, L. Massively parallel quantum chemical density matrix renormalization group method. J. Comput. Chem. 2021, 42, 534–544. [Google Scholar] [CrossRef] [PubMed]
- Larsson, H.R.; Zhai, H.; Umrigar, C.J.; Chan, G.K.L. The chromium dimer: Closing a chapter of quantum chemistry. J. Am. Chem. Soc. 2022, 144, 15932–15937. [Google Scholar] [CrossRef] [PubMed]
- Perez-Garcia, D.; Verstraete, F.; Wolf, M.M.; Cirac, J.I. Matrix Product State Representations. Quantum Inf. Comput. 2007, 7, 401–430. [Google Scholar] [CrossRef]
- Purvis, G.D.; Bartlett, R.J. A full coupled-cluster singles and doubles model: The inclusion of disconnected triples. J. Chem. Phys. 1982, 76, 1910–1918. [Google Scholar] [CrossRef]
- Čížek, J. On the Correlation Problem in Atomic and Molecular Systems. Calculation of Wavefunction Components in Ursell-Type Expansion Using Quantum-Field Theoretical Methods. J. Chem. Phys. 1966, 45, 4256–4266. [Google Scholar] [CrossRef]
- Coester, F.; Kümmel, H. Short-range correlations in nuclear wave functions. Nucl. Phys. 1960, 17, 477–485. [Google Scholar] [CrossRef]
- Shepard, R. The Multiconfiguration Self-Consistent Field Method. In Advances in Chemical Physics; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 1987; pp. 63–200. [Google Scholar] [CrossRef]
- Knowles, P.J.; Werner, H.J. An efficient second-order MC SCF method for long configuration expansions. Chem. Phys. Lett. 1985, 115, 259–267. [Google Scholar] [CrossRef]
- Jensen, H.J.A. Electron Correlation in Molecules Using Direct Second Order MCSCF. In Relativistic and Electron Correlation Effects in Molecules and Solids; Malli, G.L., Ed.; Springer: Boston, MA, USA, 1994; pp. 179–206. [Google Scholar] [CrossRef]
- Sun, Q.; Zhang, X.; Banerjee, S.; Bao, P.; Barbry, M.; Blunt, N.S.; Bogdanov, N.A.; Booth, G.H.; Chen, J.; Cui, Z.H.; et al. Recent developments in the PySCF program package. J. Chem. Phys. 2020, 153, 024109. [Google Scholar] [CrossRef] [PubMed]
- Carleo, G.; Troyer, M. Solving the quantum many-body problem with artificial neural networks. Science 2017, 355, 602–606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choo, K.; Neupert, T.; Carleo, G. Two-dimensional frustrated J1 − J2 model studied with neural network quantum states. Phys. Rev. B 2019, 100, 125124. [Google Scholar] [CrossRef] [Green Version]
- Schmitt, M.; Heyl, M. Quantum Many-Body Dynamics in Two Dimensions with Artificial Neural Networks. Phys. Rev. Lett. 2020, 125, 100503. [Google Scholar] [CrossRef] [PubMed]
- Yuan, D.; Wang, H.R.; Wang, Z.; Deng, D.L. Solving the Liouvillian Gap with Artificial Neural Networks. Phys. Rev. Lett. 2021, 126, 160401. [Google Scholar] [CrossRef]
- Moreno, J.R.; Carleo, G.; Georges, A.; Stokes, J. Fermionic wave functions from neural-network constrained hidden states. Proc. Natl. Acad. Sci. USA 2022, 119, e2122059119. [Google Scholar] [CrossRef]
- Hermann, J.; Schätzle, Z.; Noé, F. Deep-neural-network solution of the electronic Schrödinger equation. Nat. Chem. 2020, 12, 891–897. [Google Scholar] [CrossRef]
- Pfau, D.; Spencer, J.S.; Matthews, A.G.D.G.; Foulkes, W.M.C. Ab initio solution of the many-electron Schrödinger equation with deep neural networks. Phys. Rev. Res. 2020, 2, 033429. [Google Scholar] [CrossRef]
- Humeniuk, S.; Wan, Y.; Wang, L. Autoregressive neural Slater-Jastrow ansatz for variational Monte Carlo simulation. arXiv 2022, arXiv:2210.05871. [Google Scholar]
- Choo, K.; Mezzacapo, A.; Carleo, G. Fermionic neural-network states for ab-initio electronic structure. Nat. Commun. 2020, 11, 2368. [Google Scholar] [CrossRef]
- Barrett, T.D.; Malyshev, A.; Lvovsky, A. Autoregressive neural-network wavefunctions for ab initio quantum chemistry. Nat. Mach. Intell. 2022, 4, 351–358. [Google Scholar] [CrossRef]
- Zhao, T.; Stokes, J.; Veerapaneni, S. Scalable neural quantum states architecture for quantum chemistry. arXiv 2022, arXiv:2208.05637. [Google Scholar]
- Wu, D.; Rossi, R.; Vicentini, F.; Carleo, G. From Tensor Network Quantum States to Tensorial Recurrent Neural Networks. arXiv 2022, arXiv:2206.12363. [Google Scholar]
- Sharir, O.; Shashua, A.; Carleo, G. Neural tensor contractions and the expressive power of deep neural quantum states. Phys. Rev. B 2022, 106, 205136. [Google Scholar] [CrossRef]
- Glasser, I.; Pancotti, N.; August, M.; Rodriguez, I.D.; Cirac, J.I. Neural-Network Quantum States, String-Bond States, and Chiral Topological States. Phys. Rev. X 2018, 8, 011006. [Google Scholar] [CrossRef] [Green Version]
- Deng, D.L.; Li, X.; Das Sarma, S. Quantum Entanglement in Neural Network States. Phys. Rev. X 2017, 7, 021021. [Google Scholar] [CrossRef] [Green Version]
- Nomura, Y.; Imada, M. Dirac-Type Nodal Spin Liquid Revealed by Refined Quantum Many-Body Solver Using Neural-Network Wave Function, Correlation Ratio, and Level Spectroscopy. Phys. Rev. X 2021, 11, 031034. [Google Scholar] [CrossRef]
- Liang, X.; Li, M.; Xiao, Q.; An, H.; He, L.; Zhao, X.; Chen, J.; Yang, C.; Wang, F.; Qian, H.; et al. 21296 Exponentially Complex Quantum Many-Body Simulation via Scalable Deep Learning Method. arXiv 2022, arXiv:2204.07816. [Google Scholar]
- Torlai, G.; Mazzola, G.; Carrasquilla, J.; Troyer, M.; Melko, R.; Carleo, G. Neural-network quantum state tomography. Nat. Phys. 2018, 14, 447–450. [Google Scholar] [CrossRef] [Green Version]
- Hastings, W.K. Monte Carlo Sampling Methods Using Markov Chains and Their Applications. Biometrika 1970, 57, 97–109. [Google Scholar] [CrossRef]
- Sorella, S.; Capriotti, L. Green function Monte Carlo with stochastic reconfiguration: An effective remedy for the sign problem. Phys. Rev. B 2000, 61, 2599–2612. [Google Scholar] [CrossRef] [Green Version]
- Sorella, S.; Casula, M.; Rocca, D. Weak binding between two aromatic rings: Feeling the van der Waals attraction by quantum Monte Carlo methods. J. Chem. Phys. 2007, 127, 014105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vicentini, F.; Hofmann, D.; Szabó, A.; Wu, D.; Roth, C.; Giuliani, C.; Pescia, G.; Nys, J.; Vargas-Calderón, V.; Astrakhantsev, N.; et al. NetKet 3: Machine Learning Toolbox for Many-Body Quantum Systems. SciPost Phys. Codebases 2022, 7. [Google Scholar] [CrossRef]
- Zhang, W.; Xu, X.; Wu, Z.; Balachandran, V.; Poletti, D. Ground state search by local and sequential updates of neural network quantum states. arXiv 2022, arXiv:2207.10882. [Google Scholar]
- Kingma, D.P.; Ba, J. Adam: A Method for Stochastic Optimization. arXiv 2014, arXiv:1412.6980. [Google Scholar]
- Reed, A.E.; Weinstock, R.B.; Weinhold, F. Natural population analysis. J. Chem. Phys. 1985, 83, 735–746. [Google Scholar] [CrossRef]
- Ma, Y.; Ma, H. Assessment of various natural orbitals as the basis of large active space density-matrix renormalization group calculations. J. Chem. Phys. 2013, 138, 224105. [Google Scholar] [CrossRef] [PubMed]
- Zen, R.; My, L.; Tan, R.; Hébert, F.; Gattobigio, M.; Miniatura, C.; Poletti, D.; Bressan, S. Transfer learning for scalability of neural-network quantum states. Phys. Rev. E 2020, 101, 053301. [Google Scholar] [CrossRef] [PubMed]
- Hébert, F.; Zen, R.; My, L.; Tan, R.; Gattobigio, M.; Miniatura, C.; Poletti, D.; Bressan, S. Finding Quantum Critical Points with Neural-Network Quantum States. arXiv 2020, arXiv:2002.02618. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecule | RBM [23] | tanh-FCN | CCSD | FCI | |
|---|---|---|---|---|---|
| 4 | |||||
| Be | 10 | - | |||
| C | 10 | - | |||
| 20 | - | ||||
| LiH | 12 | ||||
| 16 | |||||
| 14 | |||||
| 20 | |||||
| 20 | |||||
| 30 | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, Y.; Xu, X.; Poletti, D.; Fan, Y.; Guo, C.; Shang, H. A Real Neural Network State for Quantum Chemistry. Mathematics 2023, 11, 1417. https://doi.org/10.3390/math11061417
Wu Y, Xu X, Poletti D, Fan Y, Guo C, Shang H. A Real Neural Network State for Quantum Chemistry. Mathematics. 2023; 11(6):1417. https://doi.org/10.3390/math11061417
Chicago/Turabian StyleWu, Yangjun, Xiansong Xu, Dario Poletti, Yi Fan, Chu Guo, and Honghui Shang. 2023. "A Real Neural Network State for Quantum Chemistry" Mathematics 11, no. 6: 1417. https://doi.org/10.3390/math11061417
APA StyleWu, Y., Xu, X., Poletti, D., Fan, Y., Guo, C., & Shang, H. (2023). A Real Neural Network State for Quantum Chemistry. Mathematics, 11(6), 1417. https://doi.org/10.3390/math11061417