
miR-210 Regulates Apoptotic Cell Death during Cellular Hypoxia and Reoxygenation in a Diametrically Opposite Manner



Abstract
:1. Introduction
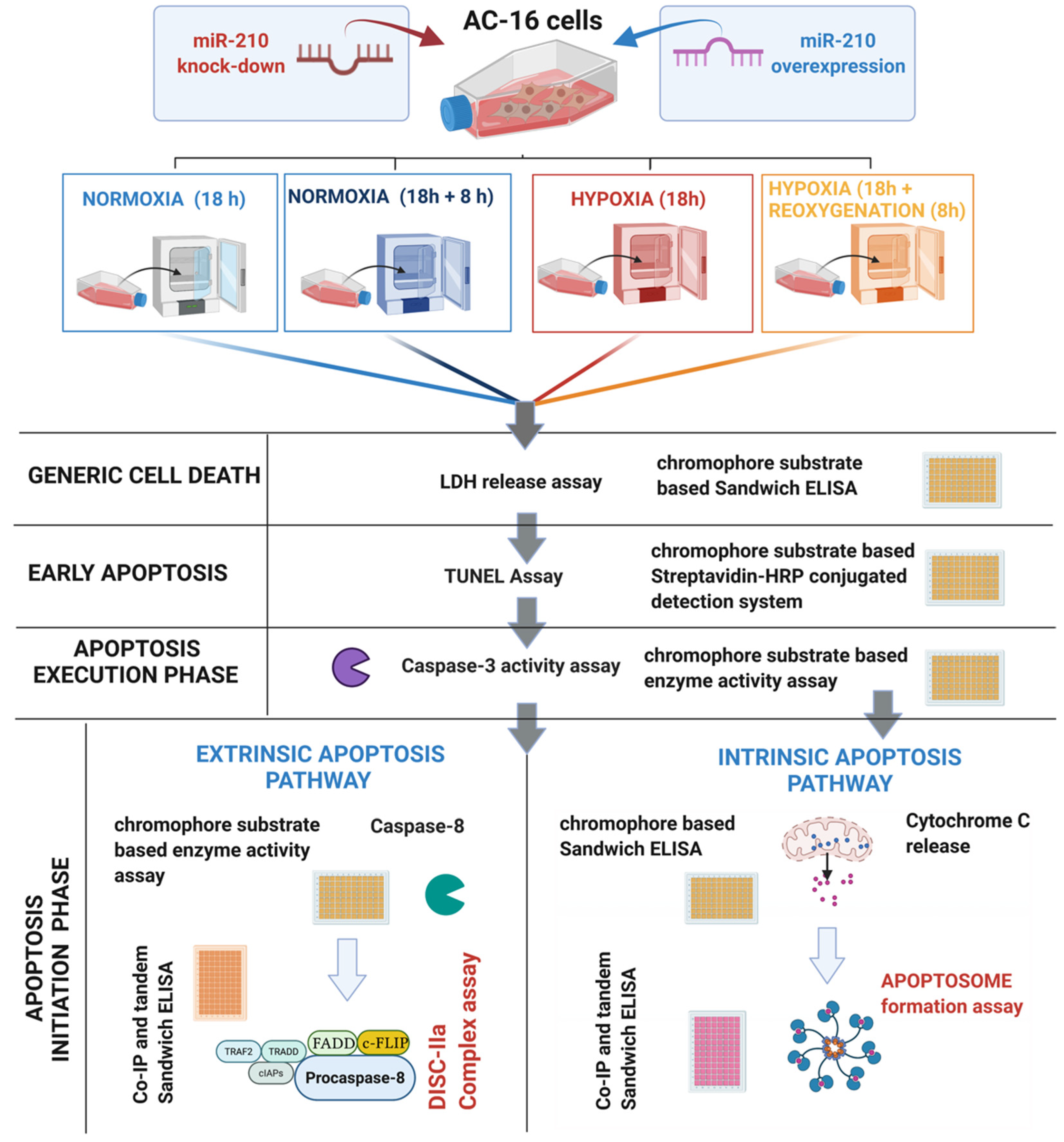
2. Materials and Methods
2.1. Cell Culture and Treatments
2.2. Western Blotting
2.3. Enzyme-Coupled miR-210 Hybridization Immunoassay
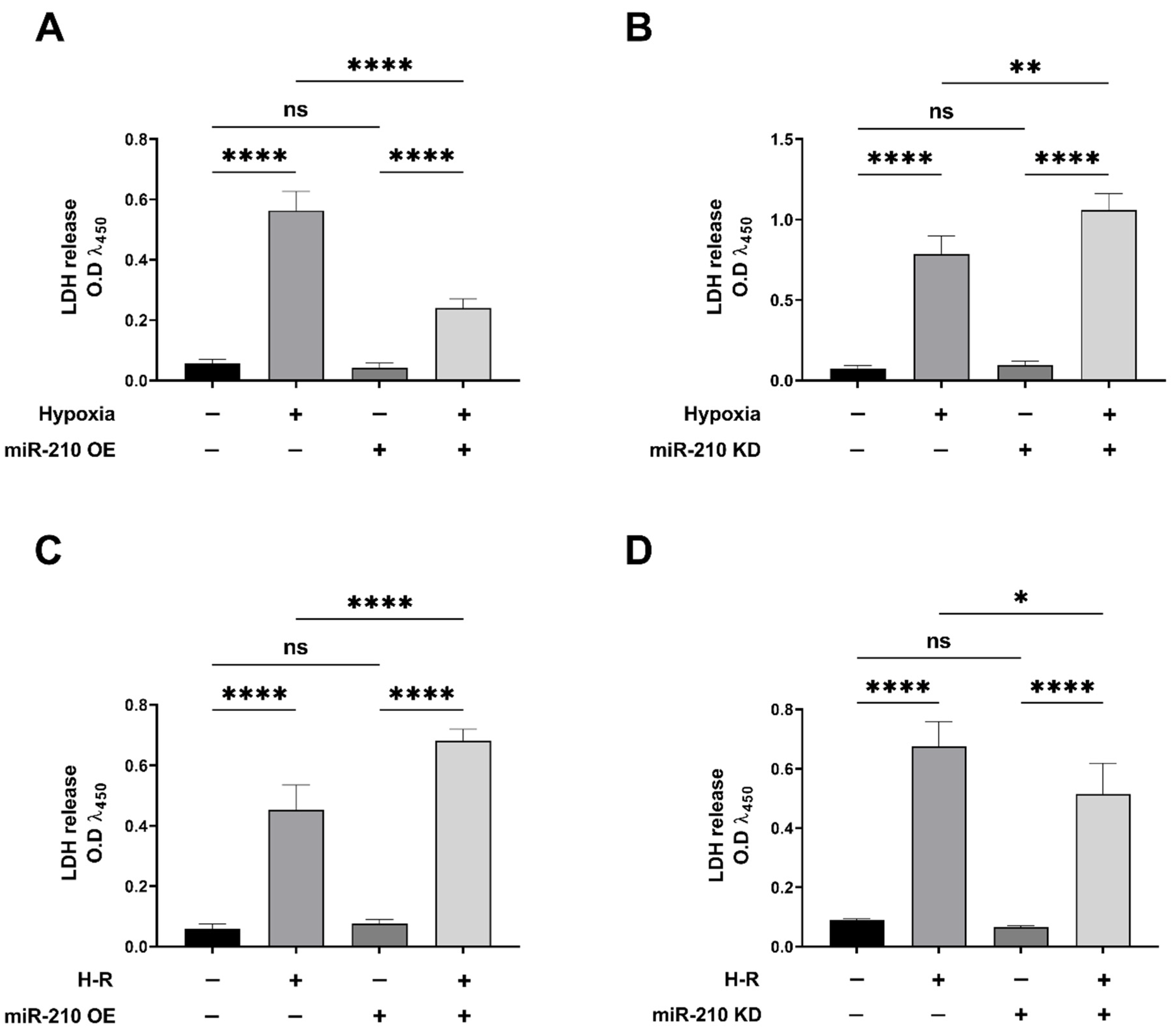
2.4. Lactate Dehydrogenase (LDH) Assay
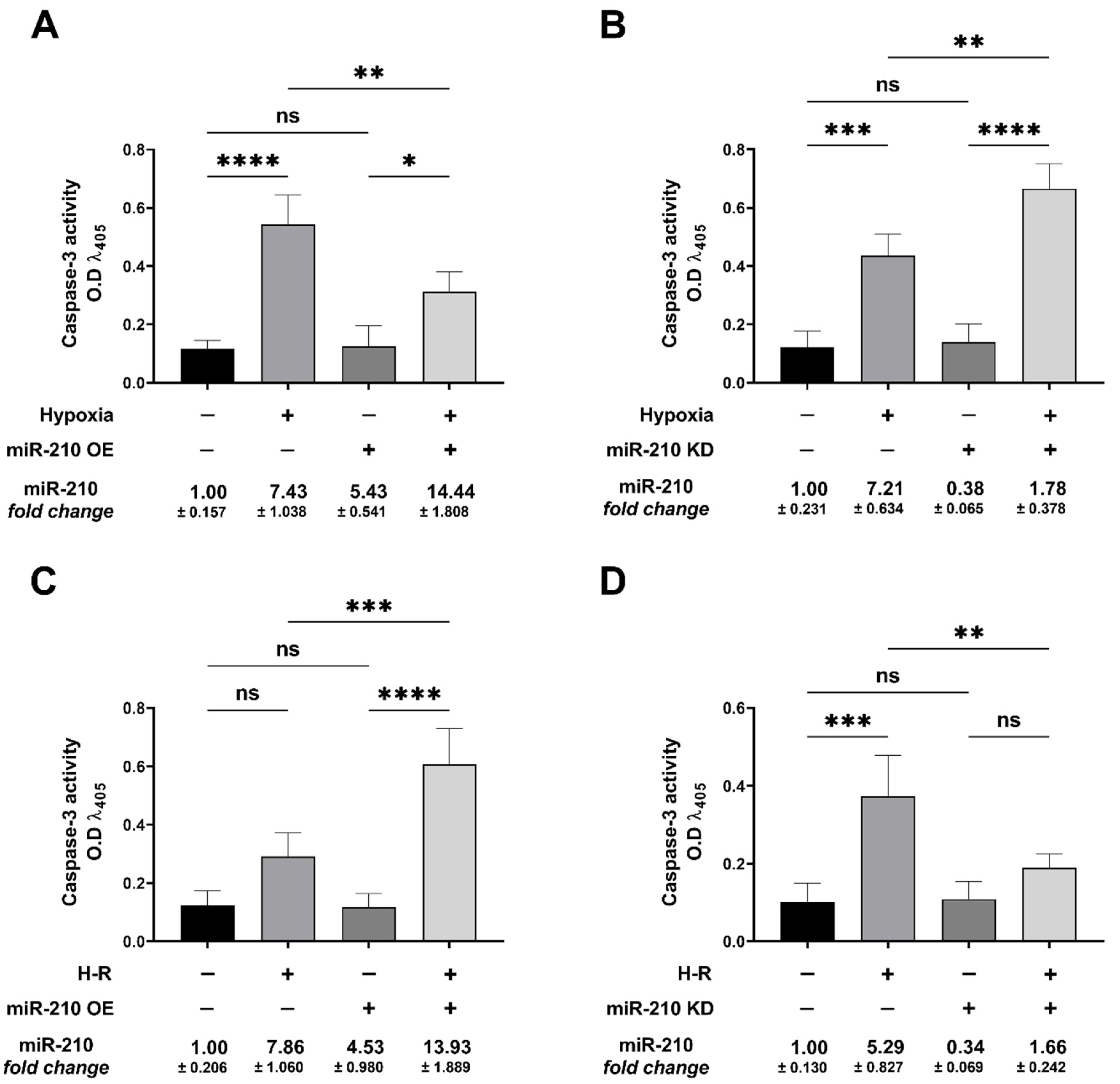
2.5. Caspase-3 Activity Assay
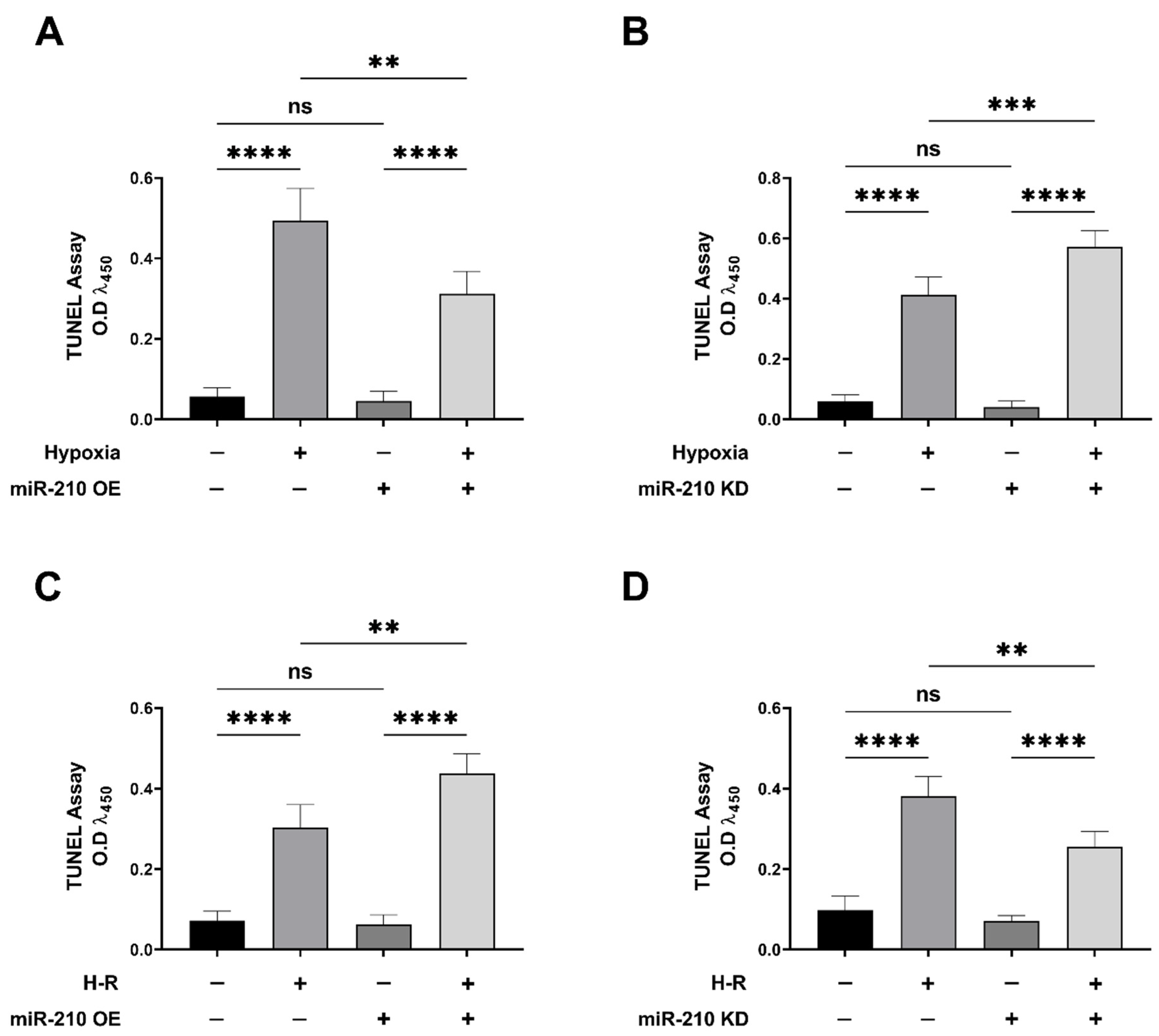
2.6. Terminal Deoxynucleotidyl Transferase dUTP Nick End Labeling (TUNEL) Assay
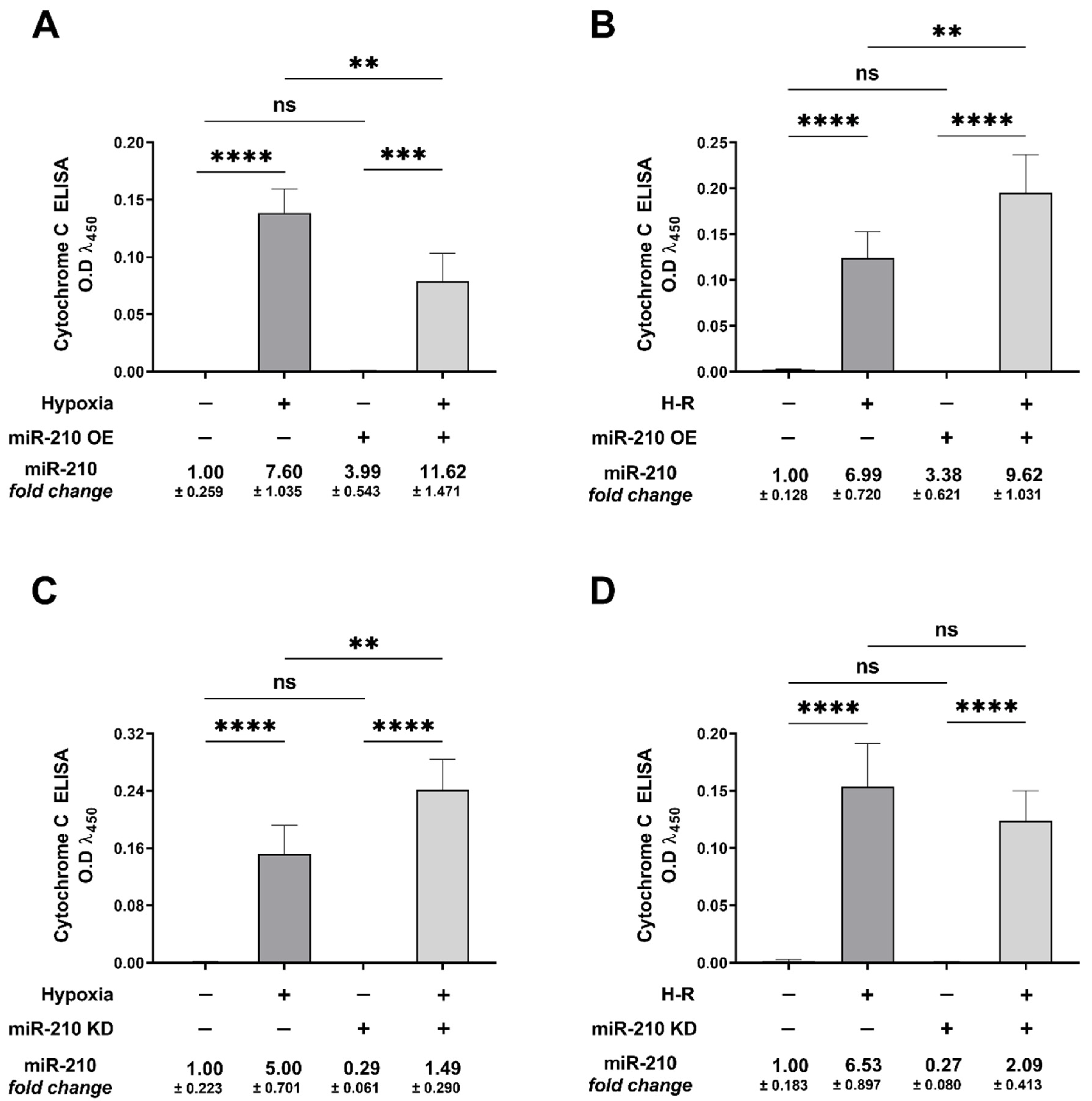
2.7. Cytochrome c Release Assay
2.8. Co-Immunoprecipitation (Co-IP) Analysis
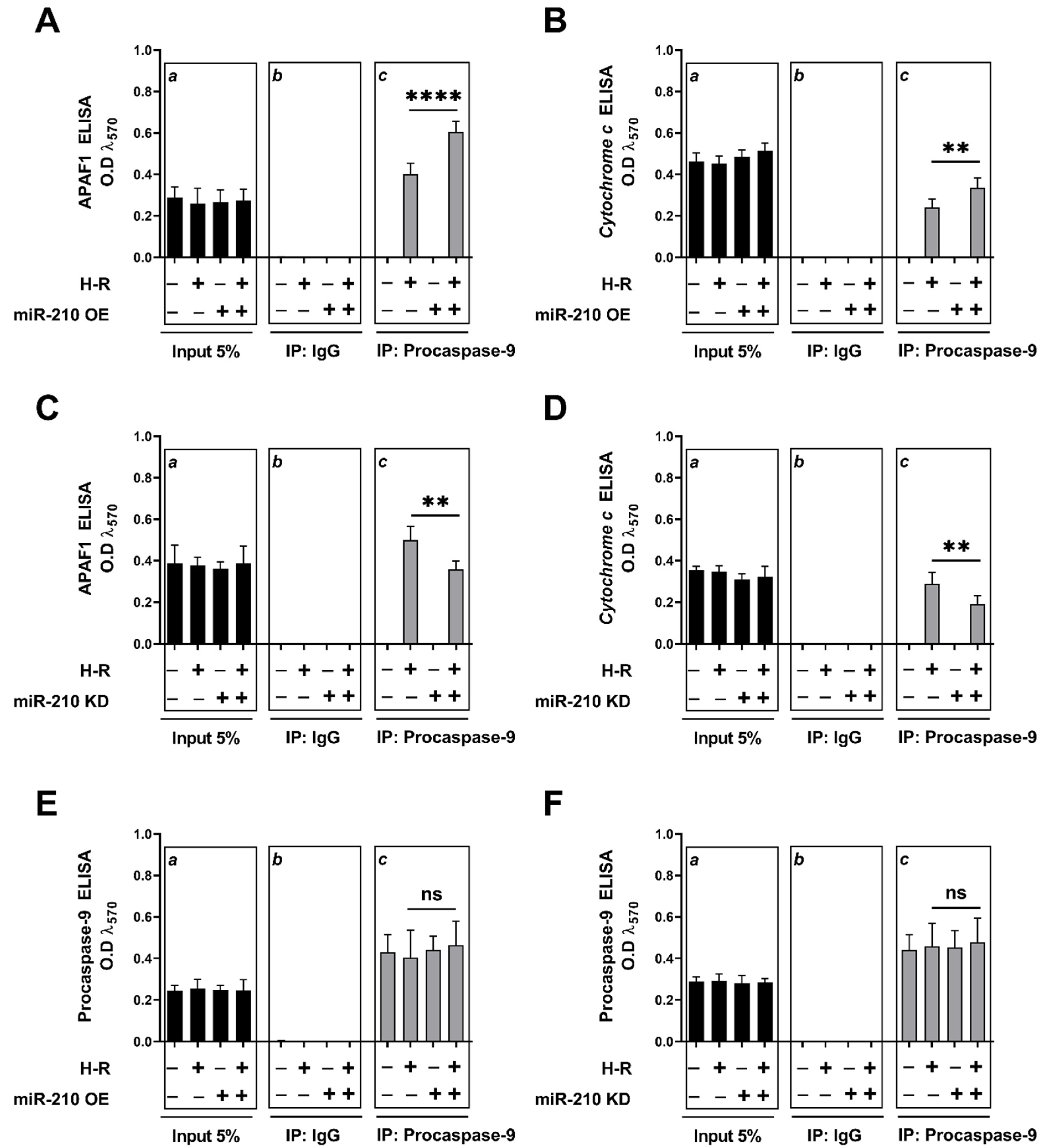
2.9. Apoptosome Complex Formation Assay by Co-Immunoprecipitation (Co-IP) and Tandem ELISA
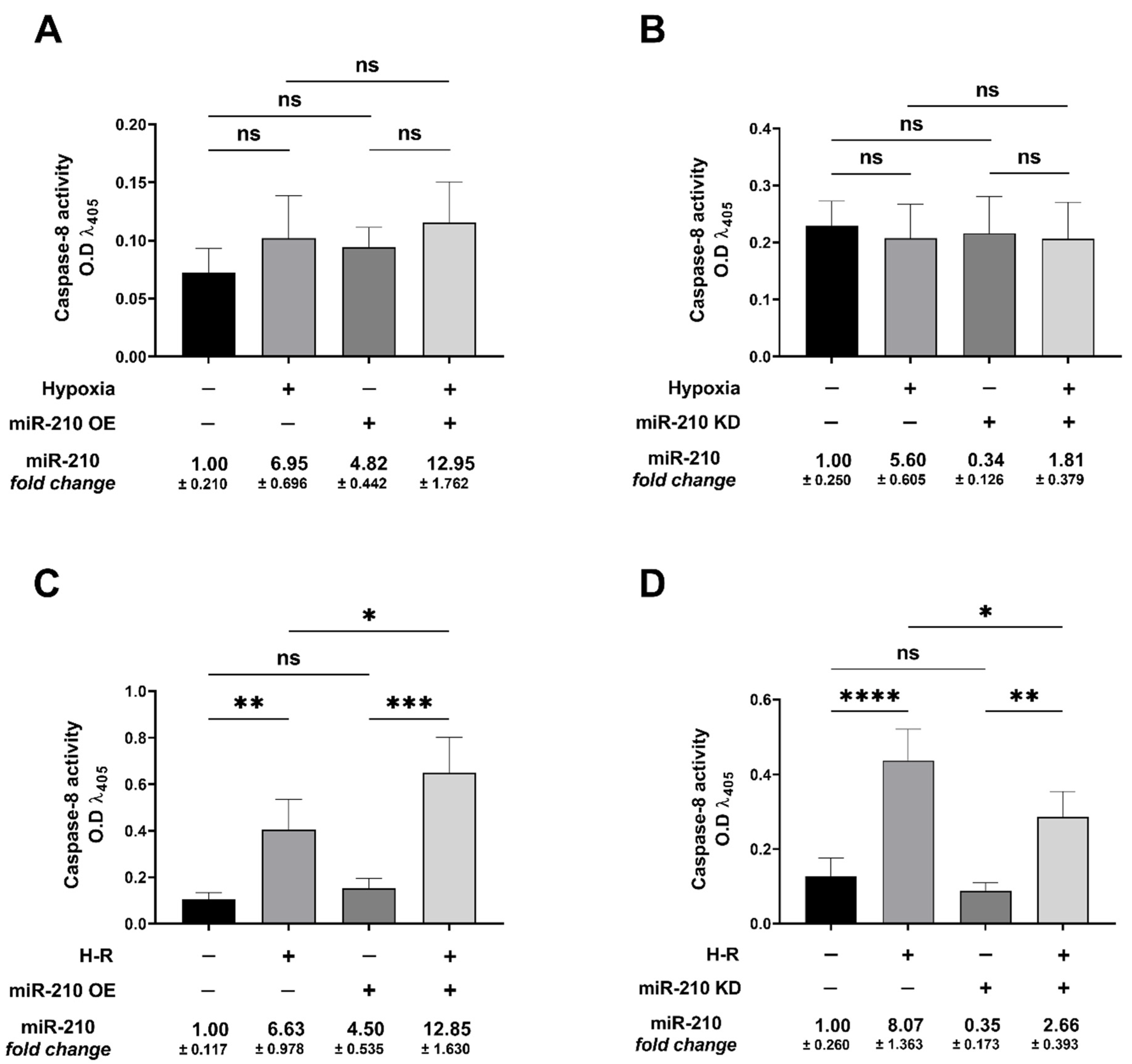
2.10. Caspase-8 Activity Assay
2.11. RIPK1 Immunoprecipitation and RIPK1-Precleared Lysate Preparation
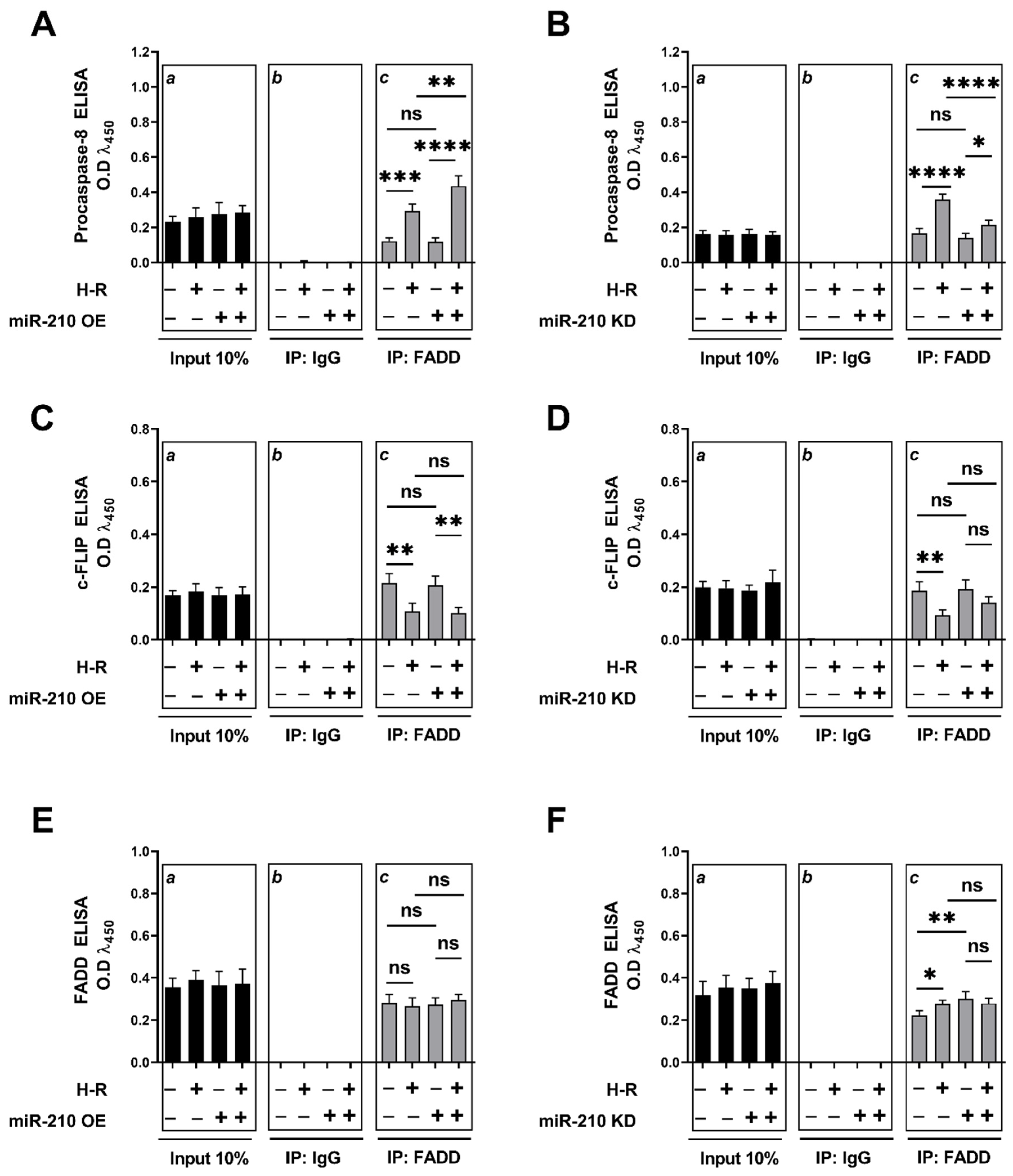
2.12. DISC-IIa Complex Formation Assay by Co-Immunoprecipitation (Co-IP) and Tandem ELISA
2.13. Statistical Analysis
3. Results
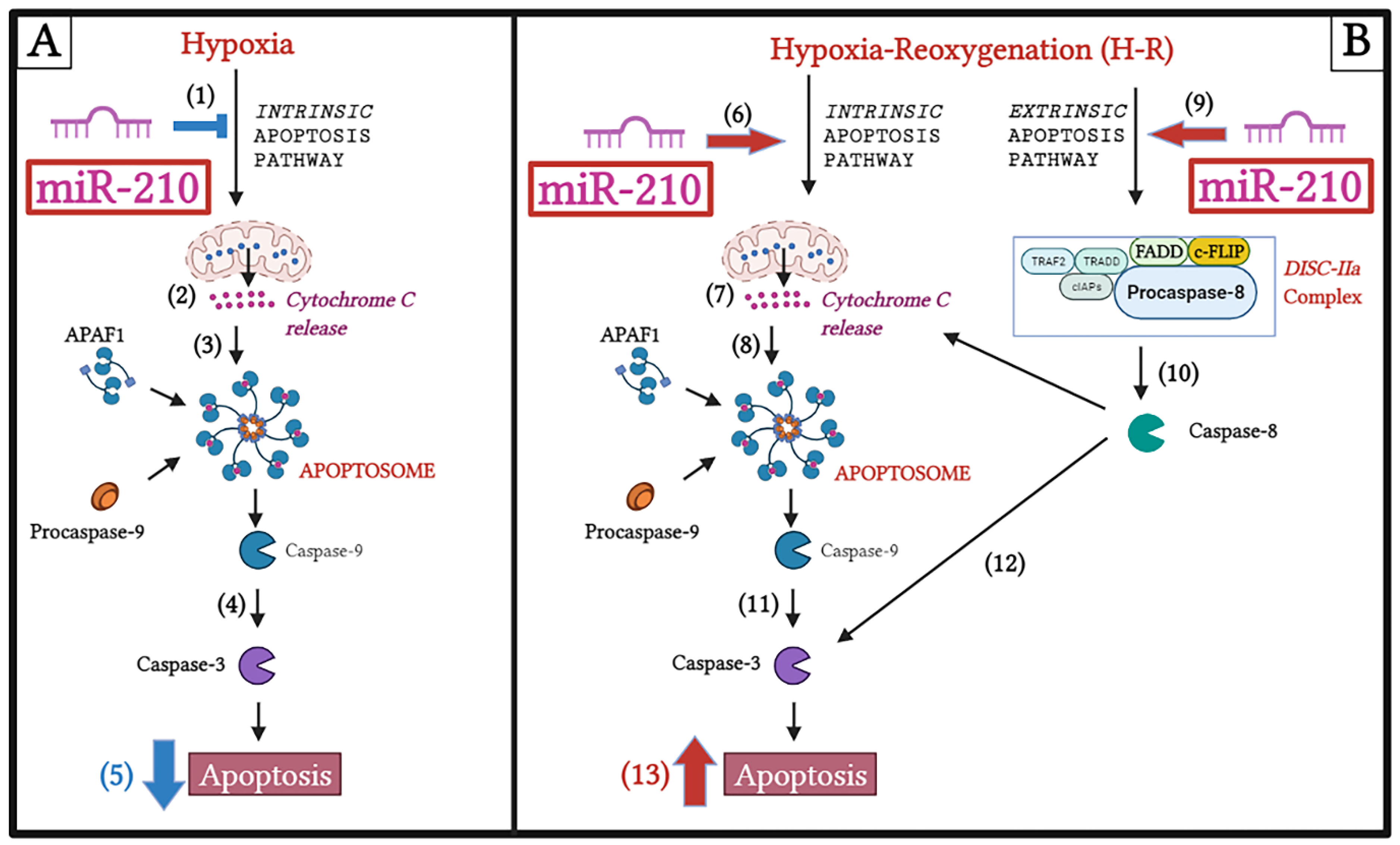
3.1. miR-210 Mitigated Apoptotic Cell Death during Hypoxia and Enhanced Apoptotic Cell Death during the Reoxygenation Phase
3.2. miR-210 Regulated the Intrinsic Apoptosis Pathway in a Diametrically Opposite Manner during the Hypoxia Phase and Reoxygenation Phase
3.3. miR-210 Regulated the Extrinsic Apoptosis Pathway during the H-R Phase but Not during the Hypoxia Phase
4. Discussion
4.1. Limitations
4.2. Future Directions
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Arnett, D.K.; Blumenthal, R.S.; Albert, M.A.; Buroker, A.B.; Goldberger, Z.D.; Hahn, E.J.; Himmelfarb, C.D.; Khera, A.; Lloyd-Jones, D.; McEvoy, J.W.; et al. 2019 ACC/AHA Guideline on the Primary Prevention of Cardiovascular Disease: Executive Summary: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Circulation 2019, 140, e563–e595. [Google Scholar] [CrossRef]
- Knuuti, J.; Wijns, W.; Saraste, A.; Capodanno, D.; Barbato, E.; Funck-Brentano, C.; Prescott, E.; Storey, R.F.; Deaton, C.; Cuisset, T.; et al. 2019 ESC Guidelines for the diagnosis and management of chronic coronary syndromes: The Task Force for the diagnosis and management of chronic coronary syndromes of the European Society of Cardiology (ESC). Eur. Heart J. 2019, 41, 407–477. [Google Scholar] [CrossRef] [PubMed]
- Virani, S.S.; Alonso, A.; Aparicio, H.J.; Benjamin, E.J.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Cheng, S.; Delling, F.N.; et al. Heart Disease and Stroke Statistics—2021 Update. Circulation 2021, 143, e254–e743. [Google Scholar] [CrossRef] [PubMed]
- Jennings, R.B.; Sommers, H.M.; Smyth, G.A.; Flack, H.A.; Linn, H. Myocardial necrosis induced by temporary occlusion of a coronary artery in the dog. Arch. Pathol. 1960, 70, 68–78. [Google Scholar] [PubMed]
- Hausenloy, D.J.; Yellon, D.M. Myocardial ischemia-reperfusion injury: A neglected therapeutic target. J. Clin. Investig. 2013, 123, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Piper, H.M.; García-Dorado, D.; Ovize, M. A fresh look at reperfusion injury. Cardiovasc. Res. 1998, 38, 291–300. [Google Scholar] [CrossRef] [Green Version]
- Yellon, D.M.; Hausenloy, D.J. Myocardial reperfusion injury. N. Engl. J. Med. 2007, 357, 1121–1135. [Google Scholar] [CrossRef]
- Braunwald, E.; Kloner, R.A. Myocardial reperfusion: A double-edged sword? J. Clin. Investig. 1985, 76, 1713–1719. [Google Scholar] [CrossRef]
- Prasad, A.; Stone, G.W.; Holmes, D.R.; Gersh, B. Reperfusion injury, microvascular dysfunction, and cardioprotection: The “dark side” of reperfusion. Circulation 2009, 120, 2105–2112. [Google Scholar] [CrossRef]
- Konstantinidis, K.; Whelan, R.S.; Kitsis, R.N. Mechanisms of cell death in heart disease. Arterioscler. Thromb Vasc. Biol. 2012, 32, 1552–1562. [Google Scholar] [CrossRef] [Green Version]
- Greco, S.; Gaetano, C.; Martelli, F. HypoxamiR Regulation and Function in Ischemic Cardiovascular Diseases. Antioxid. Redox Signal. 2013, 21, 1202–1219. [Google Scholar] [CrossRef] [Green Version]
- Chan, S.Y.; Loscalzo, J. MicroRNA-210: A unique and pleiotropic hypoxamir. Cell Cycle 2010, 9, 1072–1083. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.; Ding, L.; Bennewith, K.L.; Tong, R.T.; Welford, S.M.; Ang, K.K.; Story, M.; Le, Q.-T.; Giaccia, A.J. Hypoxia-inducible mir-210 regulates normoxic gene expression involved in tumor initiation. Mol. Cell 2009, 35, 856–867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivan, M.; Huang, X. miR-210: Fine-tuning the hypoxic response. Adv. Exp. Med. Biol. 2014, 772, 205–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bavelloni, A.; Ramazzotti, G.; Poli, A.; Piazzi, M.; Focaccia, E.; Blalock, W.; Faenza, I. MiRNA-210: A Current Overview. Anticancer Res. 2017, 37, 6511–6521. [Google Scholar] [CrossRef] [Green Version]
- Chan, Y.C.; Banerjee, J.; Choi, S.Y.; Sen, C.K. miR-210: The master hypoxamir. Microcirculation 2012, 19, 215–223. [Google Scholar] [CrossRef] [Green Version]
- Guan, Y.; Song, X.; Sun, W.; Wang, Y.; Liu, B. Effect of Hypoxia-Induced MicroRNA-210 Expression on Cardiovascular Disease and the Underlying Mechanism. Oxid. Med. Cell. Longev. 2019, 2019, 4727283. [Google Scholar] [CrossRef] [PubMed]
- Devlin, C.; Greco, S.; Martelli, F.; Ivan, M. miR-210: More than a silent player in hypoxia. IUBMB Life 2011, 63, 94–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivan, M.; Harris, A.L.; Martelli, F.; Kulshreshtha, R. Hypoxia response and microRNAs: No longer two separate worlds. J. Cell. Mol. Med. 2008, 12, 1426–1431. [Google Scholar] [CrossRef]
- Ma, X.; Wang, J.; Li, J.; Ma, C.; Chen, S.; Lei, W.; Yang, Y.; Liu, S.; Bihl, J.; Chen, C. Loading MiR-210 in Endothelial Progenitor Cells Derived Exosomes Boosts Their Beneficial Effects on Hypoxia/Reoxygeneation-Injured Human Endothelial Cells via Protecting Mitochondrial Function. Cell. Physiol. Biochem. 2018, 46, 664–675. [Google Scholar] [CrossRef]
- Puisségur, M.P.; Mazure, N.M.; Bertero, T.; Pradelli, L.; Grosso, S.; Robbe-Sermesant, K.; Maurin, T.; Lebrigand, K.; Cardinaud, B.; Hofman, V.; et al. miR-210 is overexpressed in late stages of lung cancer and mediates mitochondrial alterations associated with modulation of HIF-1 activity. Cell Death Differ. 2011, 18, 465–478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, S.Y.; Zhang, Y.-Y.; Hemann, C.; Mahoney, C.E.; Zweier, J.L.; Loscalzo, J. MicroRNA-210 controls mitochondrial metabolism during hypoxia by repressing the iron-sulfur cluster assembly proteins ISCU1/2. Cell Metab. 2009, 10, 273–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.; Li, Y.; Zhang, H.; Huang, P.; Luthra, R. Hypoxia-regulated microRNA-210 modulates mitochondrial function and decreases ISCU and COX10 expression. Oncogene 2010, 29, 4362–4368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giannakakis, A.; Sandaltzopoulos, R.; Greshock, J.; Liang, S.; Huang, J.; Hasegawa, K.; Li, C.; O’Brien-Jenkins, A.; Katsaros, D.; Weber, B.L.; et al. miR-210 links hypoxia with cell cycle regulation and is deleted in human epithelial ovarian cancer. Cancer Biol. Ther. 2008, 7, 255–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mok, Y.; Schwierzeck, V.; Thomas, D.C.; Vigorito, E.; Rayner, T.F.; Jarvis, L.B.; Prosser, H.M.; Bradley, A.; Withers, D.R.; Mårtensson, I.-L.; et al. MiR-210 is induced by Oct-2, regulates B cells, and inhibits autoantibody production. J. Immunol. 2013, 191, 3037–3048. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Sun, H.; Dai, H.; Walsh, R.M.; Imakura, M.; Schelter, J.; Burchard, J.; Dai, X.; Chang, A.N.; Diaz, R.L.; et al. MicroRNA miR-210 modulates cellular response to hypoxia through the MYC antagonist MNT. Cell Cycle 2009, 8, 2756–2768. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Lai, N.; Liao, K.; Sun, J.; Lin, Y. MicroRNA-210 regulates cell proliferation and apoptosis by targeting regulator of differentiation 1 in glioblastoma cells. Folia Neuropathol. 2015, 53, 236–244. [Google Scholar] [CrossRef]
- Minayi, N.; Alizadeh, S.; Dargahi, H.; Soleimani, M.; Khatib, Z.K.; Tayebi, B.; Mohammadian, M.; Alijani, S.; Karami, F. The Effect of miR-210 Up-regulation on Proliferation and Survival of Mouse Bone Marrow Derived Mesenchymal Stem Cell. Int. J. Hematol. Oncol. Stem Cell Res. 2014, 8, 15–23. [Google Scholar]
- Tang, T.; Yang, Z.; Zhu, Q.; Wu, Y.; Sun, K.; Alahdal, M.; Zhang, Y.; Xing, Y.; Shen, Y.; Xia, T.; et al. Up-regulation of miR-210 induced by a hypoxic microenvironment promotes breast cancer stem cell metastasis, proliferation, and self-renewal by targeting E-cadherin. FASEB J. 2018, 32, 6965–6981. [Google Scholar] [CrossRef]
- Dang, K.; Myers, K.A. The role of hypoxia-induced miR-210 in cancer progression. Int. J. Mol. Sci. 2015, 16, 6353–6372. [Google Scholar] [CrossRef]
- Kim, H.W.; Haider, H.K.; Jiang, S.; Ashraf, M. Ischemic preconditioning augments survival of stem cells via miR-210 expression by targeting caspase-8-associated protein 2. J. Biol. Chem. 2009, 284, 33161–33168. [Google Scholar] [CrossRef] [Green Version]
- Gou, D.; Ramchandran, R.; Peng, X.; Yao, L.; Kang, K.; Sarkar, J.; Wang, Z.; Zhou, G.; Raj, J.U. miR-210 has an antiapoptotic effect in pulmonary artery smooth muscle cells during hypoxia. Am. J. Physiol. Lung Cell Mol. Physiol. 2012, 303, L682–L691. [Google Scholar] [CrossRef]
- Hu, S.; Huang, M.; Li, Z.; Jia, F.; Ghosh, Z.; Lijkwan, M.A.; Fasanaro, P.; Sun, N.; Wang, X.; Martelli, F.; et al. MicroRNA-210 as a novel therapy for treatment of ischemic heart disease. Circulation 2010, 122, S124–S131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chio, C.C.; Lin, J.W.; Cheng, H.A.; Chiu, W.T.; Wang, Y.H.; Wang, J.J.; Hsing, C.H.; Chen, R.M. MicroRNA-210 targets antiapoptotic Bcl-2 expression and mediates hypoxia-induced apoptosis of neuroblastoma cells. Arch. Toxicol. 2013, 87, 459–468. [Google Scholar] [CrossRef]
- Qiu, J.; Zhou, X.Y.; Zhou, X.G.; Cheng, R.; Liu, H.Y.; Li, Y. Neuroprotective effects of microRNA-210 against oxygen-glucose deprivation through inhibition of apoptosis in PC12 cells. Mol. Med. Rep. 2013, 7, 1955–1959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, F.; Xiong, L.; Huang, X.; Zhao, T.; Wu, L.-y.; Liu, Z.-h.; Ding, X.; Liu, S.; Wu, Y.; Zhao, Y.; et al. miR-210 suppresses BNIP3 to protect against the apoptosis of neural progenitor cells. Stem Cell Res. 2013, 11, 657–667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grosso, S.; Doyen, J.; Parks, S.K.; Bertero, T.; Paye, A.; Cardinaud, B.; Gounon, P.; Lacas-Gervais, S.; Noël, A.; Pouysségur, J.; et al. MiR-210 promotes a hypoxic phenotype and increases radioresistance in human lung cancer cell lines. Cell Death Dis. 2013, 4, e544. [Google Scholar] [CrossRef]
- Arif, M.; Pandey, R.; Alam, P.; Jiang, S.; Sadayappan, S.; Paul, A.; Ahmed, R.P.H. MicroRNA-210-mediated proliferation, survival, and angiogenesis promote cardiac repair post myocardial infarction in rodents. J. Mol. Med. 2017, 95, 1369–1385. [Google Scholar] [CrossRef] [Green Version]
- Tagscherer, K.E.; Fassl, A.; Sinkovic, T.; Richter, J.; Schecher, S.; Macher-Goeppinger, S.; Roth, W. MicroRNA-210 induces apoptosis in colorectal cancer via induction of reactive oxygen. Cancer Cell Int. 2016, 16, 42. [Google Scholar] [CrossRef] [Green Version]
- Biswas, S.; Roy, S.; Banerjee, J.; Hussain, S.-R.A.; Khanna, S.; Meenakshisundaram, G.; Kuppusamy, P.; Friedman, A.; Sen, C.K. Hypoxia inducible microRNA 210 attenuates keratinocyte proliferation and impairs closure in a murine model of ischemic wounds. Proc. Natl. Acad. Sci. USA 2010, 107, 6976–6981. [Google Scholar] [CrossRef] [Green Version]
- Freshney, R.I. Culture of Animal Cells: A Manual of Basic Technique and Specialized Applications, 7th ed.; Wiley-Blackwell: Hoboken, NJ, USA, 2016. [Google Scholar]
- Burnette, W.N. “Western blotting”: Electrophoretic transfe.er of proteins from sodium dodecyl sulfate--polyacrylamide gels to unmodified nitrocellulose and radiographic detection with antibody and radioiodinated protein A. Anal. Biochem. 1981, 112, 195–203. [Google Scholar] [CrossRef]
- Towbin, H.; Staehelin, T.; Gordon, J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: Procedure and some applications. Proc. Natl. Acad. Sci. USA 1979, 76, 4350–4354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sambrook, J.; Fritsch, E.F.; Maniatis, T.; Irwin, N. Molecular Cloning: A Laboratory Manual; Cold Spring Harbor Laboratory: Cold Spring Harbor, NY, USA, 1989. [Google Scholar]
- Kurien, B.T.; Scofield, R.H. Western Blotting–Methods and Protocols; Humana Press: Totowa, NJ, USA, 2015. [Google Scholar]
- Thieme, D.; Neubauer, P.; Nies, D.H.; Grass, G. Sandwich hybridization assay for sensitive detection of dynamic changes in mRNA transcript levels in crude Escherichia coli cell extracts in response to copper ions. Appl. Environ. Microbiol. 2008, 74, 7463–7470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, K.F.; Rhodes, L.L.; Adamson, J.E.; Tyrrell, J.V.; Mountfort, D.O.; Jones, W.J. Application of a sandwich hybridisation assay for rapid detection of the Northern Pacific seastar, Asterias amurensis. N. Z. J. Mar. Freshw. Res. 2011, 45, 145–152. [Google Scholar] [CrossRef] [Green Version]
- Rautio, J.; Barken, K.B.; Lahdenperä, J.; Breitenstein, A.; Molin, S.; Neubauer, P. Sandwich hybridisation assay for quantitative detection of yeast RNAs in crude cell lysates. Microb. Cell Factories 2003, 2, 4. [Google Scholar] [CrossRef] [Green Version]
- Deverre, J.-R.; Boutet, V.; Boquet, D.; Ezan, E.; Grassi, J.; Grognet, J.-M. A competitive enzyme hybridization assay for plasma determination of phosphodiester and phosphorothioate antisense oligonucleotides. Nucleic Acids Res. 1997, 25, 3584–3589. [Google Scholar] [CrossRef] [Green Version]
- Tremblay, G.; Khalafaghian, G.; Legault, J.; Nielsen, P.; Bartlett, A. Dual ligation hybridization assay for the specific determination of oligonucleotide therapeutics. Bioanalysis 2011, 3, 499–508. [Google Scholar] [CrossRef]
- Wei, X.; Dai, G.; Marcucci, G.; Liu, Z.; Hoyt, D.; Blum, W.; Chan, K.K. A Specific Picomolar Hybridization-Based ELISA Assay for the Determination of Phosphorothioate Oligonucleotides in Plasma and Cellular Matrices. Pharm. Res. 2006, 23, 1251–1264. [Google Scholar] [CrossRef]
- Coutlee, F.; Rubalcaba, E.A.; Viscidi, R.P.; Gern, J.E.; Murphy, P.A.; Lederman, H.M. Quantitative detection of messenger RNA by solution hybridization and enzyme immunoassay. J. Biol. Chem. 1990, 265, 11601–11604. [Google Scholar] [CrossRef]
- Cohen, L.; Hartman, M.R.; Amardey-Wellington, A.; Walt, D.R. Digital direct detection of microRNAs using single molecule arrays. Nucleic Acids Res. 2017, 45, e137. [Google Scholar] [CrossRef]
- Aramburu, J.; Navas-Castillo, J.; Moreno, P.; Cambra, M. Detection of double-stranded RNA by ELISA and dot immunobinding assay using an antiserum to synthetic polynucleotides. J. Virol. Methods 1991, 33, 1–11. [Google Scholar] [CrossRef]
- Van Weemen, B.K.; Schuurs, A.H. Immunoassay using antigen-enzyme conjugates. FEBS Lett. 1971, 15, 232–236. [Google Scholar] [CrossRef] [Green Version]
- Engvall, E.; Perlmann, P. Enzyme-linked immunosorbent assay (ELISA). Quantitative assay of immunoglobulin G. Immunochemistry 1971, 8, 871–874. [Google Scholar] [CrossRef]
- Crowther, J.R. ELISA. Theory and practice. Methods Mol. Biol. 1995, 42, 218. [Google Scholar] [CrossRef]
- Engvall, E.; Perlmann, P. Enzyme-Linked Immunosorbent Assay, Elisa. III. Quantitation of Specific Antibodies by Enzyme-Labeled Anti-Immunoglobulin in Antigen-Coated Tubes. J. Immunol. 1972, 109, 129–135. [Google Scholar]
- Butler, J.E.; Ni, L.; Nessler, R.; Joshi, K.S.; Suter, M.; Rosenberg, B.; Chang, J.; Brown, W.R.; Cantarero, L.A. The physical and functional behavior of capture antibodies adsorbed on polystyrene. J. Immunol. Methods 1992, 150, 77–90. [Google Scholar] [CrossRef]
- Cold Spring Harbor Laboratory. Antibodies: A Laboratory Manual, 2nd ed.; Grenfield, E.A., Ed.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2014. [Google Scholar]
- Gosling, J. Immunoassays: A Practical Approach; Oxford University Press: Oxford, UK, 2000. [Google Scholar]
- Tarcha, P.J.; Chu, V.P.; Whittern, D. 2,3-diaminophenazine is the product from the horseradish peroxidase-catalyzed oxidation of o-phenylenediamine. Anal. Biochem. 1987, 165, 230–233. [Google Scholar] [CrossRef]
- Bovaird, J.H.; Ngo, T.T.; Lenhoff, H.M. Optimizing the o-phenylenediamine assay for horseradish peroxidase: Effects of phosphate and pH, substrate and enzyme concentrations, and stopping reagents. Clin. Chem. 1982, 28, 2423–2426. [Google Scholar] [CrossRef]
- Nicholson, D.W.; Ali, A.; Thornberry, N.A.; Vaillancourt, J.P.; Ding, C.K.; Gallant, M.; Gareau, Y.; Griffin, P.R.; Labelle, M.; Lazebnik, Y.A.; et al. Identification and inhibition of the ICE/CED-3 protease necessary for mammalian apoptosis. Nature 1995, 376, 37–43. [Google Scholar] [CrossRef]
- Liu, Z.-Q.; Du, J.-J.; Ren, J.-J.; Zhang, Z.-Y.; Guo, X.-B.; Yan, Y.-E.; Jia, X.-T.; Gu, N.-B.; Di, Z.-L.; Li, S.-Z. miR-183-96-182 clusters alleviated ox-LDL-induced vascular endothelial cell apoptosis in vitro by targeting FOXO1. RSC Adv. 2018, 8, 35031–35041. [Google Scholar] [CrossRef] [Green Version]
- Riccio, G.; Esposito, G.; Leoncini, E.; Contu, R.; Condorelli, G.; Chiariello, M.; Laccetti, P.; Hrelia, S.; D’Alessio, G.; De Lorenzo, C. Cardiotoxic effects, or lack thereof, of anti-ErbB2 immunoagents. FASEB J. 2009, 23, 3171–3178. [Google Scholar] [CrossRef] [Green Version]
- Bollo, E.; Guglielmino, R.; Sant, S.; Pregel, P.; Riondato, F.; Miniscalco, B.; Cornaglia, E.; Nebbia, C.; Dacasto, M. Biochemical, ultrastructural and molecular characterization of the triphenyltin acetate (TPTA)-induced apoptosis in primary cultures of mouse thymocytes. Cell Biol. Toxicol. 2006, 22, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Wu, Y.; Zhu, Y. Mechanism of MALAT1 preventing apoptosis of vascular endothelial cells induced by oxygen–glucose deficiency and reoxidation. Artif. Cells Nanomed. Biotechnol. 2018, 46, 798–805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorczyca, W.; Bruno, S.; Darzynkiewicz, R.; Gong, J.; Darzynkiewicz, Z. DNA strand breaks occurring during apoptosis–their early insitu detection by the terminal deoxynucleotidyl transferase and nick translation assays and prevention by serine protease inhibitors. Int. J. Oncol. 1992, 1, 639–648. [Google Scholar] [CrossRef] [PubMed]
- Medema, J.P.; Scaffidi, C.; Kischkel, F.C.; Shevchenko, A.; Mann, M.; Krammer, P.H.; Peter, M.E. FLICE is activated by association with the CD95 death-inducing signaling complex (DISC). EMBO J. 1997, 16, 2794–2804. [Google Scholar] [CrossRef] [PubMed]
- Decker, T.; Lohmann-Matthes, M.L. A quick and simple method for the quantitation of lactate dehydrogenase release in measurements of cellular cytotoxicity and tumor necrosis factor (TNF) activity. J. Immunol. Methods 1988, 115, 61–69. [Google Scholar] [CrossRef]
- Korzeniewski, C.; Callewaert, D.M. An enzyme-release assay for natural cytotoxicity. J. Immunol. Methods 1983, 64, 313–320. [Google Scholar] [CrossRef]
- Green, D.R. Apoptotic Pathways: Paper Wraps Stone Blunts Scissors. Cell 2000, 102, 8674. [Google Scholar] [CrossRef] [Green Version]
- Salvesen, G.S. Caspases: Opening the boxes and interpreting the arrows. Cell Death Differ. 2002, 9, 3–5. [Google Scholar] [CrossRef]
- Earnshaw, W.C.; Martins, L.M.; Kaufmann, S.H. Mammalian caspases: Structure, activation, substrates, and functions during apoptosis. Annu. Rev. Biochem. 1999, 68, 383–424. [Google Scholar] [CrossRef]
- Riedl, S.J.; Salvesen, G.S. The apoptosome: Signalling platform of cell death. Nat. Rev. Mol. Cell Biol. 2007, 8, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Kim, C.N.; Yang, J.; Jemmerson, R.; Wang, X. Induction of Apoptotic Program in Cell-Free Extracts: Requirement for dATP and Cytochrome c. Cell 1996, 86, 147–157. [Google Scholar] [CrossRef] [Green Version]
- Willson, J. A matter of life and death for caspase 8. Nat. Rev. Mol. Cell Biol. 2020, 21, 63. [Google Scholar] [CrossRef] [PubMed]
- Tummers, B.; Green, D.R. Caspase-8: Regulating life and death. Immunol. Rev. 2017, 277, 76–89. [Google Scholar] [CrossRef] [Green Version]
- Kischkel, F.C.; Hellbardt, S.; Behrmann, I.; Germer, M.; Pawlita, M.; Krammer, P.H.; Peter, M.E. Cytotoxicity-dependent APO-1 (Fas/CD95)-associated proteins form a death-inducing signaling complex (DISC) with the receptor. EMBO J. 1995, 14, 5579–5588. [Google Scholar] [CrossRef] [PubMed]
- Wilson, N.S.; Dixit, V.; Ashkenazi, A. Death receptor signal transducers: Nodes of coordination in immune signaling networks. Nat. Immunol. 2009, 10, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Kantari, C.; Walczak, H. Caspase-8 and Bid: Caught in the act between death receptors and mitochondria. Biochim. Biophys. Acta Mol. Cell Res. 2011, 1813, 558–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muzio, M.; Chinnaiyan, A.M.; Kischkel, F.C.; O’Rourke, K.; Shevchenko, A.; Ni, J.; Scaffidi, C.; Bretz, J.D.; Zhang, M.; Gentz, R.; et al. FLICE, A Novel FADD-Homologous ICE/CED-3–like Protease, Is Recruited to the CD95 (Fas/APO-1) Death-Inducing Signaling Complex. Cell 1996, 85, 817–827. [Google Scholar] [CrossRef] [Green Version]
- Berghe, T.V.; Linkermann, A.; Jouan-Lanhouet, S.; Walczak, H.; Vandenabeele, P. Regulated necrosis: The expanding network of non-apoptotic cell death pathways. Nat. Rev. Mol. Cell Biol. 2014, 15, 135–147. [Google Scholar] [CrossRef]
- Vandenabeele, P.; Galluzzi, L.; Vanden Berghe, T.; Kroemer, G. Molecular mechanisms of necroptosis: An ordered cellular explosion. Nat. Rev. Mol. Cell Biol. 2010, 11, 700–714. [Google Scholar] [CrossRef]
- Mandal, R.; Barrón, J.C.; Kostova, I.; Becker, S.; Strebhardt, K. Caspase-8: The double-edged sword. Biochim. Biophys. Acta Rev. Cancer 2020, 1873, 188357. [Google Scholar] [CrossRef]
- Fritsch, M.; Günther, S.D.; Schwarzer, R.; Albert, M.-C.; Schorn, F.; Werthenbach, J.P.; Schiffmann, L.M.; Stair, N.; Stocks, H.; Seeger, J.M.; et al. Caspase-8 is the molecular switch for apoptosis, necroptosis and pyroptosis. Nature 2019, 575, 683–687. [Google Scholar] [CrossRef] [PubMed]
- Newton, K.; Wickliffe, K.E.; Maltzman, A.; Dugger, D.L.; Reja, R.; Zhang, Y.; Roose-Girma, M.; Modrusan, Z.; Sagolla, M.S.; Webster, J.D.; et al. Activity of caspase-8 determines plasticity between cell death pathways. Nature 2019, 575, 679–682. [Google Scholar] [CrossRef] [PubMed]
- Feoktistova, M.; Geserick, P.; Panayotova-Dimitrova, D.; Leverkus, M. Pick your poison: The Ripoptosome, a cell death platform regulating apoptosis and necroptosis. Cell Cycle 2012, 11, 460–467. [Google Scholar] [CrossRef] [PubMed]
- Seifert, L.; Miller, G. Molecular Pathways: The Necrosome—A Target for Cancer Therapy. Clin. Cancer Res. 2017, 23, 1132–1136. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Kos, R.; Garssen, J.; Redegeld, F. Molecular Insights into the Mechanism of Necroptosis: The Necrosome as a Potential Therapeutic Target. Cells 2019, 8, 1486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srinivasula, S.M.; Ahmad, M.; Ottilie, S.; Bullrich, F.; Banks, S.; Wang, Y.; Fernandes-Alnemri, T.; Croce, C.M.; Litwack, G.; Tomaselli, K.J.; et al. FLAME-1, a Novel FADD-like Anti-apoptotic Molecule That Regulates Fas/TNFR1-induced Apoptosis. J. Biol. Chem. 1997, 272, 18542–18545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shu, H.-B.; Halpin, D.R.; Goeddel, D.V. Casper Is a FADD- and Caspase-Related Inducer of Apoptosis. Immunity 1997, 6, 751–763. [Google Scholar] [CrossRef] [Green Version]
- Irmler, M.; Thome, M.; Hahne, M.; Schneider, P.; Hofmann, K.; Steiner, V.; Bodmer, J.-L.; Schröter, M.; Burns, K.; Mattmann, C.; et al. Inhibition of death receptor signals by cellular FLIP. Nature 1997, 388, 190–195. [Google Scholar] [CrossRef]
- Hu, S.; Vincenz, C.; Ni, J.; Gentz, R.; Dixit, V.M. I-FLICE, a Novel Inhibitor of Tumor Necrosis Factor Receptor-1- and CD-95-induced Apoptosis. J. Biol. Chem. 1997, 272, 17255–17257. [Google Scholar] [CrossRef] [Green Version]
- Goltsev, Y.V.; Kovalenko, A.V.; Arnold, E.; Varfolomeev, E.E.; Brodianskii, V.M.; Wallach, D. CASH, a Novel Caspase Homologue with Death Effector Domains. J. Biol. Chem. 1997, 272, 19641–19644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsuchiya, Y.; Nakabayashi, O.; Nakano, H. FLIP the Switch: Regulation of Apoptosis and Necroptosis by cFLIP. Int. J. Mol. Sci. 2015, 16, 30321–30341. [Google Scholar] [CrossRef] [Green Version]
- Scaffidi, C.; Schmitz, I.; Krammer, P.H.; Peter, M.E. The role of c-FLIP in modulation of CD95-induced apoptosis. J. Biol. Chem. 1999, 274, 1541–1548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krueger, A.; Schmitz, I.; Baumann, S.; Krammer, P.H.; Kirchhoff, S. Cellular FLICE-inhibitory protein splice variants inhibit different steps of caspase-8 activation at the CD95 death-inducing signaling complex. J. Biol. Chem. 2001, 276, 20633–20640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tschopp, J.; Irmler, M.; Thome, M. Inhibition of fas death signals by FLIPs. Curr. Opin. Immunol. 1998, 10, 552–558. [Google Scholar] [CrossRef]
- Krueger, A.; Baumann, S.; Krammer, P.H.; Kirchhoff, S. FLICE-inhibitory proteins: Regulators of death receptor-mediated apoptosis. Mol. Cell. Biol. 2001, 21, 8247–8254. [Google Scholar] [CrossRef] [Green Version]
- Hughes, M.A.; Powley, I.R.; Jukes-Jones, R.; Horn, S.; Feoktistova, M.; Fairall, L.; Schwabe, J.W.R.; Leverkus, M.; Cain, K.; MacFarlane, M. Co-operative and Hierarchical Binding of c-FLIP and Caspase-8: A Unified Model Defines How c-FLIP Isoforms Differentially Control Cell Fate. Mol. Cell 2016, 61, 834–849. [Google Scholar] [CrossRef] [Green Version]
- Feoktistova, M.; Geserick, P.; Leverkus, M. Ripoptosome Analysis by Caspase-8 Coimmunoprecipitation. Cold Spring Harb. Protoc. 2016, 2016, pdb.prot087403. [Google Scholar] [CrossRef]
- Sanada, S.; Komuro, I.; Kitakaze, M. Pathophysiology of myocardial reperfusion injury: Preconditioning, postconditioning, and translational aspects of protective measures. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H1723–H1741. [Google Scholar] [CrossRef] [Green Version]
- Kang, P.M.; Haunstetter, A.; Aoki, H.; Usheva, A.; Izumo, S. Morphological and Molecular Characterization of Adult Cardiomyocyte Apoptosis During Hypoxia and Reoxygenation. Circ. Res. 2000, 87, 118–125. [Google Scholar] [CrossRef]
- Sendoel, A.; Hengartner, M.O. Apoptotic Cell Death Under Hypoxia. Physiology 2014, 29, 168–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimizu, S.; Eguchi, Y.; Kamiike, W.; Itoh, Y.; Hasegawa, J.; Yamabe, K.; Otsuki, Y.; Matsuda, H.; Tsujimoto, Y. Induction of apoptosis as well as necrosis by hypoxia and predominant prevention of apoptosis by Bcl-2 and Bcl-XL. Cancer Res. 1996, 56, 2161–2166. [Google Scholar] [PubMed]
- Krijnen, P.A.J.; Nijmeijer, R.; Meijer, C.J.L.M.; Visser, C.A.; Hack, C.E.; Niessen, H.W.M. Apoptosis in myocardial ischaemia and infarction. J. Clin. Pathol. 2002, 55, 801–811. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez, M.; Lucchesi, B.R.; Schaper, J. Apoptosis in myocardial infarction. Ann. Med. 2002, 34, 470–479. [Google Scholar] [CrossRef]
- Kajstura, J.; Cheng, W.; Reiss, K.; Clark, W.A.; Sonnenblick, E.H.; Krajewski, S.; Reed, J.C.; Olivetti, G.; Anversa, P. Apoptotic and necrotic myocyte cell deaths are independent contributing variables of infarct size in rats. Lab. Investig. 1996, 74, 86–107. [Google Scholar] [PubMed]
- McCully, J.D.; Wakiyama, H.; Hsieh, Y.-J.; Jones, M.; Levitsky, S. Differential contribution of necrosis and apoptosis in myocardial ischemia-reperfusion injury. Am. J. Physiol.-Heart Circ. Physiol. 2004, 286, H1923–H1935. [Google Scholar] [CrossRef]
- Buja, L.M.; Entman, M.L. Modes of Myocardial Cell Injury and Cell Death in Ischemic Heart Disease. Circulation 1998, 98, 1355–1357. [Google Scholar] [CrossRef] [Green Version]
- Saraste, A.; Pulkki, K.; Kallajoki, M.; Henriksen, K.; Parvinen, M.; Voipio-Pulkki, L.-M. Apoptosis in Human Acute Myocardial Infarction. Circulation 1997, 95, 320–323. [Google Scholar] [CrossRef]
- Olivetti, G.; Quaini, F.; Sala, R.; Lagrasta, C.; Corradi, D.; Bonacina, E.; Gambert, S.R.; Cigola, E.; Anversa, P. Acute Myocardial Infarction in Humans is Associated with Activation of Programmed Myocyte Cell Death in the Surviving Portion of the Heart. J. Mol. Cell. Cardiol. 1996, 28, 2005–2016. [Google Scholar] [CrossRef]
- Abbate, A.; Melfi, R.; Patti, G.; Baldi, F.; D’Ambrosio, A.; Manzoli, A.; Baldi, A.; Di Sciascio, G. Apoptosis in recent myocardial infarction. Clin. Ter. 2000, 151, 247–251. [Google Scholar]
- Teringova, E.; Tousek, P. Apoptosis in ischemic heart disease. J. Transl. Med. 2017, 15, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gottlieb, R.A.; Burleson, K.O.; Kloner, R.A.; Babior, B.M.; Engler, R.L. Reperfusion injury induces apoptosis in rabbit cardiomyocytes. J. Clin. Investig. 1994, 94, 1621–1628. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Chua, C.C.; Ho, Y.S.; Hamdy, R.C.; Chua, B.H. Overexpression of Bcl-2 attenuates apoptosis and protects against myocardial I/R injury in transgenic mice. Am. J. Physiol. Heart Circ. Physiol. 2001, 280, H2313–H2320. [Google Scholar] [CrossRef] [PubMed]
- Brocheriou, V.; Hagège, A.A.; Oubenaïssa, A.; Lambert, M.; Mallet, V.O.; Duriez, M.; Wassef, M.; Kahn, A.; Menasché, P.; Gilgenkrantz, H. Cardiac functional improvement by a human Bcl-2 transgene in a mouse model of ischemia/reperfusion injury. J. Gene Med. 2000, 2, 326–333. [Google Scholar] [CrossRef]
- Jeremias, I.; Kupatt, C.; Martin-Villalba, A.; Habazettl, H.; Schenkel, J.; Boekstegers, P.; Debatin, K.M. Involvement of CD95/Apo1/Fas in cell death after myocardial ischemia. Circulation 2000, 102, 915–920. [Google Scholar] [CrossRef] [Green Version]
- Lee, P.; Sata, M.; Lefer, D.J.; Factor, S.M.; Walsh, K.; Kitsis, R.N. Fas pathway is a critical mediator of cardiac myocyte death and MI during ischemia-reperfusion in vivo. Am. J. Physiol. Heart Circ. Physiol. 2003, 284, H456–H463. [Google Scholar] [CrossRef] [PubMed]
- Zaccagnini, G.; Maimone, B.; Fuschi, P.; Maselli, D.; Spinetti, G.; Gaetano, C.; Martelli, F. Overexpression of miR-210 and its significance in ischemic tissue damage. Sci. Rep. 2017, 7, 9563. [Google Scholar] [CrossRef] [Green Version]
- Yao, R.; Cooper, G.M. Requirement for phosphatidylinositol-3 kinase in the prevention of apoptosis by nerve growth factor. Science 1995, 267, 2003–2006. [Google Scholar] [CrossRef]
- Scheid, M.P.; Woodgett, J.R. PKB/AKT: Functional insights from genetic models. Nat. Rev. Mol. Cell. Biol. 2001, 2, 760–768. [Google Scholar] [CrossRef]
- Brazil, D.P.; Hemmings, B.A. Ten years of protein kinase B signalling: A hard Akt to follow. Trends Biochem. Sci. 2001, 26, 657–664. [Google Scholar] [CrossRef]
- Manning, B.D.; Toker, A. AKT/PKB Signaling: Navigating the Network. Cell 2017, 169, 381–405. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Jiang, D.; Gong, F.; Huang, Y.; Luo, Y.; Rong, Y.; Wang, J.; Ge, X.; Ji, C.; Fan, J.; et al. miR-210-5p promotes epithelial–mesenchymal transition by inhibiting PIK3R5 thereby activating oncogenic autophagy in osteosarcoma cells. Cell Death Dis. 2020, 11, 93. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Song, Y.; Wang, H.; Liu, K.; Shao, Z.; Shang, Z. MiR-210-3p-EphrinA3-PI3K/AKT axis regulates the progression of oral cancer. J. Cell. Mol. Med. 2020, 24, 4011–4022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, H.S.; Das, D.K. Role of cytokines in myocardial ischemia and reperfusion. Mediators Inflamm. 1997, 6, 175–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nian, M.; Lee, P.; Khaper, N.; Liu, P. Inflammatory Cytokines and Postmyocardial Infarction Remodeling. Circ. Res. 2004, 94, 1543–1553. [Google Scholar] [CrossRef] [PubMed]
- Toldo, S.; Mauro, A.G.; Cutter, Z.; Abbate, A. Inflammasome, pyroptosis, and cytokines in myocardial ischemia-reperfusion injury. Am. J. Physiology. Heart Circ. Physiol. 2018, 315, H1553–H1568. [Google Scholar] [CrossRef] [PubMed]
- Ong, S.-B.; Hernández-Reséndiz, S.; Crespo-Avilan, G.E.; Mukhametshina, R.T.; Kwek, X.-Y.; Cabrera-Fuentes, H.A.; Hausenloy, D.J. Inflammation following acute myocardial infarction: Multiple players, dynamic roles, and novel therapeutic opportunities. Pharmacol. Ther. 2018, 186, 73–87. [Google Scholar] [CrossRef]
- Guicciardi, M.E.; Gores, G.J. Life and death by death receptors. FASEB J. 2009, 23, 1625–1637. [Google Scholar] [CrossRef] [Green Version]
- Lavrik, I.; Golks, A.; Krammer, P.H. Death receptor signaling. J. Cell Sci. 2005, 118, 265–267. [Google Scholar] [CrossRef] [Green Version]
- Kumar, R.; Herbert, P.E.; Warrens, A.N. An introduction to death receptors in apoptosis. Int. J. Surg. 2005, 3, 268–277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashkenazi, A.; Dixit, V.M. Death receptors: Signaling and modulation. Science 1998, 281, 1305–1308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guicciardi, M.E.; Gores, G.J. The Death Receptor Family and the Extrinsic Pathway. In Essentials of Apoptosis: A Guide for Basic and Clinical Research; Yin, X.-M., Dong, Z., Eds.; Humana Press: Totowa, NJ, USA, 2003; pp. 67–84. [Google Scholar]
- Wu, R.; Zeng, J.; Yuan, J.; Deng, X.; Huang, Y.; Chen, L.; Zhang, P.; Feng, H.; Liu, Z.; Wang, Z.; et al. MicroRNA-210 overexpression promotes psoriasis-like inflammation by inducing Th1 and Th17 cell differentiation. J. Clin. Investig. 2018, 128, 2551–2568. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Ma, Q.; Li, Y.; Li, B.; Zhang, L. Inhibition of microRNA-210 suppresses pro-inflammatory response and reduces acute brain injury of ischemic stroke in mice. Exp. Neurol. 2018, 300, 41–50. [Google Scholar] [CrossRef]
- Wu, T.Y.; Leng, Q.; Tian, L.Q. The microRNA-210/Casp8ap2 Axis Alleviates Hypoxia-Induced Myocardial Injury by Regulating Apoptosis and Autophagy. Cytogenet. Genome Res. 2021, 161, 132–142. [Google Scholar] [CrossRef]
- Chen, D.; Hou, Y.; Cai, X. MiR-210-3p Enhances Cardiomyocyte Apoptosis and Mitochondrial Dysfunction by Targeting the NDUFA4 Gene in Sepsis-Induced Myocardial Dysfunction. Int. Heart J. 2021, 62, 636–646. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Yang, C.; Zhang, L.; Yang, P. MicroRNA-210 induces endothelial cell apoptosis by directly targeting PDK1 in the setting of atherosclerosis. Cell. Mol. Biol. Lett. 2017, 22, 335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shang, C.; Hong, Y.; Guo, Y.; Liu, Y.-h.; Xue, Y.-x. MiR-210 up-regulation inhibits proliferation and induces apoptosis in glioma cells by targeting SIN3A. Med. Sci. Monit. 2014, 20, 2571–2577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lv, J.X.; Zhou, J.; Tong, R.Q.; Wang, B.; Chen, X.L.; Zhuang, Y.Y.; Xia, F.; Wei, X.D. Hypoxia-induced miR-210 contributes to apoptosis of mouse spermatocyte GC-2 cells by targeting Kruppel-like factor 7. Mol. Med. Rep. 2019, 19, 271–279. [Google Scholar] [CrossRef] [Green Version]
- Luan, Y.; Zhang, X.; Zhang, Y.; Dong, Y. MicroRNA-210 Protects PC-12 Cells Against Hypoxia-Induced Injury by Targeting BNIP3. Front. Cell. Neurosci. 2017, 11, 285. [Google Scholar] [CrossRef] [Green Version]
- Bao, Q.; Jia, H.; Rong, A.; Cao, Z.; Zhang, Y. MiR-210 inhibits hypoxia-induced apoptosis of smooth muscle cells via targeting MEF2C. Int. J. Clin. Exp. Pathol. 2019, 12, 1846–1858. [Google Scholar]
- He, J.; Wu, J.; Xu, N.; Xie, W.; Li, M.; Li, J.; Jiang, Y.; Yang, B.B.; Zhang, Y. MiR-210 disturbs mitotic progression through regulating a group of mitosis-related genes. Nucleic Acids Res. 2013, 41, 498–508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, G.; Tao, Y.; Yu, W.; Schwartz, R.J. Brief report: SRF-dependent MiR-210 silences the sonic hedgehog signaling during cardiopoesis. Stem Cells 2013, 31, 2279–2285. [Google Scholar] [CrossRef] [PubMed]
- Davidson, M.M.; Nesti, C.; Palenzuela, L.; Walker, W.F.; Hernandez, E.; Protas, L.; Hirano, M.; Isaac, N.D. Novel cell lines derived from adult human ventricular cardiomyocytes. J. Mol. Cell. Cardiol. 2005, 39, 133–147. [Google Scholar] [CrossRef]
- Ren, J.; Li, X.; Dong, H.; Suo, L.; Zhang, J.; Zhang, L.; Zhang, J. miR-210-3p regulates the proliferation and apoptosis of non-small cell lung cancer cells by targeting SIN3A. Exp. Ther. Med. 2019, 18, 2565–2573. [Google Scholar] [CrossRef]
- Yang, W.; Sun, T.; Cao, J.; Liu, F.; Tian, Y.; Zhu, W. Downregulation of miR-210 expression inhibits proliferation, induces apoptosis and enhances radiosensitivity in hypoxic human hepatoma cells in vitro. Exp. Cell. Res. 2012, 318, 944–954. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Zhou, B. Cardiomyocyte proliferation: Remove brakes and push accelerators. Cell Res. 2017, 27, 959–960. [Google Scholar] [CrossRef] [Green Version]
- Rubart, M.; Field, L.J. CARDIAC REGENERATION: Repopulating the Heart. Annu. Rev. Physiol. 2006, 68, 29–49. [Google Scholar] [CrossRef] [PubMed]
- Ellison, G.M.; Vicinanza, C.; Smith, A.J.; Aquila, I.; Leone, A.; Waring, C.D.; Henning, B.J.; Stirparo, G.G.; Papait, R.; Scarfò, M.; et al. Adult c-kitpos Cardiac Stem Cells Are Necessary and Sufficient for Functional Cardiac Regeneration and Repair. Cell 2013, 154, 827–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergmann, O.; Bhardwaj, R.D.; Bernard, S.; Zdunek, S.; Barnabé-Heider, F.; Walsh, S.; Zupicich, J.; Alkass, K.; Buchholz, B.A.; Druid, H.; et al. Evidence for Cardiomyocyte Renewal in Humans. Science 2009, 324, 98–102. [Google Scholar] [CrossRef] [Green Version]
- Kikuchi, K.; Holdway, J.E.; Werdich, A.A.; Anderson, R.M.; Fang, Y.; Egnaczyk, G.F.; Evans, T.; MacRae, C.A.; Stainier, D.Y.R.; Poss, K.D. Primary contribution to zebrafish heart regeneration by gata4+ cardiomyocytes. Nature 2010, 464, 601–605. [Google Scholar] [CrossRef]
- Porrello, E.R.; Mahmoud, A.I.; Simpson, E.; Hill, J.A.; Richardson, J.A.; Olson, E.N.; Sadek, H.A. Transient Regenerative Potential of the Neonatal Mouse Heart. Science 2011, 331, 1078–1080. [Google Scholar] [CrossRef] [PubMed] [Green Version]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Control Empty Vector (pEZX-MR04-Scrambled) | miR-210 Overexpression (OE) Vector (pEZX-MR04-miR-210) | Control Empty Vector (pEZX-AM01- Scrambled) | miR-210 Decoy/Inhibition (KD) Vector (pEZX-AM01-miR-210) | |
|---|---|---|---|---|
| Normoxia, 18 h | n = 4 | n = 4 | n = 4 | n = 4 |
| Hypoxia, 18 h | n = 4 | n = 4 | n = 4 | n = 4 |
| Normoxia (18 + 8 h) | n = 4 | n = 4 | n = 4 | n = 4 |
| Hypoxia (18 h) + Reoxygenation (8 h) | n = 4 | n = 4 | n = 4 | n = 4 |
| Antibody | Application | Amount | Host | Manufacturer | Catalogue | Resource Identifier ID (RRID) |
|---|---|---|---|---|---|---|
| β-Actin | WB 1:5000 | 1 µg | Mouse | Santa Cruz Biotechnology | sc-47778 | AB_2714189 |
| APAF1 | IP | 5 µg | Rabbit | Novus Biologicals | NBP1-77000 | AB_11008194 |
| APAF1 | ELISA capture | 50 ng/well | Mouse | Santa Cruz Biotechnology | sc-65891 | AB_1119006 |
| APAF1 | ELISA detection | 50 ng/well | Rabbit | Novus Biologicals | NBP1-77000 | AB_11008194 |
| c-FLIP | ELISA capture | 50 ng/well | Mouse | Santa Cruz Biotechnology | sc-5276 | AB_627764 |
| c-FLIP | ELISA detection | 50 ng/well | Rabbit | Novus Biologicals | NBP1-77016 | AB_11024867 |
| Cytochrome c | WB 1:1000 | 5 µg | Mouse | Thermo Fisher | BMS1037 | AB_10598651 |
| Cytochrome c | ELISA capture | 20 ng/well | Mouse | Thermo Fisher | BMS1037 | AB_10598651 |
| Cytochrome c | ELISA detection | 20 ng/well | Rabbit | Cell Signaling Technology | 4280 | AB_10695410 |
| Procaspase-8/Caspase-8 | ELISA capture | 40 ng/well | Mouse | Cell Signaling Technology | 9746 | AB_2275120 |
| Procaspase-8/Caspase-8 | ELISA detection | 40 ng/well | Rabbit | Novus Biologicals | NBP1-76610 | AB_11034997 |
| Procaspase-8/Caspase-8 | WB 1:1000 | 5 µg | Mouse | Cell Signaling Technology | 9746 | AB_2275120 |
| Procaspase-9/Caspase-9 | IP | 5 µg | Rabbit | Cell Signaling Technology | 9502 | AB_2068621 |
| Procaspase-9/Caspase-9 | ELISA capture | 40 ng/well | Mouse | Cell Signaling Technology | 9508 | AB_2068620 |
| Procaspase-9/Caspase-9 | ELISA detection | 40 ng/well | Rabbit | Novus Biologicals | NBP1-76961 | AB_11034844 |
| FADD | IP | 5 µg | Mouse | Sigma Aldrich/Merck Life Science | F8053 | AB_476989 |
| FADD | ELISA capture | 30 ng/well | Rabbit | Cell Signaling Technology | 2782 | AB_2100484 |
| FADD | ELISA detection | 30 ng/well | Mouse | BioVision/VWR | 3039-100/ 10005-490 | AB_2100612 |
| Goat Anti-Mouse IgG (H + L)-HRP Conjugate | 1:5000 | 1 µg | Goat | Bio-Rad | 1706516 | AB_11125547 |
| Goat Anti-Mouse IgG-AP Conjugate | 1:5000 | N/A € | Goat | Bio-Rad | 1706520 | AB_11125348 |
| Goat Anti-Rabbit IgG (H + L)-HRP Conjugate | 1:5000 | 1 µg | Goat | Bio-Rad | 1706515 | AB_11125142 |
| Goat Anti-Rabbit IgG-AP Conjugate | 1:20,000 | N/A € | Goat | Sigma Aldrich/Merck Life Science | A3687 | AB_258103 |
| LDH | ELISA capture | 30 ng/well | Mouse | Santa Cruz Biotechnology | sc-133123 | AB_2134964 |
| LDH-A | ELISA detection | 30 ng/well | Rabbit | Novus Biologicals | NBP1-48336 | AB_10011099 |
| LDH-B | ELISA detection | 30 ng/well | Rabbit | Novus Biologicals | NBP2-38131 | N/A |
| Mouse IgG | IP | 5 µg | Mouse | Santa Cruz Biotechnology | sc-2025 | AB_737182 |
| Rabbit IgG | IP | 5-10 µg | Rabbit | Santa Cruz Biotechnology | sc-3888 | AB_737196 |
| RIPK1 | IP | 10 µg | Rabbit | Cell Signaling Technology | 3493 | AB_2305314 |
| RIPK1 | ELISA capture | 5 µg | Mouse | Santa Cruz Biotechnology | sc-133102 | AB_1568814 |
| RIPK1 | ELISA detection | 50 ng/well | Rabbit | Sigma Aldrich/Merck Life Science | SAB3500420 | AB_10643987 |
| TOM20 | 1:5000 | 1 µg | Mouse | Thermo Fisher Scientific | MA5-34964 | AB_2848869 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marwarha, G.; Røsand, Ø.; Scrimgeour, N.; Slagsvold, K.H.; Høydal, M.A. miR-210 Regulates Apoptotic Cell Death during Cellular Hypoxia and Reoxygenation in a Diametrically Opposite Manner. Biomedicines 2022, 10, 42. https://doi.org/10.3390/biomedicines10010042
Marwarha G, Røsand Ø, Scrimgeour N, Slagsvold KH, Høydal MA. miR-210 Regulates Apoptotic Cell Death during Cellular Hypoxia and Reoxygenation in a Diametrically Opposite Manner. Biomedicines. 2022; 10(1):42. https://doi.org/10.3390/biomedicines10010042
Chicago/Turabian StyleMarwarha, Gurdeep, Øystein Røsand, Nathan Scrimgeour, Katrine Hordnes Slagsvold, and Morten Andre Høydal. 2022. "miR-210 Regulates Apoptotic Cell Death during Cellular Hypoxia and Reoxygenation in a Diametrically Opposite Manner" Biomedicines 10, no. 1: 42. https://doi.org/10.3390/biomedicines10010042
APA StyleMarwarha, G., Røsand, Ø., Scrimgeour, N., Slagsvold, K. H., & Høydal, M. A. (2022). miR-210 Regulates Apoptotic Cell Death during Cellular Hypoxia and Reoxygenation in a Diametrically Opposite Manner. Biomedicines, 10(1), 42. https://doi.org/10.3390/biomedicines10010042