
Neurogenic Inflammation: The Participant in Migraine and Recent Advancements in Translational Research





Abstract
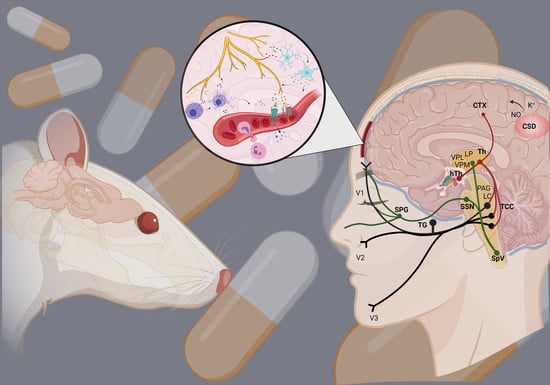
:
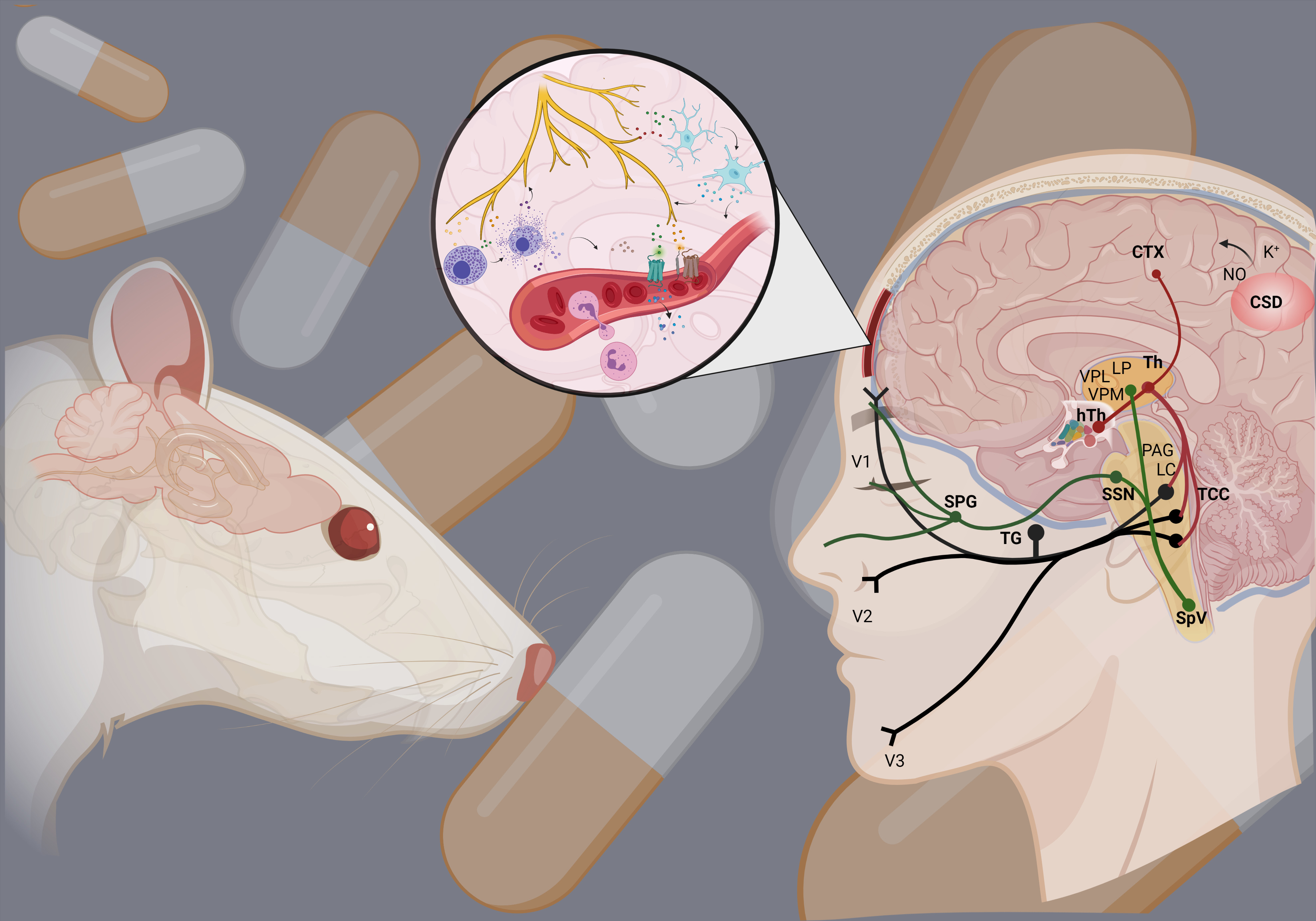
1. Introduction
2. Dura Mater in Migraine
3. Neuropeptides and Neurotransmitters
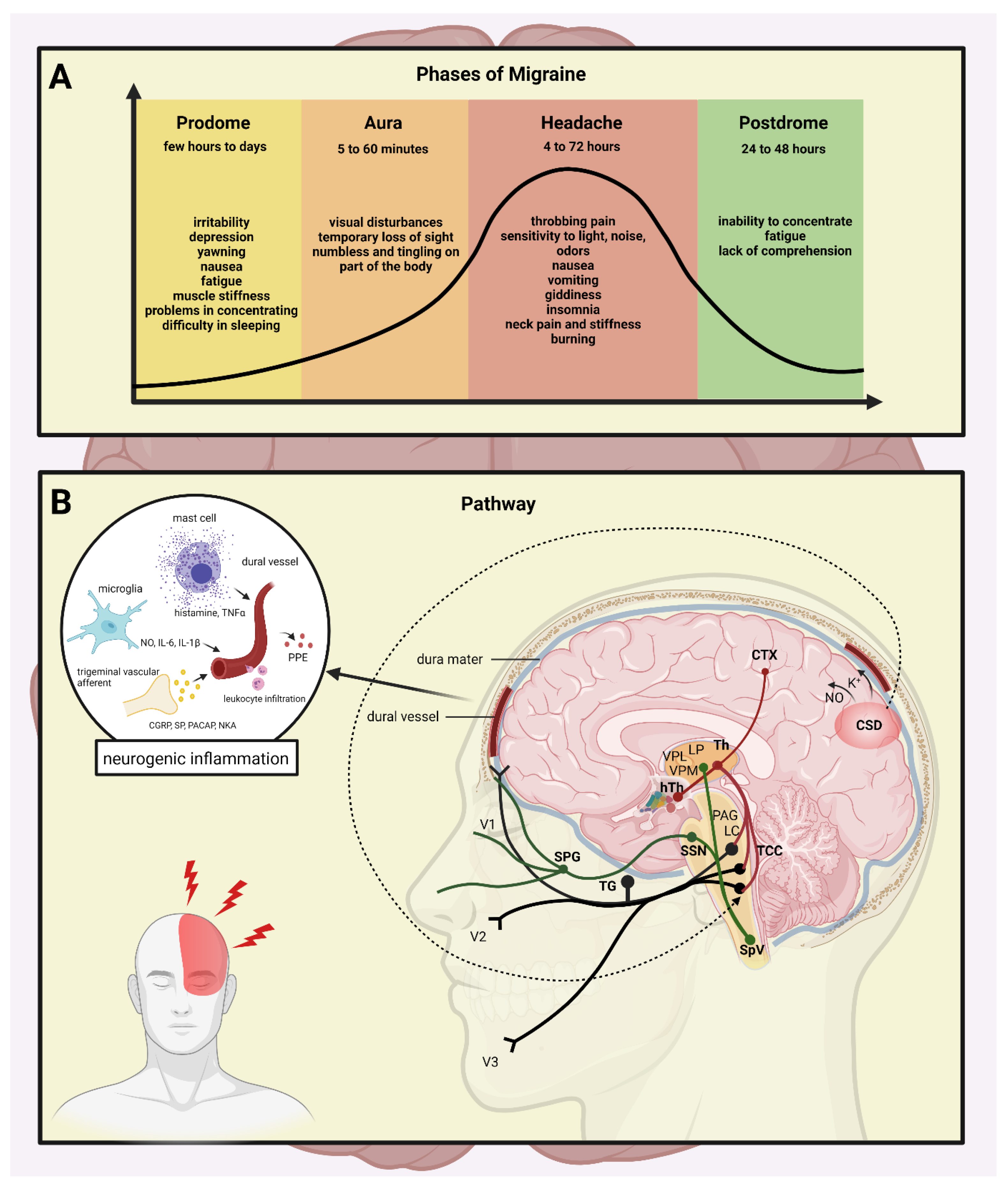
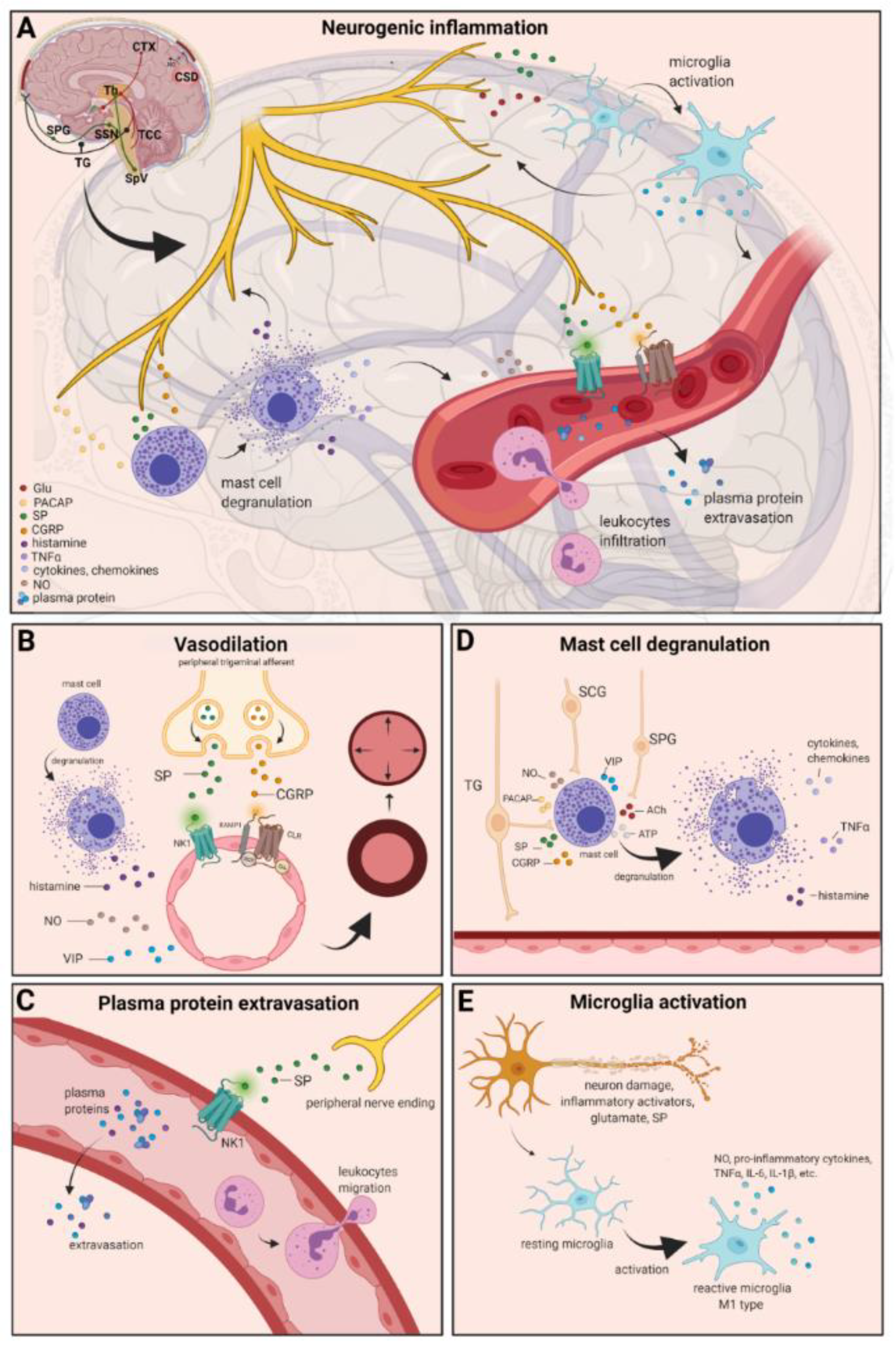
4. Neurogenic Inflammation
4.1. Vasodilation
4.2. Plasma Protein Extravasation
4.3. Mast Cell Degranulation
4.4. Microglia Activation
4.5. Cytokines, Chemokines
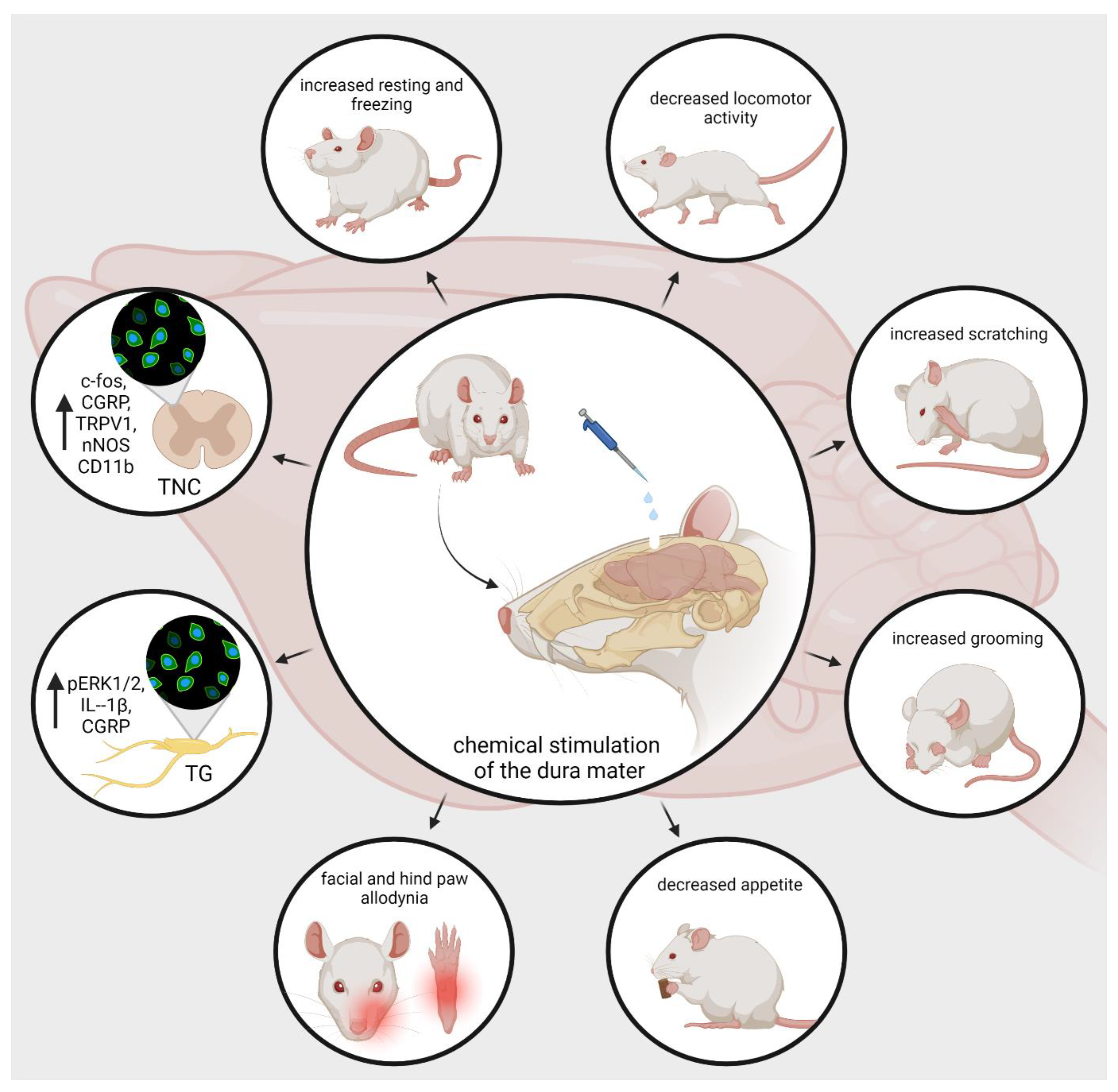
5. Animal Models of Neurogenic Inflammation
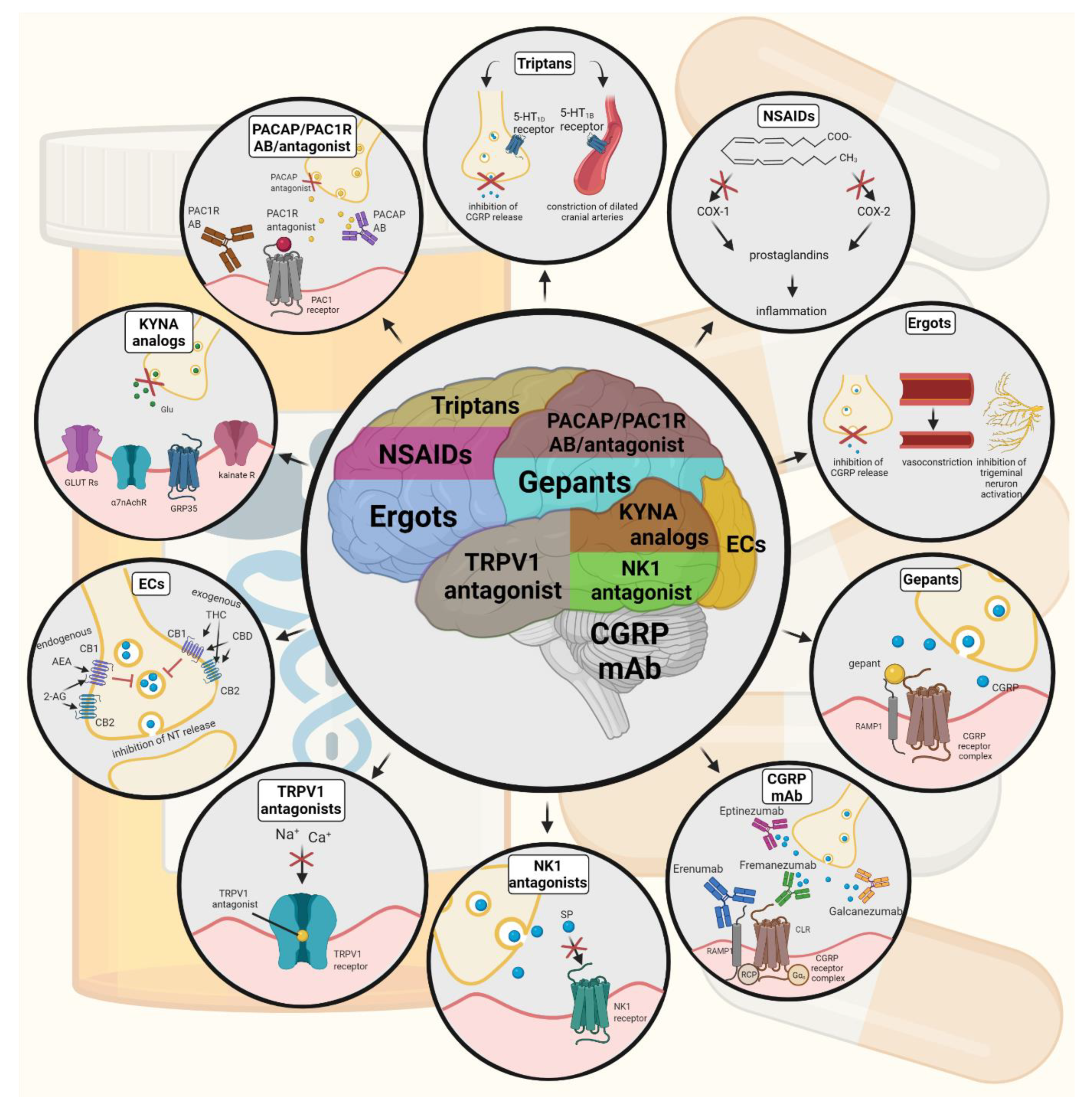
6. Current Treatments and Advances in Preclinical Research
7. Conclusions and Future Perspective
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| α7AchR | alpha-7 nicotinic receptor |
| 2-AG | 2-arachidonoylglycerol |
| AEA | anandamide |
| ATP | adenosine triphosphate |
| cAMP | cyclic adenosine monophosphate |
| CB1 | cannabinoid receptor type 1 |
| CBD | cannabidiol |
| CFA | Complete Freund’s adjuvant |
| CGRP | calcitonin gene-related peptide |
| CNS | central nervous system |
| CSD | cortical spreading depression |
| CSF | cerebrospinal fluid |
| DR | nucleus raphe dorsalis |
| DRG | dorsal root ganglion |
| GLUT R | glutamate receptors |
| GPR35 | G protein-coupled receptor 35 |
| 5-HT1B/1D | 5-hydroxytryptamine receptors |
| IDO | indoleamine 2,3-dioxygenase |
| IS | inflammatory soup |
| KMO | kynurenine 3-monooxygenase |
| KP | tryptophan-kynurenine metabolic pathway |
| KYNA | kynurenic acid |
| KYNU | kynureninase |
| LC | locus coeruleus |
| miRs | microRNAs |
| NGF | nerve-growth factor |
| NI | neurogenic inflammation |
| NK1 | neurokinin 1 |
| NKA | neurokinin A |
| nNOS | neuronal nitric oxide synthase |
| NO | nitric oxide |
| NPY | neuropeptide Y |
| NRM | nucleus raphe magnus |
| NSAIDs | nonsteroidal anti-inflammatory agents |
| NT | neurotransmitter |
| PAC1R | pituitary adenylate cyclase 1 receptor |
| PACAP | pituitary adenylate cyclase-activating polypeptide |
| PAG | the periaqueductal grey matter |
| PEA | palmitoylethanolamide |
| pERK | phosphorylated extracellular signal-regulated kinase |
| PKA | protein kinase A |
| PKC | protein kinase C |
| PLC | phospholipase C |
| PPE | plasma protein extravasation |
| SP | substance P |
| TACR1 | tachykinin Receptor 1 |
| TG | trigeminal ganglion |
| THC | tetrahidrokannabinol |
| TNC | caudal trigeminal nucleus |
| TNFα | tumor necrosis factor alpha |
| TRPV1 | transient receptor potential vanilloid-1 receptor |
| TS | trigeminal system |
| VIP | vasoactive intestinal peptide |
| VPAC1/2 | vasoactive intestinal peptide receptor 1/2 |
References
- Steiner, T.J.; Stovner, L.J.; Vos, T. GBD 2015: Migraine is the third cause of disability in under 50s. J. Headache Pain 2016, 17, 104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- GBD 2016 Disease and Injury Incidence and Prevalence Collaborators. Global, regional, and national incidence, prevalence, and years lived with disability for 328 diseases and injuries for 195 countries, 1990-2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet 2017, 390, 1211–1259. [Google Scholar] [CrossRef] [Green Version]
- Gazerani, P.; Cairns, B.E. Sex-Specific Pharmacotherapy for Migraine: A Narrative Review. Front. Neurosci. 2020, 14, 222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerbo, R.; Pesare, M.; Aurilia, C.; Rondelli, V.; Barbanti, P. Socio–economic costs of migraine. J. Headache Pain 2001, 2, s15–s19. [Google Scholar] [CrossRef] [Green Version]
- Mayans, L.; Walling, A. Acute Migraine Headache: Treatment Strategies. Am. Fam. Physician 2018, 97, 243–251. [Google Scholar]
- Headache Classification Committee of the International Headache Society (IHS). The International Classification of Headache Disorders, 3rd edition (beta version). Cephalalgia. 2013, 33, 629–808. [Google Scholar] [CrossRef] [Green Version]
- Headache Classification Subcommittee of the International Headache Society. The International Classification of Headache Disorders: 2nd edition. Cephalalgia 2004, 24 (Suppl. S1), 9–160. [Google Scholar] [CrossRef]
- Giffin, N.J.; Ruggiero, L.; Lipton, R.B.; Silberstein, S.D.; Tvedskov, J.F.; Olesen, J.; Altman, J.; Goadsby, P.J.; Macrae, A. Premonitory symptoms in migraine: An electronic diary study. Neurology 2003, 60, 935–940. [Google Scholar] [CrossRef]
- Lai, J.; Dilli, E. Migraine Aura: Updates in Pathophysiology and Management. Curr. Neurol. Neurosci. Rep. 2020, 20, 17. [Google Scholar] [CrossRef] [PubMed]
- van Dongen, R.M.; Haan, J. Symptoms related to the visual system in migraine. F1000Research 2019, 8, F1000. [Google Scholar] [CrossRef]
- Pescador Ruschel, M.A.; De Jesus, O. Migraine Headache. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA. Available online: https://www.ncbi.nlm.nih.gov/books/NBK560787/ (accessed on 27 October 2021).
- Chen, P.K.; Wang, S.J. Non-headache symptoms in migraine patients. F1000Research 2018, 7, 188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goadsby, P.J.; Holland, P.R.; Martins-Oliveira, M.; Hoffmann, J.; Schankin, C.; Akerman, S. Pathophysiology of Migraine: A Disorder of Sensory Processing. Physiol. Rev. 2017, 97, 553–622. [Google Scholar] [CrossRef] [PubMed]
- Edvinsson, L. Tracing neural connections to pain pathways with relevance to primary headaches. Cephalalgia 2011, 31, 737–747. [Google Scholar] [CrossRef]
- Fusco, B.M.; Barzoi, G.; Agrò, F. Repeated intranasal capsaicin applications to treat chronic migraine. Br. J. Anaesth 2003, 90, 812. [Google Scholar] [CrossRef] [Green Version]
- Cross, S.A. Pathophysiology of pain. Mayo. Clin. Proc. 1994, 69, 375–383. [Google Scholar] [CrossRef]
- Matharu, M.S.; Bartsch, T.; Ward, N.; Frackowiak, R.S.; Weiner, R.; Goadsby, P.J. Central neuromodulation in chronic migraine patients with suboccipital stimulators: A PET study. Brain 2004, 127 Pt 1, 220–230. [Google Scholar] [CrossRef] [Green Version]
- Vincent, M.; Hadjikhani, N. The cerebellum and migraine. Headache 2007, 47, 820–833. [Google Scholar] [CrossRef] [Green Version]
- Ray, B.S.; Wolff, H.G. Experimental studies on headache: Pain sensitive structures of the head and their significance in headache. Headache Arch. Surg. 1940, 41, 813–856. [Google Scholar] [CrossRef]
- Tunis, M.M.; Wolff, H.G. Studies on headache; long-term observations of the reactivity of the cranial arteries in subjects with vascular headache of the migraine type. AMA Arch. Neurol. Psychiatry 1953, 70, 551–557. [Google Scholar] [CrossRef]
- Leao, A.A.P.; Morison, R.S. Propagation of spreading cortical depression. J. Neurophysiol. 1945, 8, 33–45. [Google Scholar] [CrossRef]
- Ayata, C. Cortical spreading depression triggers migraine attack: Pro. Headache 2010, 50, 725–730. [Google Scholar] [CrossRef]
- Lauritzen, M. Pathophysiology of the migraine aura. The spreading depression theory. Brain 1994, 117, 199–210. [Google Scholar] [CrossRef]
- Olesen, J.; Goadsby, P.J. Synthesis of migraine mechanisms. In The Headaches, 2nd ed.; Olesen, J., Tfelt-Hansen, P., Welch, K.M.A., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2000; pp. 331–336. [Google Scholar]
- Moskowitz, M.A. Defining a pathway to discovery from bench to bedside: The trigeminovascular system and sensitization. Headache 2008, 48, 688–690. [Google Scholar] [CrossRef] [PubMed]
- Weiller, C.; May, A.; Limmroth, V.; Jüptner, M.; Kaube, H.; Schayck, R.V.; Coenen, H.H.; Diener, H.C. Brain stem activation in spontaneous human migraine attacks. Nat. Med. 1995, 1, 658–660. [Google Scholar] [CrossRef]
- Edvinsson, L.; Goadsby, P.J. Neuropeptides in the Cerebral Circulation: Relevance to Headache. Cephalalgia 1995, 15, 272–276. [Google Scholar] [CrossRef] [PubMed]
- Akerman, S.; Holland, P.R.; Goadsby, P.J. Diencephalic and brainstem mechanisms in migraine. Nat. Rev. Neurosci. 2011, 12, 570–584. [Google Scholar] [CrossRef]
- Fleetwood-Walker, S.M.; Hope, P.J.; Mitchell, R. Antinociceptive actions of descending dopaminergic tracts on cat and rat dorsal horn somatosensory neurones. J. Physiol. 1988, 399, 335–348. [Google Scholar] [CrossRef]
- Settle, M. The hypothalamus. Neonatal Netw. 2000, 19, 9–14. [Google Scholar] [CrossRef]
- Witten, A.; Marotta, D.; Cohen-Gadol, A. Developmental innervation of cranial dura mater and migraine headache: A narrative literature review. Headache 2021, 61, 569–575. [Google Scholar] [CrossRef] [PubMed]
- Rozen, T.; Swidan, S.Z. Elevation of CSF tumor necrosis factor alpha levels in new daily persistent headache and treatment refractory chronic migraine. Headache 2007, 47, 1050–1055. [Google Scholar] [CrossRef]
- Dowgjallo, N. Über die Nerven der harten Hirnhaut des Menschen und einiger Säuger. Z. Anat. Entwickl. Gesch. 1929, 89, 453–466. [Google Scholar] [CrossRef]
- Grzybowski, J. L’innervation de la dure-mére cranienne chez l’homme. Arch. Anat. Histol. Embryol. 1931, 14, 387–428. [Google Scholar]
- Penfield, W.; McNaughton, F. Dural headache and innervation of the dura mater. Arch. NeurPsych. 1940, 44, 43–75. [Google Scholar] [CrossRef]
- Steiger, H.J.; Meakin, C.J. The meningeal representation in the trigeminal ganglion--an experimental study in the cat. Headache 1984, 24, 305–309. [Google Scholar] [CrossRef] [PubMed]
- Uddman, R.; Hara, H.; Edvinsson, L. Neuronal pathways to the rat middle meningeal artery revealed by retrograde tracing and immunocytochemistry. J. Auton. Nerv. Syst. 1989, 26, 69–75. [Google Scholar] [CrossRef]
- Strassman, A.M.; Raymond, S.A.; Burstein, R. Sensitization of meningeal sensory neurons and the origin of headaches. Nature 1996, 384, 560–564. [Google Scholar] [CrossRef] [PubMed]
- Burstein, R.; Yamamura, H.; Malick, A.; Strassman, A.M. Chemical stimulation of the intracranial dura induces enhanced responses to facial stimulation in brain stem trigeminal neurons. J. Neurophysiol. 1998, 79, 964–982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amenta, F.; Sancesario, G.; Ferrante, F.; Cavallotti, C. Acetylcholinesterase-containing nerve fibers in the dura mater of guinea pig, mouse, and rat. J. Neural. Transm. 1980, 47, 237–242. [Google Scholar] [CrossRef] [PubMed]
- Schueler, M.; Neuhuber, W.L.; De Col, R.; Messlinger, K. Innervation of rat and human dura mater and pericranial tissues in the parieto-temporal region by meningeal afferents. Headache 2014, 54, 996–1009. [Google Scholar] [CrossRef]
- Ebersberger, A.; Averbeck, B.; Messlinger, K.; Reeh, P.W. Release of substance P, calcitonin gene-related peptide and prostaglandin E2 from rat dura mater encephali following electrical and chemical stimulation in vitro. Neuroscience 1999, 89, 901–907. [Google Scholar] [CrossRef]
- Lv, X.; Wu, Z.; Li, Y. Innervation of the cerebral dura mater. Neuroradiol. J. 2014, 27, 293–298. [Google Scholar] [CrossRef] [Green Version]
- Asghar, M.S.; Hansen, A.E.; Amin, F.M.; van der Geest, R.J.; Koning, P.V.; Larsson, H.B.; Olesen, J.; Ashina, M. Evidence for a vascular factor in migraine. Ann. Neurol. 2011, 69, 635–645. [Google Scholar] [CrossRef]
- Holzer, P. Neurogenic vasodilatation and plasma leakage in the skin. Gen. Pharmacol. 1998, 30, 5–11. [Google Scholar] [CrossRef]
- Ottosson, A.; Edvinsson, L. Release of histamine from dural mast cells by substance P and calcitonin gene-related peptide. Cephalalgia 1997, 17, 166–174. [Google Scholar] [CrossRef]
- Lennerz, J.K.; Rühle, V.; Ceppa, E.P.; Neuhuber, W.L.; Bunnett, N.W.; Grady, E.F.; Messlinger, K. Calcitonin receptor-like receptor (CLR), receptor activity-modifying protein 1 (RAMP1), and calcitonin gene-related peptide (CGRP) immunoreactivity in the rat trigeminovascular system: Differences between peripheral and central CGRP receptor distribution. J. Comp. Neurol. 2008, 507, 1277–1299. [Google Scholar] [CrossRef]
- Raddant, A.C.; Russo, A.F. Calcitonin gene-related peptide in migraine: Intersection of peripheral inflammation and central modulation. Expert Rev. Mol. Med. 2011, 13, e36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hökfelt, T.; Kellerth, J.O.; Nilsson, G.; Pernow, B. Substance p: Localization in the central nervous system and in some primary sensory neurons. Science 1975, 190, 889–890. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro-da-Silva, A.; Hökfelt, T. Neuroanatomical localisation of Substance P in the CNS and sensory neurons. Neuropeptides 2000, 34, 256–271. [Google Scholar] [CrossRef] [PubMed]
- Snijdelaar, D.G.; Dirksen, R.; Slappendel, R.; Crul, B.J. Substance P. Eur. J. Pain 2000, 4, 121–135. [Google Scholar] [CrossRef] [PubMed]
- Graefe, S.; Mohiuddin, S.S. Biochemistry, Substance P. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA. Available online: https://www.ncbi.nlm.nih.gov/books/NBK554583/ (accessed on 27 October 2021).
- Killingsworth, C.R.; Shore, S.A.; Alessandrini, F.; Dey, R.D.; Paulauskis, J.D. Rat alveolar macrophages express preprotachykinin gene-I mRNA-encoding tachykinins. Am. J. Physiol. 1997, 273, L1073–L1081. [Google Scholar] [CrossRef] [PubMed]
- Weinstock, J.V.; Blum, A.; Walder, J.; Walder, R. Eosinophils from granulomas in murine schistosomiasis mansoni produce substance P. J. Immunol. 1988, 141, 961–966. [Google Scholar]
- Holzer, P.; Holzer-Petsche, U. Tachykinins in the gut. Part II. Roles in neural excitation, secretion and inflammation. Pharmacol. Ther. 1997, 73, 219–263. [Google Scholar] [CrossRef]
- Pernow, B. Substance P. Pharmacol. Rev. 1983, 35, 85–141. [Google Scholar]
- Gibbins, I.L.; Furness, J.B.; Costa, M.; MacIntyre, I.; Hillyard, C.J.; Girgis, S. Co-localization of calcitonin gene-related peptide-like immunoreactivity with substance P in cutaneous, vascular and visceral sensory neurons of guinea pigs. Neurosci. Lett. 1985, 57, 125–130. [Google Scholar] [CrossRef]
- Battaglia, G.; Rustioni, A. Coexistence of glutamate and substance P in dorsal root ganglion neurons of the rat and monkey. J. Comp. Neurol. 1988, 277, 302–312. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, R. Understanding migraine: Potential role of neurogenic inflammation. Ann. Indian Acad. Neurol. 2016, 19, 175–182. [Google Scholar] [CrossRef]
- Jansen-Olesen, I.; Hougaard Pedersen, S. PACAP and its receptors in cranial arteries and mast cells. J. Headache Pain 2018, 19, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eftekhari, S.; Warfvinge, K.; Blixt, F.W.; Edvinsson, L. Differentiation of nerve fibers storing CGRP and CGRP receptors in the peripheral trigeminovascular system. J. Pain 2013, 14, 1289–1303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nielsen, H.S.; Hannibal, J.; Fahrenkrug, J. Embryonic expression of pituitary adenylate cyclase-activating polypeptide in sensory and autonomic ganglia and in spinal cord of the rat. J. Comp. Neurol. 1998, 394, 403–415. [Google Scholar] [CrossRef]
- Jansen-Olesen, I.; Baun, M.; Amrutkar, D.V.; Ramachandran, R.; Christophersen, D.V.; Olesen, J. PACAP-38 but not VIP induces release of CGRP from trigeminal nucleus caudalis via a receptor distinct from the PAC1 receptor. Neuropeptides 2014, 48, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Uddman, R.; Tajti, J.; Hou, M.; Sundler, F.; Edvinsson, L. Neuropeptide expression in the human trigeminal nucleus caudalis and in the cervical spinal cord C1 and C2. Cephalalgia 2002, 22, 112–116. [Google Scholar] [CrossRef] [Green Version]
- Hashimoto, H.; Shintani, N.; Baba, A. New insights into the central PACAPergic system from the phenotypes in PACAP- and PACAP receptor-knockout mice. Ann. N. Y. Acad. Sci. 2006, 1070, 75–89. [Google Scholar] [CrossRef] [PubMed]
- Martin, M.; Otto, C.; Santamarta, M.T.; Torrecilla, M.; Pineda, J.; Schütz, G.; Maldonado, R. Morphine withdrawal is modified in pituitary adenylate cyclase-activating polypeptide type I-receptor-deficient mice. Brain Res. Mol. Brain Res. 2003, 110, 109–118. [Google Scholar] [CrossRef]
- Missig, G.; Roman, C.W.; Vizzard, M.A.; Braas, K.M.; Hammack, S.E.; May, V. Parabrachial nucleus (PBn) pituitary adenylate cyclase activating polypeptide (PACAP) signaling in the amygdala: Implication for the sensory and behavioral effects of pain. Neuropharmacology 2014, 86, 38–48. [Google Scholar] [CrossRef] [Green Version]
- Kaiser, E.A.; Russo, A.F. CGRP and migraine: Could PACAP play a role too? Neuropeptides 2013, 47, 451–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Danielsen, N.; Sundler, F.; Mulder, H. Pituitary adenylate cyclase-activating peptide is upregulated in sensory neurons by inflammation. Neuroreport 1998, 9, 2833–2836. [Google Scholar] [CrossRef]
- Kilinc, E.; Firat, T.; Tore, F.; Kiyan, A.; Kukner, A.; Tunçel, N. Vasoactive Intestinal peptide modulates c-Fos activity in the trigeminal nucleus and dura mater mast cells in sympathectomized rats. J. Neurosci. Res. 2015, 93, 644–650. [Google Scholar] [CrossRef]
- Ohhashi, T.; Olschowka, J.A.; Jacobowitz, D.M. Vasoactive intestinal peptide inhibitory innervation in bovine mesenteric lymphatics. A histochemical and pharmacological study. Circ. Res. 1983, 53, 535–538. [Google Scholar] [CrossRef] [Green Version]
- Kakurai, M.; Fujita, N.; Murata, S.; Furukawa, Y.; Demitsu, T.; Nakagawa, H. Vasoactive intestinal peptide regulates its receptor expression and functions of human keratinocytes via type I vasoactive intestinal peptide receptors. J. Invest. Dermatol. 2001, 116, 743–749. [Google Scholar] [CrossRef] [Green Version]
- Cernuda-Morollón, E.; Martínez-Camblor, P.; Ramón, C.; Larrosa, D.; Serrano-Pertierra, E.; Pascual, J. CGRP and VIP levels as predictors of efficacy of Onabotulinumtoxin type A in chronic migraine. Headache 2014, 54, 987–995. [Google Scholar] [CrossRef]
- Pellesi, L.; Al-Karagholi, M.A.; Chaudhry, B.A.; Lopez, C.L.; Snellman, J.; Hannibal, J.; Amin, F.M.; Ashina, M. Two-hour infusion of vasoactive intestinal polypeptide induces delayed headache and extracranial vasodilation in healthy volunteers. Cephalalgia 2020, 40, 1212–1223. [Google Scholar] [CrossRef] [PubMed]
- Caterina, M.J.; Leffler, A.; Malmberg, A.B.; Martin, W.J.; Trafton, J.; Petersen-Zeitz, K.R.; Koltzenburg, M.; Basbaum, A.I.; Julius, D. Impaired nociception and pain sensation in mice lacking the capsaicin receptor. Science 2000, 288, 306–313. [Google Scholar] [CrossRef] [PubMed]
- Quartu, M.; Serra, M.P.; Boi, M.; Poddighe, L.; Picci, C.; Demontis, R.; Del Fiacco, M. TRPV1 receptor in the human trigeminal ganglion and spinal nucleus: Immunohistochemical localization and comparison with the neuropeptides CGRP and SP. J. Anat. 2016, 229, 755–767. [Google Scholar] [CrossRef] [PubMed]
- Dux, M.; Rosta, J.; Messlinger, K. TRP Channels in the Focus of Trigeminal Nociceptor Sensitization Contributing to Primary Headaches. Int. J. Mol. Sci. 2020, 21, 342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bevan, S.; Quallo, T.; Andersson, D.A. TRPV1. Handb. Exp. Pharmacol. 2014, 222, 207–245. [Google Scholar] [CrossRef]
- Meents, J.E.; Neeb, L.; Reuter, U. TRPV1 in migraine pathophysiology. Trends Mol. Med. 2010, 16, 153–159. [Google Scholar] [CrossRef]
- Yuan, H.; Silberstein, S.D. Histamine and Migraine. Headache 2018, 58, 184–193. [Google Scholar] [CrossRef]
- Castillo, J.; Martínez, F.; Corredera, E.; Lema, M.; Noya, M. Migraña e histamina: Determinación de histidina en plasma y líquido cefalorraquídeo durante crisis de migraña [Migraine and histamine: Determining histidine in plasma and cerebrospinal fluid during migraine attacks]. Rev. Neurol. 1995, 23, 749–751. [Google Scholar]
- Heatley, R.V.; Denburg, J.A.; Bayer, N.; Bienenstock, J. Increased plasma histamine levels in migraine patients. Clin. Allergy 1982, 12, 145–149. [Google Scholar] [CrossRef]
- Foreman, J.C.; Jordan, C.C.; Oehme, P.; Renner, H. Structure-activity relationships for some substance P-related peptides that cause wheal and flare reactions in human skin. J. Physiol. 1983, 335, 449–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosa, A.C.; Fantozzi, R. The role of histamine in neurogenic inflammation. Br. J. Pharmacol. 2013, 170, 38–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goadsby, P.J.; Knight, Y.E.; Hoskin, K.L.; Butler, P. Stimulation of an intracranial trigeminally-innervated structure selectively increases cerebral blood flow. Brain Res. 1997, 751, 247–252. [Google Scholar] [CrossRef]
- Ji, R.R.; Xu, Z.Z.; Gao, Y.J. Emerging targets in neuroinflammation-driven chronic pain. Nat. Rev. Drug Discov. 2014, 13, 533–548. [Google Scholar] [CrossRef] [Green Version]
- Goltz, F. Uber gefasserweiternde nerven. Pflueger Arch. Ges. Physiol. 1874, 9, 185. [Google Scholar] [CrossRef]
- Bayliss, W.M. On the origin from the spinal cord of the vaso-dilator fibres of the hind-limb, and on the nature of these fibres. J. Physiol. 1901, 26, 173–209. [Google Scholar] [CrossRef]
- Dalessio, D.J. Vascular permeability and vasoactive substances: Their relationship to migraine. Adv. Neurol. 1974, 4, 395–401. [Google Scholar]
- Moskowitz, M.A. The neurobiology of vascular head pain. Ann. Neurol. 1984, 16, 157–168. [Google Scholar] [CrossRef]
- Ramachandran, R. Neurogenic inflammation and its role in migraine. Semin. Immunopathol. 2018, 40, 301–314. [Google Scholar] [CrossRef]
- Bolay, H.; Reuter, U.; Dunn, A.K.; Huang, Z.; Boas, D.A.; Moskowitz, M.A. Intrinsic brain activity triggers trigeminal meningeal afferents in a migraine model. Nat. Med. 2002, 8, 136–142. [Google Scholar] [CrossRef]
- Xanthos, D.N.; Sandkühler, J. Neurogenic neuroinflammation: Inflammatory CNS reactions in response to neuronal activity. Nat. Rev. Neurosci. 2014, 15, 43–53. [Google Scholar] [CrossRef]
- Tajti, J.; Szok, D.; Majláth, Z.; Tuka, B.; Csáti, A.; Vécsei, L. Migraine and neuropeptides. Neuropeptides 2015, 52, 19–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berger, R.J.; Zuccarello, M.; Keller, J.T. Nitric oxide synthase immunoreactivity in the rat dura mater. Neuroreport 1994, 5, 519–521. [Google Scholar] [CrossRef]
- Wallace, J.L. Nitric oxide as a regulator of inflammatory processes. Mem. Inst. Oswaldo Cruz 2005, 100 (Suppl. 1), 5–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellamy, J.; Bowen, E.J.; Russo, A.F.; Durham, P.L. Nitric oxide regulation of calcitonin gene-related peptide gene expression in rat trigeminal ganglia neurons. Eur. J. Neurosci. 2006, 23, 2057–2066. [Google Scholar] [CrossRef] [Green Version]
- Strecker, T.; Dux, M.; Messlinger, K. Increase in meningeal blood flow by nitric oxide--interaction with calcitonin gene-related peptide receptor and prostaglandin synthesis inhibition. Cephalalgia 2002, 22, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Leston, J.M. Anatomie fonctionnelle du nerf trijumeau [Functional anatomy of the trigeminal nerve]. Neurochirurgie 2009, 55, 99–112. (In French) [Google Scholar] [CrossRef] [PubMed]
- Pedersen, S.F.; Owsianik, G.; Nilius, B. TRP channels: An overview. Cell Calcium 2005, 38, 233–252. [Google Scholar] [CrossRef] [PubMed]
- Planells-Cases, R.; Garcìa-Sanz, N.; Morenilla-Palao, C.; Ferrer-Montiel, A. Functional aspects and mechanisms of TRPV1 involvement in neurogenic inflammation that leads to thermal hyperalgesia. Pflugers Arch. 2005, 451, 151–159. [Google Scholar] [CrossRef]
- Bhave, G.; Zhu, W.; Wang, H.; Brasier, D.J.; Oxford, G.S.; Gereau, R.W., Iv. cAMP-dependent protein kinase regulates desensitization of the capsaicin receptor (VR1) by direct phosphorylation. Neuron 2002, 35, 721–731. [Google Scholar] [CrossRef] [Green Version]
- Crandall, M.; Kwash, J.; Yu, W.; White, G. Activation of protein kinase C sensitizes human VR1 to capsaicin and to moderate decreases in pH at physiological temperatures in Xenopus oocytes. Pain 2002, 98, 109–117. [Google Scholar] [CrossRef]
- Premkumar, L.S.; Ahern, G.P. Induction of vanilloid receptor channel activity by protein kinase C. Nature 2000, 408, 985–990. [Google Scholar] [CrossRef] [PubMed]
- Vellani, V.; Mapplebeck, S.; Moriondo, A.; Davis, J.B.; McNaughton, P.A. Protein kinase C activation potentiates gating of the vanilloid receptor VR1 by capsaicin, protons, heat and anandamide. J. Physiol. 2001, 534 Pt 3, 813–825. [Google Scholar] [CrossRef]
- Vergnolle, N.; Cenac, N.; Altier, C.; Cellars, L.; Chapman, K.; Zamponi, G.W.; Materazzi, S.; Nassini, R.; Liedtke, W.; Cattaruzza, F.; et al. A role for transient receptor potential vanilloid 4 in tonicity-induced neurogenic inflammation. Br. J. Pharmacol. 2010, 159, 1161–1173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perini, F.; D’Andrea, G.; Galloni, E.; Pignatelli, F.; Billo, G.; Alba, S.; Bussone, G.; Toso, V. Plasma cytokine levels in migraineurs and controls. Headache 2005, 45, 926–931. [Google Scholar] [CrossRef]
- Sarchielli, P.; Alberti, A.; Baldi, A.; Coppola, F.; Rossi, C.; Pierguidi, L.; Floridi, A.; Calabresi, P. Proinflammatory cytokines, adhesion molecules, and lymphocyte integrin expression in the internal jugular blood of migraine patients without aura assessed ictally. Headache 2006, 46, 200–207. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, B.; Dussor, G. Neurovascular contributions to migraine: Moving beyond vasodilation. Neuroscience 2016, 338, 130–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breier, G.; Risau, W. The role of vascular endothelial growth factor in blood vessel formation. Trends Cell Biol. 1996, 6, 454–456. [Google Scholar] [CrossRef]
- Palmer, R.M.; Ashton, D.S.; Moncada, S. Vascular endothelial cells synthesize nitric oxide from L-arginine. Nature 1988, 333, 664–666. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Sozzani, S.; Introna, M. Endothelial activation by cytokines. Ann. N. Y. Acad. Sci. 1997, 832, 93–116. [Google Scholar] [CrossRef]
- Yonehara, N.; Yoshimura, M. Effect of nitric oxide on substance P release from the peripheral endings of primary afferent neurons. Neurosci. Lett. 1999, 271, 199–201. [Google Scholar] [CrossRef]
- Price, T.J.; Louria, M.D.; Candelario-Soto, D.; Dussor, G.O.; Jeske, N.A.; Patwardhan, A.M.; Diogenes, A.; Trott, A.A.; Hargreaves, K.M.; Flores, C.M. Treatment of trigeminal ganglion neurons in vitro with NGF, GDNF or BDNF: Effects on neuronal survival, neurochemical properties and TRPV1-mediated neuropeptide secretion. BMC Neurosci. 2005, 6, 4. [Google Scholar] [CrossRef] [Green Version]
- Miyoshi, H.; Nakaya, Y. Calcitonin gene-related peptide activates the K+ channels of vascular smooth muscle cells via adenylate cyclase. Basic Res. Cardiol. 1995, 90, 332–336. [Google Scholar] [CrossRef] [PubMed]
- Hanko, J.; Hardebo, J.E.; Kåhrström, J.; Owman, C.; Sundler, F. Calcitonin gene-related peptide is present in mammalian cerebrovascular nerve fibres and dilates pial and peripheral arteries. Neurosci. Lett. 1985, 57, 91–95. [Google Scholar] [CrossRef]
- Wilkins, B.W.; Chung, L.H.; Tublitz, N.J.; Wong, B.J.; Minson, C.T. Mechanisms of vasoactive intestinal peptide-mediated vasodilation in human skin. J. Appl. Physiol. (1985) 2004, 97, 1291–1298. [Google Scholar] [CrossRef] [Green Version]
- Williamson, D.J.; Hargreaves, R.J. Neurogenic inflammation in the context of migraine. Microsc. Res. Tech. 2001, 53, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Markowitz, S.; Saito, K.; Moskowitz, M.A. Neurogenically mediated plasma extravasation in dura mater: Effect of ergot alkaloids. A possible mechanism of action in vascular headache. Cephalalgia 1988, 8, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Lundy, F.T.; Linden, G.J. Neuropeptides and neurogenic mechanisms in oral and periodontal inflammation. Crit. Rev. Oral Biol. Med. 2004, 15, 82–98. [Google Scholar] [CrossRef]
- De Swert, K.O.; Joos, G.F. Extending the understanding of sensory neuropeptides. Eur. J. Pharmacol. 2006, 533, 171–181. [Google Scholar] [CrossRef]
- Koyuncu Irmak, D.; Kilinc, E.; Tore, F. Shared Fate of Meningeal Mast Cells and Sensory Neurons in Migraine. Front. Cell Neurosci. 2019, 13, 136. [Google Scholar] [CrossRef] [Green Version]
- Theoharides, T.C.; Spanos, C.; Pang, X.; Alferes, L.; Ligris, K.; Letourneau, R.; Rozniecki, J.J.; Webster, E.; Chrousos, G.P. Stress-induced intracranial mast cell degranulation: A corticotropin-releasing hormone-mediated effect. Endocrinology 1995, 136, 5745–5750. [Google Scholar] [CrossRef]
- Rozniecki, J.J.; Dimitriadou, V.; Lambracht-Hall, M.; Pang, X.; Theoharides, T.C. Morphological and functional demonstration of rat dura mater mast cell-neuron interactions in vitro and in vivo. Brain Res. 1999, 849, 1–15. [Google Scholar] [CrossRef]
- Theoharides, T.C.; Donelan, J.; Kandere-Grzybowska, K.; Konstantinidou, A. The role of mast cells in migraine pathophysiology. Brain Res. Brain Res. Rev. 2005, 49, 65–76. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, L.B. Mediators of human mast cells and human mast cell subsets. Ann. Allergy 1987, 58, 226–235. [Google Scholar]
- Aich, A.; Afrin, L.B.; Gupta, K. Mast Cell-Mediated Mechanisms of Nociception. Int. J. Mol. Sci. 2015, 16, 29069–29092. [Google Scholar] [CrossRef]
- Theoharides, T.C.; Alysandratos, K.D.; Angelidou, A.; Delivanis, D.A.; Sismanopoulos, N.; Zhang, B.; Asadi, S.; Vasiadi, M.; Weng, Z.; Miniati, A.; et al. Mast cells and inflammation. Biochim. Biophys. Acta. 2012, 1822, 21–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karatas, H.; Erdener, S.E.; Gursoy-Ozdemir, Y.; Lule, S.; Eren-Koçak, E.; Sen, Z.D.; Dalkara, T. Spreading depression triggers headache by activating neuronal Panx1 channels. Science 2013, 339, 1092–1095. [Google Scholar] [CrossRef]
- Zhao, J.; Levy, D. Modulation of intracranial meningeal nociceptor activity by cortical spreading depression: A reassessment. J. Neurophysiol. 2015, 113, 2778–2785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kempuraj, D.; Selvakumar, G.P.; Thangavel, R.; Ahmed, M.E.; Zaheer, S.; Raikwar, S.P.; Iyer, S.S.; Bhagavan, S.M.; Beladakere-Ramaswamy, S.; Zaheer, A. Mast Cell Activation in Brain Injury, Stress, and Post-traumatic Stress Disorder and Alzheimer’s Disease Pathogenesis. Front. Neurosci. 2017, 11, 703. [Google Scholar] [CrossRef] [Green Version]
- Baldwin, A.L. Mast cell activation by stress. Methods Mol. Biol. 2006, 315, 349–360. [Google Scholar] [CrossRef] [PubMed]
- Sauro, K.M.; Becker, W.J. The stress and migraine interaction. Headache 2009, 49, 1378–1386. [Google Scholar] [CrossRef] [PubMed]
- Radat, F. Stress et migraine [Stress and migraine]. Rev. Neurol (Paris) 2013, 169, 406–412. [Google Scholar] [CrossRef]
- Davalos, D.; Grutzendler, J.; Yang, G.; Kim, J.V.; Zuo, Y.; Jung, S.; Littman, D.R.; Dustin, M.L.; Gan, W.B. ATP mediates rapid microglial response to local brain injury in vivo. Nat. Neurosci. 2005, 8, 752–758. [Google Scholar] [CrossRef]
- Hanisch, U.K.; Kettenmann, H. Microglia: Active sensor and versatile effector cells in the normal and pathologic brain. Nat. Neurosci. 2007, 10, 1387–1394. [Google Scholar] [CrossRef]
- Colton, C.A.; Gilbert, D.L. Production of superoxide anions by a CNS macrophage, the microglia. FEBS Lett. 1987, 223, 284–288. [Google Scholar] [CrossRef] [Green Version]
- de Vries, H.E.; Blom-Roosemalen, M.C.; van Oosten, M.; de Boer, A.G.; van Berkel, T.J.; Breimer, D.D.; Kuiper, J. The influence of cytokines on the integrity of the blood-brain barrier in vitro. J. Neuroimmunol. 1996, 64, 37–43. [Google Scholar] [CrossRef]
- Färber, K.; Kettenmann, H. Physiology of microglial cells. Brain Res. Brain Res. Rev. 2005, 48, 133–143. [Google Scholar] [CrossRef]
- Pannasch, U.; Färber, K.; Nolte, C.; Blonski, M.; Yan Chiu, S.; Messing, A.; Kettenmann, H. The potassium channels Kv1.5 and Kv1.3 modulate distinct functions of microglia. Mol. Cell Neurosci. 2006, 33, 401–411. [Google Scholar] [CrossRef] [PubMed]
- Pocock, J.M.; Kettenmann, H. Neurotransmitter receptors on microglia. Trends Neurosci. 2007, 30, 527–535. [Google Scholar] [CrossRef] [PubMed]
- Taylor, D.L.; Jones, F.; Kubota, E.S.; Pocock, J.M. Stimulation of microglial metabotropic glutamate receptor mGlu2 triggers tumor necrosis factor alpha-induced neurotoxicity in concert with microglial-derived Fas ligand. J. Neurosci. 2005, 25, 2952–2964. [Google Scholar] [CrossRef] [Green Version]
- Biber, K.; Neumann, H.; Inoue, K.; Boddeke, H.W. Neuronal ‘On’ and ‘Off’ signals control microglia. Trends Neurosci. 2007, 30, 596–602. [Google Scholar] [CrossRef]
- Koyama, Y. Endothelin ETB Receptor-Mediated Astrocytic Activation: Pathological Roles in Brain Disorders. Int. J. Mol. Sci. 2021, 22, 4333. [Google Scholar] [CrossRef] [PubMed]
- Fiebich, B.L.; Schleicher, S.; Butcher, R.D.; Craig, A.; Lieb, K. The neuropeptide substance P activates p38 mitogen-activated protein kinase resulting in IL-6 expression independently from NF-kappa B. J. Immunol. 2000, 165, 5606–5611. [Google Scholar] [CrossRef] [Green Version]
- Lin, R.C. Reactive astrocytes express substance-P immunoreactivity in the adult forebrain after injury. Neuroreport 1995, 7, 310–312. [Google Scholar] [CrossRef]
- Carthew, H.L.; Ziebell, J.M.; Vink, R. Substance P-induced changes in cell genesis following diffuse traumatic brain injury. Neuroscience 2012, 214, 78–83. [Google Scholar] [CrossRef]
- Bruno, P.P.; Carpino, F.; Carpino, G.; Zicari, A. An overview on immune system and migraine. Eur. Rev. Med. Pharmacol. Sci. 2007, 11, 245–248. [Google Scholar]
- Turner, M.D.; Nedjai, B.; Hurst, T.; Pennington, D.J. Cytokines and chemokines: At the crossroads of cell signalling and inflammatory disease. Biochim. Biophys. Acta. 2014, 1843, 2563–2582. [Google Scholar] [CrossRef] [Green Version]
- Edvinsson, L.; Haanes, K.A.; Warfvinge, K. Does inflammation have a role in migraine? Nat. Rev. Neurol. 2019, 15, 483–490. [Google Scholar] [CrossRef]
- Roach, D.R.; Bean, A.G.; Demangel, C.; France, M.P.; Briscoe, H.; Britton, W.J. TNF regulates chemokine induction essential for cell recruitment, granuloma formation, and clearance of mycobacterial infection. J. Immunol. 2002, 168, 4620–4627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conti, P.; D’Ovidio, C.; Conti, C.; Gallenga, C.E.; Lauritano, D.; Caraffa, A.; Kritas, S.K.; Ronconi, G. Progression in migraine: Role of mast cells and pro-inflammatory and anti-inflammatory cytokines. Eur. J. Pharmacol. 2019, 844, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Empl, M.; Sostak, P.; Riedel, M.; Schwarz, M.; Müller, N.; Förderreuther, S.; Straube, A. Decreased sTNF-RI in migraine patients? Cephalalgia 2003, 23, 55–58. [Google Scholar] [CrossRef] [PubMed]
- Mueller, L.; Gupta, A.K.; Stein, T.P. Deficiency of tumor necrosis factor alpha in a subclass of menstrual migraineurs. Headache 2001, 41, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Christopherson, K., II; Hromas, R. Chemokine regulation of normal and pathologic immune responses. Stem Cells 2001, 19, 388–396. [Google Scholar] [CrossRef] [PubMed]
- Tsay, H.J.; Liu, H.K.; Kuo, Y.H.; Chiu, C.S.; Liang, C.C.; Chung, C.W.; Chen, C.C.; Chen, Y.P.; Shiao, Y.J. EK100 and Antro-din C Improve Brain Amyloid Pathology in APP/PS1 Transgenic Mice by Promoting Microglial and Perivascular Clearance Pathways. Int. J. Mol. Sci. 2021, 22, 10413. [Google Scholar] [CrossRef] [PubMed]
- Lam, S.; Hartmann, N.; Benfeitas, R.; Zhang, C.; Arif, M.; Turkez, H.; Uhlén, M.; Englert, C.; Knight, R.; Mardinoglu, A. Systems Analysis Reveals Ageing-Related Perturbations in Retinoids and Sex Hormones in Alzheimer’s and Parkinson’s Diseases. Biomedicines 2021, 9, 1310. [Google Scholar] [CrossRef] [PubMed]
- Hsu, Y.L.; Hung, H.S.; Tsai, C.W.; Liu, S.P.; Chiang, Y.T.; Kuo, Y.H.; Shyu, W.C.; Lin, S.Z.; Fu, R.H. Peiminine Reduces ARTS-Mediated Degradation of XIAP by Modulating the PINK1/Parkin Pathway to Ameliorate 6-Hydroxydopamine Toxicity and α-Synuclein Accumulation in Parkinson’s Disease Models In Vivo and In Vitro. Int. J. Mol. Sci. 2021, 22, 10240. [Google Scholar] [CrossRef] [PubMed]
- Smagin, D.A.; Kovalenko, I.L.; Galyamina, A.G.; Belozertseva, I.V.; Tamkovich, N.V.; Baranov, K.O.; Kudryavtseva, N.N. Chronic Lithium Treatment Affects Anxious Behaviors and the Expression of Serotonergic Genes in Midbrain Raphe Nuclei of Defeated Male Mice. Biomedicines 2021, 9, 1293. [Google Scholar] [CrossRef]
- Bezerra, F.; Niemietz, C.; Schmidt, H.H.J.; Zibert, A.; Guo, S.; Monia, B.P.; Gonçalves, P.; Saraiva, M.J.; Almeida, M.R. In Vitro and In Vivo Effects of SerpinA1 on the Modulation of Transthyretin Proteolysis. Int. J. Mol. Sci. 2021, 22, 9488. [Google Scholar] [CrossRef]
- Lee, G.A.; Lin, Y.K.; Lai, J.H.; Lo, Y.C.; Yang, Y.S.H.; Ye, S.Y.; Lee, C.J.; Wang, C.C.; Chiang, Y.H.; Tseng, S.H. Maternal Immune Activation Causes Social Behavior Deficits and Hypomyelination in Male Rat Offspring with an Autism-Like Microbiota Profile. Brain Sci. 2021, 11, 1085. [Google Scholar] [CrossRef]
- Garro-Martínez, E.; Fullana, M.N.; Florensa-Zanuy, E.; Senserrich, J.; Paz, V.; Ruiz-Bronchal, E.; Adell, A.; Castro, E.; Díaz, Á.; Pazos, Á.; et al. mTOR Knockdown in the Infralimbic Cortex Evokes a Depressive-Like State in Mouse. Int. J. Mol. Sci. 2021, 22, 8671. [Google Scholar] [CrossRef]
- Santana-Santana, M.; Bayascas, J.R.; Giménez-Llort, L. Sex-Dependent Signatures, Time Frames and Longitudinal Fine-Tuning of the Marble Burying Test in Normal and AD-Pathological Aging Mice. Biomedicines 2021, 9, 994. [Google Scholar] [CrossRef]
- Abuaish, S.; Al-Otaibi, N.M.; Abujamel, T.S.; Alzahrani, S.A.; Alotaibi, S.M.; AlShawakir, Y.A.; Aabed, K.; El-Ansary, A. Fecal Transplant and Bifidobacterium Treatments Modulate Gut Clostridium Bacteria and Rescue Social Impairment and Hippocampal BDNF Expression in a Rodent Model of Autism. Brain Sci. 2021, 11, 1038. [Google Scholar] [CrossRef] [PubMed]
- Phebus, L.A.; Johnson, K.W. Dural inflammation model of migraine pain. Curr. Protoc. Neurosci. 2001, Chapter 9, Unit9.1. [Google Scholar] [CrossRef]
- Andreou, A.P.; Summ, O.; Charbit, A.R.; Romero-Reyes, M.; Goadsby, P.J. Animal models of headache: From bedside to bench and back to bedside. Expert Rev. Neurother. 2010, 10, 389–411. [Google Scholar] [CrossRef] [PubMed]
- Lukács, M.; Haanes, K.A.; Majláth, Z.; Tajti, J.; Vécsei, L.; Warfvinge, K.; Edvinsson, L. Dural administration of inflammatory soup or Complete Freund’s Adjuvant induces activation and inflammatory response in the rat trigeminal ganglion. J. Headache Pain 2015, 16, 564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ebersberger, A.; Ringkamp, M.; Reeh, P.W.; Handwerker, H.O. Recordings from brain stem neurons responding to chemical stimulation of the subarachnoid space. J. Neurophysiol. 1997, 77, 3122–3133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laborc, K.F.; Spekker, E.; Bohár, Z.; Szűcs, M.; Nagy-Grócz, G.; Fejes-Szabó, A.; Vécsei, L.; Párdutz, Á. Trigeminal activation patterns evoked by chemical stimulation of the dura mater in rats. J. Headache Pain 2020, 21, 101. [Google Scholar] [CrossRef] [PubMed]
- Spekker, E.; Laborc, K.F.; Bohár, Z.; Nagy-Grócz, G.; Fejes-Szabó, A.; Szűcs, M.; Vécsei, L.; Párdutz, Á. Effect of dural inflammatory soup application on activation and sensitization markers in the caudal trigeminal nucleus of the rat and the modulatory effects of sumatriptan and kynurenic acid. J. Headache Pain 2021, 22, 17. [Google Scholar] [CrossRef]
- Wieseler, J.; Ellis, A.; McFadden, A.; Stone, K.; Brown, K.; Cady, S.; Bastos, L.F.; Sprunger, D.; Rezvani, N.; Johnson, K.; et al. Supradural inflammatory soup in awake and freely moving rats induces facial allodynia that is blocked by putative immune modulators. Brain Res. 2017, 1664, 87–94. [Google Scholar] [CrossRef]
- Oshinsky, M.L.; Gomonchareonsiri, S. Episodic dural stimulation in awake rats: A model for recurrent headache. Headache 2007, 47, 1026–1036. [Google Scholar] [CrossRef] [Green Version]
- Melo-Carrillo, A.; Lopez-Avila, A. A chronic animal model of migraine, induced by repeated meningeal nociception, characterized by a behavioral and pharmacological approach. Cephalalgia 2013, 33, 1096–1105. [Google Scholar] [CrossRef]
- Ferrari, M.D. Migraine. Lancet 1998, 351, 1043–1051. [Google Scholar] [CrossRef]
- Malick, A.; Jakubowski, M.; Elmquist, J.K.; Saper, C.B.; Burstein, R. A neurohistochemical blueprint for pain-induced loss of appetite. Proc. Natl. Acad. Sci. USA 2001, 98, 9930–9935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wieseler, J.; Ellis, A.; Sprunger, D.; Brown, K.; McFadden, A.; Mahoney, J.; Rezvani, N.; Maier, S.F.; Watkins, L.R. A novel method for modeling facial allodynia associated with migraine in awake and freely moving rats. J. Neurosci. Methods 2010, 185, 236–245. [Google Scholar] [CrossRef] [Green Version]
- Chen, N.; Su, W.; Cui, S.H.; Guo, J.; Duan, J.C.; Li, H.X.; He, L. A novel large animal model of recurrent migraine established by repeated administration of inflammatory soup into the dura mater of the rhesus monkey. Neural Regen. Res. 2019, 14, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Saxena, P.R.; De Vries, P.; Villalón, C.M. 5-HT1-like receptors: A time to bid goodbye. Trends Pharmacol. Sci. 1998, 19, 311–316. [Google Scholar] [CrossRef]
- Buzzi, M.G.; Moskowitz, M.A. The antimigraine drug, sumatriptan (GR43175), selectively blocks neurogenic plasma extravasation from blood vessels in dura mater. Br. J. Pharmacol. 1990, 99, 202–206. [Google Scholar] [CrossRef] [Green Version]
- Cutrer, F.M.; Yu, X.J.; Ayata, G.; Moskowitz, M.A.; Waeber, C. Effects of PNU-109,291, a selective 5-HT1D receptor agonist, on electrically induced dural plasma extravasation and capsaicin-evoked c-fos immunoreactivity within trigeminal nucleus caudalis. Neuropharmacology 1999, 38, 1043–1053. [Google Scholar] [CrossRef]
- Tanaka, M.; Török, N.; Vécsei, L. Are 5-HT1 receptor agonists effective anti-migraine drugs? Expert Opin. Pharmacother. 2021, 22, 1221–1225. [Google Scholar] [CrossRef]
- Moskowitz, M.A. Neurogenic versus vascular mechanisms of sumatriptan and ergot alkaloids in migraine. Trends Pharmacol. Sci. 1992, 13, 307–311. [Google Scholar] [CrossRef]
- Pardutz, A.; Schoenen, J. NSAIDs in the Acute Treatment of Migraine: A Review of Clinical and Experimental Data. Pharmaceuticals 2010, 3, 1966–1987. [Google Scholar] [CrossRef]
- Gallelli, L.; Avenoso, T.; Falcone, D.; Palleria, C.; Peltrone, F.; Esposito, M.; De Sarro, G.; Carotenuto, M.; Guidetti, V. Effects of acetaminophen and ibuprofen in children with migraine receiving preventive treatment with magnesium. Headache 2014, 54, 313–324. [Google Scholar] [CrossRef]
- Goadsby, P.J.; Lipton, R.B.; Ferrari, M.D. Migraine--current understanding and treatment. N. Engl. J. Med. 2002, 346, 257–270. [Google Scholar] [CrossRef] [Green Version]
- Tso, A.R.; Goadsby, P.J. Anti-CGRP Monoclonal Antibodies: The Next Era of Migraine Prevention? Curr. Treat. Options Neurol. 2017, 19, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, H.; Lauritsen, C.G.; Kaiser, E.A.; Silberstein, S.D. CGRP Monoclonal Antibodies for Migraine: Rationale and Progress. BioDrugs 2017, 31, 487–501. [Google Scholar] [CrossRef]
- Negro, A.; Martelletti, P. Novel synthetic treatment options for migraine. Expert Opin. Pharmacother. 2021, 22, 907–922. [Google Scholar] [CrossRef]
- Olesen, J.; Diener, H.C.; Husstedt, I.W.; Goadsby, P.J.; Hall, D.; Meier, U.; Pollentier, S.; Lesko, L.M. Calcitonin gene-related peptide receptor antagonist BIBN 4096 BS for the acute treatment of migraine. N. Engl. J. Med. 2004, 350, 1104–1110. [Google Scholar] [CrossRef] [Green Version]
- Peroutka, S.J. Neurogenic inflammation and migraine: Implications for the therapeutics. Mol. Interv. 2005, 5, 304–311. [Google Scholar] [CrossRef] [PubMed]
- Zimmer, A.; Zimmer, A.M.; Baffi, J.; Usdin, T.; Reynolds, K.; König, M.; Palkovits, M.; Mezey, E. Hypoalgesia in mice with a targeted deletion of the tachykinin 1 gene. Proc. Natl. Acad. Sci. USA 1998, 95, 2630–2635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, Y.Q.; Mantyh, P.W.; Carlson, E.J.; Gillespie, A.M.; Epstein, C.J.; Basbaum, A.I. Primary afferent tachykinins are required to experience moderate to intense pain. Nature 1998, 392, 390–394. [Google Scholar] [CrossRef]
- Lee, W.S.; Moussaoui, S.M.; Moskowitz, M.A. Blockade by oral or parenteral RPR 100893 (a non-peptide NK1 receptor antagonist) of neurogenic plasma protein extravasation within guinea-pig dura mater and conjunctiva. Br. J. Pharmacol. 1994, 112, 920–924. [Google Scholar] [CrossRef] [Green Version]
- Goldstein, D.J.; Wang, O.; Saper, J.R.; Stoltz, R.; Silberstein, S.D.; Mathew, N.T. Ineffectiveness of neurokinin-1 antagonist in acute migraine: A crossover study. Cephalalgia 1997, 17, 785–790. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, D.J.; Offen, W.W.; Klein, E.G.; Phebus, L.A.; Hipskind, P.; Johnson, K.W.; Ryan, R.E., Jr. Lanepitant, an NK-1 antagonist, in migraine prevention. Cephalalgia 2001, 21, 102–106. [Google Scholar] [CrossRef] [PubMed]
- Herrstedt, J.; Muss, H.B.; Warr, D.G.; Hesketh, P.J.; Eisenberg, P.D.; Raftopoulos, H.; Grunberg, S.M.; Gabriel, M.; Rodgers, A.; Hustad, C.M.; et al. Efficacy and tolerability of aprepitant for the prevention of chemotherapy-induced nausea and emesis over multiple cycles of moderately emetogenic chemotherapy. Cancer 2005, 104, 1548–1555. [Google Scholar] [CrossRef]
- Jara-Oseguera, A.; Simon, S.A.; Rosenbaum, T. TRPV1: On the road to pain relief. Curr. Mol. Pharmacol. 2008, 1, 255–269. [Google Scholar] [CrossRef] [PubMed]
- Leimuranta, P.; Khiroug, L.; Giniatullin, R. Emerging Role of (Endo)Cannabinoids in Migraine. Front. Pharmacol. 2018, 9, 420. [Google Scholar] [CrossRef]
- Tsou, K.; Brown, S.; Sañudo-Peña, M.C.; Mackie, K.; Walker, J.M. Immunohistochemical distribution of cannabinoid CB1 receptors in the rat central nervous system. Neuroscience 1998, 83, 393–411. [Google Scholar] [CrossRef]
- Matsuda, L.A.; Lolait, S.J.; Brownstein, M.J.; Young, A.C.; Bonner, T.I. Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature 1990, 346, 561–564. [Google Scholar] [CrossRef] [PubMed]
- Russo, E.B. Clinical endocannabinoid deficiency (CECD): Can this concept explain therapeutic benefits of cannabis in migraine, fibromyalgia, irritable bowel syndrome and other treatment-resistant conditions? Neuro. Endocrinol. Lett. 2004, 25, 31–39. [Google Scholar]
- Greco, R.; Demartini, C.; Zanaboni, A.M.; Piomelli, D.; Tassorelli, C. Endocannabinoid System and Migraine Pain: An Update. Front. Neurosci. 2018, 12, 172. [Google Scholar] [CrossRef]
- Cupini, L.M.; Bari, M.; Battista, N.; Argiró, G.; Finazzi-Agró, A.; Calabresi, P.; Maccarrone, M. Biochemical changes in endocannabinoid system are expressed in platelets of female but not male migraineurs. Cephalalgia 2006, 26, 277–281. [Google Scholar] [CrossRef]
- Akerman, S.; Kaube, H.; Goadsby, P.J. Anandamide is able to inhibit trigeminal neurons using an in vivo model of trigeminovascular-mediated nociception. J. Pharmacol. Exp. Ther. 2004, 309, 56–63. [Google Scholar] [CrossRef] [Green Version]
- Nagy-Grócz, G.; Bohár, Z.; Fejes-Szabó, A.; Laborc, K.F.; Spekker, E.; Tar, L.; Vécsei, L.; Párdutz, Á. Nitroglycerin increases serotonin transporter expression in rat spinal cord but anandamide modulated this effect. J. Chem. Neuroanat. 2017, 85, 13–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tajti, J.; Tuka, B.; Botz, B.; Helyes, Z.; Vécsei, L. Role of pituitary adenylate cyclase-activating polypeptide in nociception and migraine. CNS Neurol. Disord. Drug Targets 2015, 14, 540–553. [Google Scholar] [CrossRef] [Green Version]
- Rubio-Beltrán, E.; Correnti, E.; Deen, M. PACAP38 and PAC1 receptor blockade: A new target for headache? J. Headache Pain 2018, 19, 64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, M.; Vécsei, L. Monitoring the kynurenine system: Concentrations, ratios or what else? Adv. Clin. Exp. Med. 2021, 30, 775–778. [Google Scholar] [CrossRef] [PubMed]
- Török, N.; Tanaka, M.; Vécsei, L. Searching for Peripheral Biomarkers in Neurodegenerative Diseases: The Tryptophan-Kynurenine Metabolic Pathway. Int. J. Mol. Sci. 2020, 21, 9338. [Google Scholar] [CrossRef]
- Liao, C.; de Molliens, M.P.; Schneebeli, S.T.; Brewer, M.; Song, G.; Chatenet, D.; Braas, K.M.; May, V.; Li, J. Targeting the PAC1 Receptor for Neurological and Metabolic Disorders. Curr. Top. Med. Chem. 2019, 19, 1399–1417. [Google Scholar] [CrossRef]
- Fejes, A.; Párdutz, Á.; Toldi, J.; Vécsei, L. Kynurenine metabolites and migraine: Experimental studies and therapeutic perspectives. Curr. Neuropharmacol. 2011, 9, 376–387. [Google Scholar] [CrossRef] [Green Version]
- Tajti, J.; Majlath, Z.; Szok, D.; Csati, A.; Toldi, J.; Fülöp, F.; Vécsei, L. Novel kynurenic acid analogues in the treatment of migraine and neurodegenerative disorders: Preclinical studies and pharmaceutical design. Curr. Pharm. Des. 2015, 21, 2250–2258. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, M.; Toldi, J.; Vécsei, L. Exploring the Etiological Links behind Neurodegenerative Diseases: Inflammatory Cytokines and Bioactive Kynurenines. Int. J. Mol. Sci. 2020, 21, 2431. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, M.; Tóth, F.; Polyák, H.; Szabó, Á.; Mándi, Y.; Vécsei, L. Immune Influencers in Action: Metabolites and Enzymes of the Tryptophan-Kynurenine Metabolic Pathway. Biomedicines 2021, 9, 734. [Google Scholar] [CrossRef]
- Tanaka, M.; Török, N.; Tóth, F.; Szabó, Á.; Vécsei, L. Co-Players in Chronic Pain: Neuroinflammation and the Trypto-phan-Kynurenine Metabolic Pathway. Biomedicines 2021, 9, 897. [Google Scholar] [CrossRef]
- Curto, M.; Lionetto, L.; Negro, A.; Capi, M.; Fazio, F.; Giamberardino, M.A.; Simmaco, M.; Nicoletti, F.; Martelletti, P. Altered kynurenine pathway metabolites in serum of chronic migraine patients. J. Headache Pain 2015, 17, 47. [Google Scholar] [CrossRef] [Green Version]
- Sarchielli, P.; Di Filippo, M.; Nardi, K.; Calabresi, P. Sensitization, glutamate, and the link between migraine and fibromyalgia. Curr. Pain Headache Rep. 2007, 11, 343–351. [Google Scholar] [CrossRef]
- Nagy-Grócz, G.; Laborc, K.F.; Veres, G.; Bajtai, A.; Bohár, Z.; Zádori, D.; Fejes-Szabó, A.; Spekker, E.; Vécsei, L.; Párdutz, Á. The Effect of Systemic Nitroglycerin Administration on the Kynurenine Pathway in the Rat. Front. Neurol. 2017, 14, 278. [Google Scholar] [CrossRef]
- Mándi, Y.; Vécsei, L. The kynurenine system and immunoregulation. J. Neural Trans. 2012, 119, 197–209. [Google Scholar] [CrossRef] [PubMed]
- Lukács, M.; Tajti, J.; Fülöp, F.; Toldi, J.; Edvinsson, L.; Vécsei, L. Migraine, Neurogenic Inflammation, Drug Development - Pharmacochemical Aspects. Curr. Med. Chem. 2017, 24, 3649–3665. [Google Scholar] [CrossRef] [PubMed]
- Jovanovic, F.; Candido, K.D.; Knezevic, N.N. The Role of the Kynurenine Signaling Pathway in Different Chronic Pain Conditions and Potential Use of Therapeutic Agents. Int. J. Mol. Sci. 2020, 21, 6045. [Google Scholar] [CrossRef] [PubMed]
- Hesselink, J.M.K. New targets in pain, non-neuronal cells, and the role of palmitoylethanolamide. Open Pain J. 2012, 5, 2–23. [Google Scholar] [CrossRef] [Green Version]
- Chirchiglia, D.; Paventi, S.; Seminara, P.; Cione, E.; Gallelli, L. N-Palmitoyl Ethanol Amide Pharmacological Treatment in Patients With Nonsurgical Lumbar Radiculopathy. J. Clin. Pharmacol. 2018, 58, 733–739. [Google Scholar] [CrossRef] [PubMed]
- Chirchiglia, D.; Della Torre, A.; Signorelli, F.; Volpentesta, G.; Guzzi, G.; Stroscio, C.A.; Deodato, F.; Gabriele, D.; Lavano, A. Administration of palmitoylethanolamide in combination with topiramate in the preventive treatment of nummular headache. Int Med. Case Rep. J. 2016, 18, 193–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chirchiglia, D.; Cione, E.; Caroleo, M.C.; Wang, M.; Di Mizio, G.; Faedda, N.; Giacolini, T.; Siviglia, S.; Guidetti, V.; Gallelli, L. Effects of Add-On Ultramicronized N-Palmitol Ethanol Amide in Patients Suffering of Migraine With Aura: A Pilot Study. Front. Neurol. 2018, 17, 674. [Google Scholar] [CrossRef] [PubMed]
- Karakurum Göksel, B. The Use of Complementary and Alternative Medicine in Patients with Migraine. Noro. Psikiyatri Arsivi 2013, 50 (Suppl. 1), S41–S46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tafuri, E.; Santovito, D.; de Nardis, V.; Marcantonio, P.; Paganelli, C.; Affaitati, G.; Bucci, M.; Mezzetti, A.; Giamberardino, M.A.; Cipollone, F. MicroRNA profiling in migraine without aura: Pilot study. Ann. Med. 2015, 47, 468–473. [Google Scholar] [CrossRef] [PubMed]
- Gallelli, L.; Cione, E.; Peltrone, F.; Siviglia, S.; Verano, A.; Chirchiglia, D.; Zampogna, S.; Guidetti, V.; Sammartino, L.; Montana, A.; et al. Hsa-miR-34a-5p and hsa-miR-375 as Biomarkers for Monitoring the Effects of Drug Treatment for Migraine Pain in Children and Adolescents: A Pilot Study. J. Clin. Med. 2019, 27, 928. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Neuropeptides/Neurotransmitters | Receptors | Migraine/Neurogenic Inflammation-Related Functions | References |
|---|---|---|---|
| CGRP | CLR, RAMP1 | craniocervical vasodilation, peripheral and central sensitization, neuron-glia interaction, involved plasma extravasation, mast cell degranulation, | Asghar et al., 2011 [44], Holzer, 1998 [45], Ottosson and Edvinsson, 1997 [46], Lennerz et al., 2008 [47], Raddant and Russo, 2011 [48] |
| SP | NK1 | craniocervical vasodilation, plasma protein extravasation, cytokines, oxygen radicals, and histamine release | Hökfelt et al., 1975 [49], Ribeiro-da-Silva and Hökfelt, 2000 [50], Snijdelaar et al., 2000 [51], Graefe and Mohiuddin, 2021 [52], Killingsworth et al., 1997 [53], Weinstock et al., 1988 [54], Holzer and Holzer-Petsche, 1997 [55], Pernow, 1983 [56], Gibbins et al., 1985 [57], Battaglia and Rustioni, 1988 [58], Malhotra, 2016 [59] |
| PACAP | PAC1, VPAC1, VPAC2 | craniocervical vasodilation, peripheral and central sensitization | Jansen-Olesen and Hougaard Pedersen, 2018 [60], Eftekhari et al., 2013 [61], Nielsen et al., 1998 [62], Jansen-Olesen et al., 2014 [63], Uddman et al., 2002 [64], Hashimoto et al., 2006 [65], Martin et al., 2003 [66], Missig et al., 2014 [67], Kaiser and Russo, 2013 [68], Zhang et al., 1998 [69] |
| VIP | VPAC1, VPAC2 | craniocervical vasodilation, mast cell degranulation, IL-6 and IL-8 production | Kilinc et al., 2015 [70], Ohhashi et al., 1983 [71], Kakurai et al., 2001 [72], Cernuda-Morollón et al., 2014 [73], Pellesi et al., 2020 [74] |
| - | TRPV1 | vasodilation, peripheral and central sensitization, neuropeptide release (SP, CGRP) initiate neurogenic inflammation | Caterina et al., 2000 [75], Quartu et al., 2016 [76], Dux et al., 2020 [77], Bevan et al., 2014 [78], Meents et al., 2010 [79] |
| histamine | H1–4R | vasodilation, mediate SP and glutamate release | Yuan and Silberstein, 2018 [80], Castillo et al., 1995 [81], Heatley et al., 1982 [82], Foreman et al., 1983 [83], Rosa and Fantozzi, 2013 [84] |
| Drug Class | Drug | Target | FDA Appoved |
|---|---|---|---|
| NSAIDs | Acetylsalicylic acid | COX1–2 | yes |
| Ibuprofen | yes | ||
| Diclofenac potassium | yes | ||
| Paracetamol | yes | ||
| Triptans | Sumatriptan | 5-HT1D receptor | yes |
| Zolmitriptan | yes | ||
| Almotriptan | yes | ||
| Rizatriptan | yes | ||
| Frovatriptan | yes | ||
| Naratriptan | yes | ||
| Eletriptan | 5-HT1B/1D receptor | yes | |
| Ditans | Lasmiditan | 5-HT1F receptor | yes |
| Gepants | Ubrogepant | CGRP receptor | yes |
| Rimegepant | yes | ||
| Atogepant | no | ||
| Vazegepant | no | ||
| Ergot alkaloids | Ergotamine tartrate | α-adrenergic receptor,5-HT receptors | yes |
| CGRP/CGRP receptor monoclonal antibody | Erenumab | CGRP receptor | yes |
| Eptinezumab | CGRP ligand | yes | |
| Fremanezumab | yes | ||
| Galcenezumab | yes | ||
| NK1R antagonists | Aprepitant | NK1 receptor | yes |
| PACAP/PAC1 receptor monoclonal antibody | ALD1910 | PACAP38 | no |
| AMG-301 | PAC1 receptor | no | |
| Endocannabinoids | 2-Arachidonoylglycerol | CB1 receptor | no |
| Anandamide | CB1 receptor | no |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Spekker, E.; Tanaka, M.; Szabó, Á.; Vécsei, L. Neurogenic Inflammation: The Participant in Migraine and Recent Advancements in Translational Research. Biomedicines 2022, 10, 76. https://doi.org/10.3390/biomedicines10010076
Spekker E, Tanaka M, Szabó Á, Vécsei L. Neurogenic Inflammation: The Participant in Migraine and Recent Advancements in Translational Research. Biomedicines. 2022; 10(1):76. https://doi.org/10.3390/biomedicines10010076
Chicago/Turabian StyleSpekker, Eleonóra, Masaru Tanaka, Ágnes Szabó, and László Vécsei. 2022. "Neurogenic Inflammation: The Participant in Migraine and Recent Advancements in Translational Research" Biomedicines 10, no. 1: 76. https://doi.org/10.3390/biomedicines10010076
APA StyleSpekker, E., Tanaka, M., Szabó, Á., & Vécsei, L. (2022). Neurogenic Inflammation: The Participant in Migraine and Recent Advancements in Translational Research. Biomedicines, 10(1), 76. https://doi.org/10.3390/biomedicines10010076