
The Role of Gut Microbiota in Heart Failure: When Friends Become Enemies

 ,
,  , ,
, ,  ,
, 
Abstract
:1. Introduction
2. HF and Inflammation
3. Impact of Immunity on LV Function and Remodeling
4. The Interplay between Immune Response and HF: Main Actors of Innate and Adaptive Immunity
5. Perspectives of Immune Modulation in Heart Failure
6. Gut Microbiota Composition and HF
7. Gut Dysbiosis, Inflammation and Cardiovascular Diseases
8. Gut Microbiota, Innate Immunity and HF
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Ni, H.; Xu, J. Recent Trends in Heart Failure-related Mortality: United States, 2000–2014. NCHS Data Brief 2015, 231, 1–8. [Google Scholar]
- Shirazi, L.F.; Bissett, J.; Romeo, F.; Mehta, J.L. Role of Inflammation in Heart Failure. Curr. Atheroscler. Rep. 2017, 19, 27. [Google Scholar] [CrossRef] [PubMed]
- Paulus, W.J.; Tschöpe, C. A novel paradigm for heart failure with preserved ejection fraction: Comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J. Am. Coll. Cardiol. 2013, 62, 263–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhakal, B.P.; Kim, C.H.; Al-Kindi, S.G.; Oliveira, G.H. Heart failure in systemic lupus erythematosus. Trends Cardiovasc. Med. 2018, 28, 187–197. [Google Scholar] [CrossRef]
- Cianci, R.; Franza, L.; Massaro, M.G.; Borriello, R.; Tota, A.; Pallozzi, M.; De Vito, F.; Gambassi, G. The Crosstalk between Gut Microbiota, Intestinal Immunological Niche and Visceral Adipose Tissue as a New Model for the Pathogenesis of Metabolic and Inflammatory Diseases: The Paradigm of Type 2 Diabetes Mellitus. Curr. Med. Chem. 2022, 29, 3189–3201. [Google Scholar] [CrossRef]
- Purchiaroni, F.; Tortora, A.; Gabrielli, M.; Bertucci, F.; Gigante, G.; Ianiro, G.; Ojetti, V.; Scarpellini, E.; Gasbarrini, A. The role of intestinal microbiota and the immune system. Eur. Rev. Med. Pharmacol. Sci. 2013, 17, 323–333. [Google Scholar]
- Jin, M.; Qian, Z.; Yin, J.; Xu, W.; Zhou, X. The role of intestinal microbiota in cardiovascular disease. J. Cell. Mol. Med. 2019, 23, 2343–2350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDonagh, T.A.; Metra, M. 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur. Heart J. 2021, 42, 3599–3726. [Google Scholar] [CrossRef] [PubMed]
- De Jong, K.A.; Lopaschuk, G.D. Complex Energy Metabolic Changes in Heart Failure with Preserved Ejection Fraction and Heart Failure with Reduced Ejection Fraction. Can. J. Cardiol. 2017, 33, 860–871. [Google Scholar] [CrossRef] [PubMed]
- Oren, O.; Goldberg, S. Heart Failure with Preserved Ejection Fraction: Diagnosis and Management. Am. J. Med. 2017, 130, 510–516. [Google Scholar] [CrossRef] [PubMed]
- Heidenreich, P.A.; Bozkurt, B.; Aguilar, D. 2022 AHA/ACC/HFSA Guideline for the Management of Heart Failure: A Report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. Circulation 2022, 145, e895–e1032. [Google Scholar] [CrossRef]
- Sani, C.M.; Pogue, E.P.L.; Hrabia, J.B.; Zayachkowski, A.G.; Zawadka, M.M.; Poniatowski, A.G.; Długosz, D.; Leśniak, W.; Kruszelnicka, O.; Chyrchel, B.; et al. Association between low-grade chronic inflammation and depressed left atrial compliance in heart failure with preserved ejection fraction: A retrospective analysis. Folia Med. Crac. 2018, 58, 45–55. [Google Scholar] [CrossRef]
- Riehle, C.; Bauersachs, J. Key inflammatory mechanisms underlying heart failure. Herz 2019, 44, 96–106. [Google Scholar] [CrossRef] [Green Version]
- Levine, B.; Kalman, J.; Mayer, L.; Fillit, H.M.; Packer, M. Elevated circulating levels of tumor necrosis factor in severe chronic heart failure. N. Engl. J. Med. 1990, 323, 236–241. [Google Scholar] [CrossRef]
- Nishida, K.; Otsu, K. Sterile Inflammation and Degradation Systems in Heart Failure. Circ. J. 2017, 81, 622–628. [Google Scholar] [CrossRef] [Green Version]
- Nozaki, N.; Yamaguchi, S.; Yamaoka, M.; Okuyama, M.; Nakamura, H.; Tomoike, H. Enhanced expression and shedding of tumor necrosis factor (TNF) receptors from mononuclear leukocytes in human heart failure. J. Mol. Cell. Cardiol. 1998, 30, 2003–2012. [Google Scholar] [CrossRef]
- Kurtzhals, J.A.; Adabayeri, V.; Goka, B.Q.; Akanmori, B.D.; Oliver-Commey, J.O.; Nkrumah, F.K.; Behr, C.; Hviid, L. Low plasma concentrations of interleukin 10 in severe malarial anaemia compared with cerebral and uncomplicated malaria. Lancet 1998, 351, 1768–1772. [Google Scholar] [CrossRef]
- Yamaoka, Y.; Kita, M.; Kodama, T.; Sawai, N.; Imanishi, J. Helicobacter pylori cagA gene and expression of cytokine messenger RNA in gastric mucosa. Gastroenterology 1996, 110, 1744–1752. [Google Scholar] [CrossRef]
- Van der Poll, T.; Coyle, S.M.; Barbosa, K.; Braxton, C.C.; Lowry, S.F. Epinephrine inhibits tumor necrosis factor-alpha and potentiates interleukin 10 production during human endotoxemia. J. Clin. Investig. 1996, 97, 713–719. [Google Scholar] [CrossRef] [Green Version]
- Chao, L.; Lei, H.; Fei, J. A meta-analysis of interleukin-10-1082 promoter genetic polymorphism associated with atherosclerotic risk. Neurol. India 2014, 62, 130–136. [Google Scholar] [CrossRef]
- Wykretowicz, A.; Furmaniuk, J.; Smielecki, J.; Deskur-Smielecka, E.; Szczepanik, A.; Banaszak, A.; Wysocki, H. The oxygen stress index and levels of circulating interleukin-10 and interleukin-6 in patients with chronic heart failure. Int. J. Cardiol. 2004, 94, 283–287. [Google Scholar] [CrossRef]
- Deng, M.C. A peripheral blood transcriptome biomarker test to diagnose functional recovery potential in advanced heart failure. Biomark. Med. 2018, 12, 619–635. [Google Scholar] [CrossRef]
- Maeda, K.; Tsutamoto, T.; Wada, A.; Mabuchi, N.; Hayashi, M.; Tsutsui, T.; Ohnishi, M.; Sawaki, M.; Fujii, M.; Matsumoto, T.; et al. High levels of plasma brain natriuretic peptide and interleukin-6 after optimized treatment for heart failure are independent risk factors for morbidity and mortality in patients with congestive heart failure. J. Am. Coll. Cardiol. 2000, 36, 1587–1593. [Google Scholar] [CrossRef] [Green Version]
- Ponikowski, P.; Voors, A.A.; Anker, S.D.; Bueno, H.; Cleland, J.G.F.; Coats, A.J.S.; Falk, V.; González-Juanatey, J.R.; Harjola, V.P.; Jankowska, E.A.; et al. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: The Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC) Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur. Heart J. 2016, 37, 2129–2200. [Google Scholar] [CrossRef] [Green Version]
- Kuster, G.M.; Pfister, O. Chronic heart failure: Advances in pharmacological treatment and future perspectives. Swiss Med. Wkly. 2019, 149, w20036. [Google Scholar] [CrossRef] [Green Version]
- Bytyçi, I.; Bajraktari, G. Mortality in heart failure patients. Anatol. J. Cardiol. 2015, 15, 63–68. [Google Scholar] [CrossRef] [Green Version]
- Bloom, M.W.; Greenberg, B.; Jaarsma, T.; Januzzi, J.L.; Lam, C.S.P.; Maggioni, A.P.; Trochu, J.N.; Butler, J. Heart failure with reduced ejection fraction. Nat. Rev. Dis. Primers 2017, 3, 17058. [Google Scholar] [CrossRef]
- Røe, Å.T.; Sjaastad, I.; Louch, W.E. Heart failure with preserved ejection fraction. Tidsskr. Den Nor. Laegeforen. Tidsskr. Prakt. Med. Ny Raekke 2017, 137. [Google Scholar] [CrossRef] [Green Version]
- Shah, S.J. Precision Medicine for Heart Failure with Preserved Ejection Fraction: An Overview. J. Cardiovasc. Transl. Res. 2017, 10, 233–244. [Google Scholar] [CrossRef]
- Mann, D.L. Innate immunity and the failing heart: The cytokine hypothesis revisited. Circ. Res. 2015, 116, 1254–1268. [Google Scholar] [CrossRef] [Green Version]
- Tschöpe, C.; Bock, C.T.; Kasner, M.; Noutsias, M.; Westermann, D.; Schwimmbeck, P.L.; Pauschinger, M.; Poller, W.C.; Kühl, U.; Kandolf, R.; et al. High prevalence of cardiac parvovirus B19 infection in patients with isolated left ventricular diastolic dysfunction. Circulation 2005, 111, 879–886. [Google Scholar] [CrossRef] [Green Version]
- Tromp, J.; Lim, S.L.; Tay, W.T.; Teng, T.K.; Chandramouli, C.; Ouwerkerk, W.; Wander, G.S.; Sawhney, J.P.S.; Yap, J.; MacDonald, M.R.; et al. Microvascular Disease in Patients with Diabetes with Heart Failure and Reduced Ejection Versus Preserved Ejection Fraction. Diabetes Care 2019, 42, 1792–1799. [Google Scholar] [CrossRef] [PubMed]
- Varga, I.; Kyselovič, J.; Galfiova, P.; Danisovic, L. The Non-cardiomyocyte Cells of the Heart. Their Possible Roles in Exercise-Induced Cardiac Regeneration and Remodeling. Adv. Exp. Med. Biol. 2017, 999, 117–136. [Google Scholar] [CrossRef]
- Carrillo-Salinas, F.J.; Ngwenyama, N.; Anastasiou, M.; Kaur, K.; Alcaide, P. Heart Inflammation: Immune Cell Roles and Roads to the Heart. Am. J. Pathol. 2019, 189, 1482–1494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeBerge, M.; Shah, S.J.; Wilsbacher, L.; Thorp, E.B. Macrophages in Heart Failure with Reduced versus Preserved Ejection Fraction. Trends Mol. Med. 2019, 25, 328–340. [Google Scholar] [CrossRef] [PubMed]
- Bajpai, G.; Schneider, C.; Wong, N.; Bredemeyer, A.; Hulsmans, M.; Nahrendorf, M.; Epelman, S.; Kreisel, D.; Liu, Y.; Itoh, A.; et al. The human heart contains distinct macrophage subsets with divergent origins and functions. Nat. Med. 2018, 24, 1234–1245. [Google Scholar] [CrossRef] [PubMed]
- González, G.E.; Rhaleb, N.E.; D’Ambrosio, M.A.; Nakagawa, P.; Liao, T.D.; Peterson, E.L.; Leung, P.; Dai, X.; Janic, B.; Liu, Y.H.; et al. Cardiac-deleterious role of galectin-3 in chronic angiotensin II-induced hypertension. Am. J. Physiol. Heart Circ. Physiol. 2016, 311, H1287–H1296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hulsmans, M.; Sager, H.B.; Roh, J.D.; Valero-Muñoz, M.; Houstis, N.E.; Iwamoto, Y.; Sun, Y.; Wilson, R.M.; Wojtkiewicz, G.; Tricot, B.; et al. Cardiac macrophages promote diastolic dysfunction. J. Exp. Med. 2018, 215, 423–440. [Google Scholar] [CrossRef] [PubMed]
- Puhl, S.L.; Steffens, S. Neutrophils in Post-myocardial Infarction Inflammation: Damage vs. Resolution? Front. Cardiovasc. Med. 2019, 6, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horckmans, M.; Ring, L.; Duchene, J.; Santovito, D.; Schloss, M.J.; Drechsler, M.; Weber, C.; Soehnlein, O.; Steffens, S. Neutrophils orchestrate post-myocardial infarction healing by polarizing macrophages towards a reparative phenotype. Eur. Heart J. 2017, 38, 187–197. [Google Scholar] [CrossRef] [Green Version]
- Konishi, T.; Funayama, N.; Yamamoto, T.; Morita, T.; Hotta, D.; Nishihara, H.; Tanaka, S. Prognostic Value of Eosinophil to Leukocyte Ratio in Patients with ST-Elevation Myocardial Infarction Undergoing Primary Percutaneous Coronary Intervention. J. Atheroscler. Thromb. 2017, 24, 827–840. [Google Scholar] [CrossRef] [Green Version]
- Jiang, P.; Wang, D.Z.; Ren, Y.L.; Cai, J.P.; Chen, B.X. Significance of eosinophil accumulation in the thrombus and decrease in peripheral blood in patients with acute coronary syndrome. Coron. Artery Dis. 2015, 26, 101–106. [Google Scholar] [CrossRef] [PubMed]
- Frosali, S.; Pagliari, D.; Gambassi, G.; Landolfi, R.; Pandolfi, F.; Cianci, R. How the Intricate Interaction among Toll-Like Receptors, Microbiota, and Intestinal Immunity Can Influence Gastrointestinal Pathology. J. Immunol. Res. 2015, 2015, 489821. [Google Scholar] [CrossRef]
- Shishido, T.; Nozaki, N.; Yamaguchi, S.; Shibata, Y.; Nitobe, J.; Miyamoto, T.; Takahashi, H.; Arimoto, T.; Maeda, K.; Yamakawa, M.; et al. Toll-like receptor-2 modulates ventricular remodeling after myocardial infarction. Circulation 2003, 108, 2905–2910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, L.; Feng, Z. The Role of Toll-Like Receptor Signaling in the Progression of Heart Failure. Mediat. Inflamm. 2018, 2018, 9874109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arslan, F.; Keogh, B.; McGuirk, P.; Parker, A.E. TLR2 and TLR4 in ischemia reperfusion injury. Mediat. Inflamm. 2010, 2010, 704202. [Google Scholar] [CrossRef] [Green Version]
- Földes, G.; von Haehling, S.; Okonko, D.O.; Jankowska, E.A.; Poole-Wilson, P.A.; Anker, S.D. Fluvastatin reduces increased blood monocyte Toll-like receptor 4 expression in whole blood from patients with chronic heart failure. Int. J. Cardiol. 2008, 124, 80–85. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, N.; Noma, T.; Ishihara, Y.; Miyauchi, Y.; Takabatake, W.; Oomizu, S.; Yamaoka, G.; Ishizawa, M.; Namba, T.; Murakami, K.; et al. Prognostic value of circulating regulatory T cells for worsening heart failure in heart failure patients with reduced ejection fraction. Int. Heart J. 2014, 55, 271–277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahmati, Z.; Amirzargar, A.A.; Saadati, S.; Rahmani, F.; Mahmoudi, M.J.; Rahnemoon, Z.; Eskandari, V.; Gorzin, F.; Hedayat, M.; Rezaei, N. Association of levels of interleukin 17 and T-helper 17 count with symptom severity and etiology of chronic heart failure: A case-control study. Croat. Med. J. 2018, 59, 139–148. [Google Scholar] [CrossRef] [Green Version]
- Pandolfi, F.; Cianci, R.; Pagliari, D.; Landolfi, R.; Cammarota, G. Cellular mediators of inflammation: Tregs and TH17 cells in gastrointestinal diseases. Mediat. Inflamm. 2009, 2009, 132028. [Google Scholar] [CrossRef] [Green Version]
- Santos-Zas, I.; Lemarié, J.; Tedgui, A.; Ait-Oufella, H. Adaptive Immune Responses Contribute to Post-ischemic Cardiac Remodeling. Front. Cardiovasc. Med. 2018, 5, 198. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.L.; Hsiao, Y.W.; Tsai, Y.N.; Lin, S.F.; Liu, S.H.; Lin, Y.J.; Lo, L.W.; Chung, F.P.; Chao, T.F.; Hu, Y.F.; et al. Interleukin-17 enhances cardiac ventricular remodeling via activating MAPK pathway in ischemic heart failure. J. Mol. Cell. Cardiol. 2018, 122, 69–79. [Google Scholar] [CrossRef]
- Xu, L.; Yan, J.; Zhang, F.; Zhou, C.; Fan, T.; Chen, X.; Cui, X.; Zhou, H.; Liang, Y. Use of Inflammatory Biomarkers and Real-Time Cardiac Catheterisation to Evaluate the Left Ventricular Diastolic Function in Patients with Diastolic Heart Failure. Heart Lung Circ. 2021, 30, 396–403. [Google Scholar] [CrossRef]
- Ong, S.; Ligons, D.L.; Barin, J.G.; Wu, L.; Talor, M.V.; Diny, N.; Fontes, J.A.; Gebremariam, E.; Kass, D.A.; Rose, N.R.; et al. Natural killer cells limit cardiac inflammation and fibrosis by halting eosinophil infiltration. Am. J. Pathol. 2015, 185, 847–861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anzai, A.; Anzai, T.; Nagai, S.; Maekawa, Y.; Naito, K.; Kaneko, H.; Sugano, Y.; Takahashi, T.; Abe, H.; Mochizuki, S.; et al. Regulatory role of dendritic cells in postinfarction healing and left ventricular remodeling. Circulation 2012, 125, 1234–1245. [Google Scholar] [CrossRef] [Green Version]
- Tang, T.T.; Ding, Y.J.; Liao, Y.H.; Yu, X.; Xiao, H.; Xie, J.J.; Yuan, J.; Zhou, Z.H.; Liao, M.Y.; Yao, R.; et al. Defective circulating CD4CD25+Foxp3+CD127(low) regulatory T-cells in patients with chronic heart failure. Cell. Physiol. Biochem. 2010, 25, 451–458. [Google Scholar] [CrossRef]
- Matsumoto, K.; Ogawa, M.; Suzuki, J.; Hirata, Y.; Nagai, R.; Isobe, M. Regulatory T lymphocytes attenuate myocardial infarction-induced ventricular remodeling in mice. Int. Heart J. 2011, 52, 382–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Organ, C.L.; Otsuka, H.; Bhushan, S.; Wang, Z.; Bradley, J.; Trivedi, R.; Polhemus, D.J.; Tang, W.H.; Wu, Y.; Hazen, S.L.; et al. Choline Diet and Its Gut Microbe-Derived Metabolite, Trimethylamine N-Oxide, Exacerbate Pressure Overload-Induced Heart Failure. Circ. Heart Fail. 2016, 9, e002314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bujak, M.; Frangogiannis, N.G. The role of TGF-beta signaling in myocardial infarction and cardiac remodeling. Cardiovasc. Res. 2007, 74, 184–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yue, Y.; Meng, K.; Pu, Y.; Zhang, X. Transforming growth factor beta (TGF-β) mediates cardiac fibrosis and induces diabetic cardiomyopathy. Diabetes Res. Clin. Pract. 2017, 133, 124–130. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Hou, L.; Kwak, D.; Fassett, J.; Xu, X.; Chen, A.; Chen, W.; Blazar, B.R.; Xu, Y.; Hall, J.L.; et al. Increasing Regulatory T Cells with Interleukin-2 and Interleukin-2 Antibody Complexes Attenuates Lung Inflammation and Heart Failure Progression. Hypertension 2016, 68, 114–122. [Google Scholar] [CrossRef] [Green Version]
- Gorzin, F.; Amirzargar, A.A.; Mahmoudi, M.J.; Rahnemoon, Z.; Najmi Varzaneh, F.; Hedayat, M.; Sadati, S.; Eskandari, V.; Rahmati, Z.; Rezaei, N. FOXP3, RORγt and IL-10 cytokine profile in chronic heart failure. Bratisl. Lek. Listy 2017, 118, 637–641. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Bian, Z.; Lu, P.; Karas, R.H.; Bao, L.; Cox, D.; Hodgin, J.; Shaul, P.W.; Thoren, P.; Smithies, O.; et al. Abnormal vascular function and hypertension in mice deficient in estrogen receptor beta. Science 2002, 295, 505–508. [Google Scholar] [CrossRef]
- Bansal, S.S.; Ismahil, M.A.; Goel, M.; Patel, B.; Hamid, T.; Rokosh, G.; Prabhu, S.D. Activated T Lymphocytes are Essential Drivers of Pathological Remodeling in Ischemic Heart Failure. Circ. Heart Fail. 2017, 10, e003688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Norling, L.V.; Cooper, D. Cardiac Dysfunction in Rheumatoid Arthritis: The Role of Inflammation. Cells 2021, 10, 881. [Google Scholar] [CrossRef]
- Hanna, A.; Frangogiannis, N.G. Inflammatory Cytokines and Chemokines as Therapeutic Targets in Heart Failure. Cardiovasc. Drugs Ther. 2020, 34, 849–863. [Google Scholar] [CrossRef]
- Lewis, G.A.; Dodd, S.; Clayton, D.; Bedson, E.; Eccleson, H.; Schelbert, E.B.; Naish, J.H.; Jimenez, B.D.; Williams, S.G.; Cunnington, C.; et al. Pirfenidone in heart failure with preserved ejection fraction: A randomized phase 2 trial. Nat. Med. 2021, 27, 1477–1482. [Google Scholar] [CrossRef] [PubMed]
- Ishiyama, Y.; Hoshide, S. Constipation-induced pressor effects as triggers for cardiovascular events. J. Clin. Hypertens. 2019, 21, 421–425. [Google Scholar] [CrossRef] [Green Version]
- Rogler, G.; Rosano, G. The heart and the gut. Eur. Heart J. 2014, 35, 426–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xanthopoulos, A.; Starling, R.C.; Kitai, T.; Triposkiadis, F. Heart Failure and Liver Disease: Cardiohepatic Interactions. JACC Heart Fail. 2019, 7, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Luedde, M.; Winkler, T.; Heinsen, F.A.; Rühlemann, M.C.; Spehlmann, M.E.; Bajrovic, A.; Lieb, W.; Franke, A.; Ott, S.J.; Frey, N. Heart failure is associated with depletion of core intestinal microbiota. ESC Heart Fail. 2017, 4, 282–290. [Google Scholar] [CrossRef] [PubMed]
- Kamo, T.; Akazawa, H. Dysbiosis and compositional alterations with aging in the gut microbiota of patients with heart failure. PLoS ONE 2017, 12, e0174099. [Google Scholar] [CrossRef]
- Pasini, E.; Aquilani, R.; Testa, C.; Baiardi, P.; Angioletti, S.; Boschi, F.; Verri, M.; Dioguardi, F. Pathogenic Gut Flora in Patients with Chronic Heart Failure. JACC Heart Fail. 2016, 4, 220–227. [Google Scholar] [CrossRef]
- Cannon, C.P.; Braunwald, E.; McCabe, C.H.; Grayston, J.T.; Muhlestein, B.; Giugliano, R.P.; Cairns, R.; Skene, A.M. Antibiotic treatment of Chlamydia pneumoniae after acute coronary syndrome. N. Engl. J. Med. 2005, 352, 1646–1654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marques, F.Z. Missing Heritability of Hypertension and Our Microbiome. Circulation 2018, 138, 1381–1383. [Google Scholar] [CrossRef] [PubMed]
- Jama, H.A.; Kaye, D.M.; Marques, F.Z. The gut microbiota and blood pressure in experimental models. Curr. Opin. Nephrol. Hypertens. 2019, 28, 97–104. [Google Scholar] [CrossRef]
- Ganesh, B.P.; Nelson, J.W.; Eskew, J.R.; Ganesan, A.; Ajami, N.J.; Petrosino, J.F.; Bryan, R.M., Jr.; Durgan, D.J. Prebiotics, Probiotics, and Acetate Supplementation Prevent Hypertension in a Model of Obstructive Sleep Apnea. Hypertension 2018, 72, 1141–1150. [Google Scholar] [CrossRef]
- Tang, T.W.H.; Chen, H.C.; Chen, C.Y.; Yen, C.Y.T.; Lin, C.J.; Prajnamitra, R.P.; Chen, L.L.; Ruan, S.C.; Lin, J.H.; Lin, P.J.; et al. Loss of Gut Microbiota Alters Immune System Composition and Cripples Postinfarction Cardiac Repair. Circulation 2019, 139, 647–659. [Google Scholar] [CrossRef]
- Zhao, P.; Zhao, S.; Tian, J.; Liu, X. Significance of Gut Microbiota and Short-Chain Fatty Acids in Heart Failure. Nutrients 2022, 14, 3758. [Google Scholar] [CrossRef]
- Kazemian, N.; Mahmoudi, M.; Halperin, F. Gut microbiota and cardiovascular disease: Opportunities and challenges. Microbiome 2020, 8, 36. [Google Scholar] [CrossRef] [Green Version]
- Calkin, A.C.; Tontonoz, P. Transcriptional integration of metabolism by the nuclear sterol-activated receptors LXR and FXR. Nat. Rev. Mol. Cell Biol. 2012, 13, 213–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordon, J.W.; Shaw, J.A.; Kirshenbaum, L.A. Multiple facets of NF-κB in the heart: To be or not to NF-κB. Circ. Res. 2011, 108, 1122–1132. [Google Scholar] [CrossRef] [PubMed]
- Pu, J.; Yuan, A.; Shan, P.; Gao, E.; Wang, X.; Wang, Y.; Lau, W.B.; Koch, W.; Ma, X.L.; He, B. Cardiomyocyte-expressed farnesoid-X-receptor is a novel apoptosis mediator and contributes to myocardial ischaemia/reperfusion injury. Eur. Heart J. 2013, 34, 1834–1845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neuschwander-Tetri, B.A.; Loomba, R.; Sanyal, A.J.; Lavine, J.E.; Van Natta, M.L.; Abdelmalek, M.F.; Chalasani, N.; Dasarathy, S.; Diehl, A.M.; Hameed, B.; et al. Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): A multicentre, randomised, placebo-controlled trial. Lancet 2015, 385, 956–965. [Google Scholar] [CrossRef] [Green Version]
- Han, C.Y. Update on FXR Biology: Promising Therapeutic Target? Int. J. Mol. Sci. 2018, 19, 2069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, T.; Dutta, R.R.; Velagala, V.R. Analyzing the Complicated Connection between Intestinal Microbiota and Cardiovascular Diseases. Cureus 2022, 14, e28165. [Google Scholar] [CrossRef]
- Lu, D.; Zou, X.; Zhang, H. The Relationship between Atrial Fibrillation and Intestinal Flora with Its Metabolites. Front. Cardiovasc. Med. 2022, 9, 948755. [Google Scholar] [CrossRef]
- Huang, K.; Wang, Y.; Bai, Y. Gut microbiota and metabolites in atrial fibrillation patients and their changes after catheter ablation. Microbiol. Spectr. 2022, 10, e01077-21. [Google Scholar] [CrossRef]
- Zhao, Y.; Wang, Z. Gut microbiome and cardiovascular disease. Curr. Opin. Cardiol. 2020, 35, 207–218. [Google Scholar] [CrossRef]
- Kitai, T.; Kirsop, J.; Tang, W.H. Exploring the Microbiome in Heart Failure. Curr. Heart Fail. Rep. 2016, 13, 103–109. [Google Scholar] [CrossRef] [Green Version]
- Lekawanvijit, S. Role of Gut-Derived Protein-Bound Uremic Toxins in Cardiorenal Syndrome and Potential Treatment Modalities. Circ. J. 2015, 79, 2088–2097. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Li, H.Y.; Hu, X.M.; Zhang, Y.; Zhang, S.Y. Current understanding of gut microbiota alterations and related therapeutic intervention strategies in heart failure. Chin. Med. J. 2019, 132, 1843–1855. [Google Scholar] [CrossRef] [PubMed]
- Al-Rubaye, H.; Perfetti, G.; Kaski, J.C. The Role of Microbiota in Cardiovascular Risk: Focus on Trimethylamine Oxide. Curr. Probl. Cardiol. 2019, 44, 182–196. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Liu, J.; Zhou, B.; Wang, S. Microbial metabolites and heart failure: Friends or enemies? Front. Microbiol. 2022, 13, 956516. [Google Scholar] [CrossRef]
- Otto, C.M. Heartbeat: The gut microbiota and heart failure. Heart 2016, 102, 811–812. [Google Scholar] [CrossRef] [Green Version]
- Tang, W.H.; Wang, Z.; Fan, Y.; Levison, B.; Hazen, J.E.; Donahue, L.M.; Wu, Y.; Hazen, S.L. Prognostic value of elevated levels of intestinal microbe-generated metabolite trimethylamine-N-oxide in patients with heart failure: Refining the gut hypothesis. J. Am. Coll. Cardiol. 2014, 64, 1908–1914. [Google Scholar] [CrossRef] [Green Version]
- Bu, J.; Wang, Z. Cross-Talk between Gut Microbiota and Heart via the Routes of Metabolite and Immunity. Gastroenterol. Res. Pract. 2018, 2018, 6458094. [Google Scholar] [CrossRef]
- Zhu, W.; Gregory, J.C.; Org, E.; Buffa, J.A.; Gupta, N.; Wang, Z.; Li, L.; Fu, X.; Wu, Y.; Mehrabian, M.; et al. Gut Microbial Metabolite TMAO Enhances Platelet Hyperreactivity and Thrombosis Risk. Cell 2016, 165, 111–124. [Google Scholar] [CrossRef] [Green Version]
- Tang, W.W.; Hazen, S.L. Dietary metabolism, gut microbiota and acute heart failure. Heart 2016, 102, 813–814. [Google Scholar] [CrossRef] [Green Version]
- Stubbs, J.R.; House, J.A.; Ocque, A.J.; Zhang, S.; Johnson, C.; Kimber, C.; Schmidt, K.; Gupta, A.; Wetmore, J.B.; Nolin, T.D.; et al. Serum Trimethylamine-N-Oxide is Elevated in CKD and Correlates with Coronary Atherosclerosis Burden. J. Am. Soc. Nephrol. JASN 2016, 27, 305–313. [Google Scholar] [CrossRef] [Green Version]
- Tang, W.H.; Wang, Z.; Shrestha, K.; Borowski, A.G.; Wu, Y.; Troughton, R.W.; Klein, A.L.; Hazen, S.L. Intestinal microbiota-dependent phosphatidylcholine metabolites, diastolic dysfunction, and adverse clinical outcomes in chronic systolic heart failure. J. Card. Fail. 2015, 21, 91–96. [Google Scholar] [CrossRef] [Green Version]
- Trøseid, M.; Ueland, T.; Hov, J.R.; Svardal, A.; Gregersen, I.; Dahl, C.P.; Aakhus, S.; Gude, E.; Bjørndal, B.; Halvorsen, B.; et al. Microbiota-dependent metabolite trimethylamine-N-oxide is associated with disease severity and survival of patients with chronic heart failure. J. Intern. Med. 2015, 277, 717–726. [Google Scholar] [CrossRef]
- Li, W.; Huang, A.; Zhu, H.; Liu, X.; Huang, X.; Huang, Y.; Cai, X.; Lu, J.; Huang, Y. Gut microbiota-derived trimethylamine N-oxide is associated with poor prognosis in patients with heart failure. Med. J. Aust. 2020, 213, 374–379. [Google Scholar] [CrossRef]
- Zabell, A.; Tang, W.H. Targeting the Microbiome in Heart Failure. Curr. Treat. Options Cardiovasc. Med. 2017, 19, 27. [Google Scholar] [CrossRef]
- Jiang, Y.R.; Du, J.Y.; Wang, D.D.; Yang, X. miRNA-130a improves cardiac function by down-regulating TNF-α expression in a rat model of heart failure. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 8454–8461. [Google Scholar] [CrossRef]
- Zhou, X.; Li, J.; Guo, J.; Geng, B.; Ji, W.; Zhao, Q.; Li, J.; Liu, X.; Liu, J.; Guo, Z.; et al. Gut-dependent microbial translocation induces inflammation and cardiovascular events after ST-elevation myocardial infarction. Microbiome 2018, 6, 66. [Google Scholar] [CrossRef] [Green Version]
- Awoyemi, A.; Trøseid, M.; Arnesen, H.; Solheim, S.; Seljeflot, I. Markers of metabolic endotoxemia as related to metabolic syndrome in an elderly male population at high cardiovascular risk: A cross-sectional study. Diabetol. Metab. Syndr. 2018, 10, 59. [Google Scholar] [CrossRef] [Green Version]
- Kain, V.; Van Der Pol, W.; Mariappan, N.; Ahmad, A.; Eipers, P.; Gibson, D.L.; Gladine, C.; Vigor, C.; Durand, T.; Morrow, C.; et al. Obesogenic diet in aging mice disrupts gut microbe composition and alters neutrophil:lymphocyte ratio, leading to inflamed milieu in acute heart failure. FASEB J. 2019, 33, 6456–6469. [Google Scholar] [CrossRef]
- Pasini, E.; Aquilani, R.; Corsetti, G.; Dioguardi, F.S. Malnutrition and Gut Flora Dysbiosis: Specific Therapies for Emerging Comorbidities in Heart Failure. BioMed Res. Int. 2015, 2015, 382585. [Google Scholar] [CrossRef] [Green Version]
- Katsimichas, T.; Antonopoulos, A.S.; Katsimichas, A.; Ohtani, T.; Sakata, Y.; Tousoulis, D. The intestinal microbiota and cardiovascular disease. Cardiovasc. Res. 2019, 115, 1471–1486. [Google Scholar] [CrossRef]
- Kummen, M.; Mayerhofer, C.C.K.; Vestad, B.; Broch, K.; Awoyemi, A.; Storm-Larsen, C.; Ueland, T.; Yndestad, A.; Hov, J.R.; Trøseid, M. Gut Microbiota Signature in Heart Failure Defined from Profiling of 2 Independent Cohorts. J. Am. Coll. Cardiol. 2018, 71, 1184–1186. [Google Scholar] [CrossRef]
- Cui, X.; Ye, L.; Li, J.; Jin, L.; Wang, W.; Li, S.; Bao, M.; Wu, S.; Li, L.; Geng, B.; et al. Metagenomic and metabolomic analyses unveil dysbiosis of gut microbiota in chronic heart failure patients. Sci. Rep. 2018, 8, 635. [Google Scholar] [CrossRef]
- Gutiérrez-Calabrés, E.; Ortega-Hernández, A.; Modrego, J.; Gómez-Gordo, R.; Caro-Vadillo, A.; Rodríguez-Bobada, C.; González, P.; Gómez-Garre, D. Gut Microbiota Profile Identifies Transition from Compensated Cardiac Hypertrophy to Heart Failure in Hypertensive Rats. Hypertension 2020, 76, 1545–1554. [Google Scholar] [CrossRef]
- Haiser, H.J.; Gootenberg, D.B.; Chatman, K.; Sirasani, G.; Balskus, E.P.; Turnbaugh, P.J. Predicting and manipulating cardiac drug inactivation by the human gut bacterium Eggerthella lenta. Science 2013, 341, 295–298. [Google Scholar] [CrossRef] [Green Version]
- Zhernakova, A.; Kurilshikov, A.; Bonder, M.J.; Tigchelaar, E.F.; Schirmer, M.; Vatanen, T.; Mujagic, Z.; Vila, A.V.; Falony, G.; Vieira-Silva, S.; et al. Population-based metagenomics analysis reveals markers for gut microbiome composition and diversity. Science 2016, 352, 565–569. [Google Scholar] [CrossRef] [Green Version]
- Masenga, S.K.; Hamooya, B.; Hangoma, J. Recent advances in modulation of cardiovascular diseases by the gut microbiota. J Hum. Hypertens. 2022, 1–8. [Google Scholar] [CrossRef]
- Wu, H.; Hu, T.; Hao, H.; Hill, M.A. Inflammatory bowel disease and cardiovascular diseases: A concise review. Eur. Heart J. Open 2022, 2, oeab029. [Google Scholar] [CrossRef]
- Bunu, D.M.; Timofte, C.E.; Ciocoiu, M.; Floria, M. Cardiovascular Manifestations of Inflammatory Bowel Disease: Pathogenesis, Diagnosis, and Preventive Strategies. Gastroenterol. Res. Pract. 2019, 13, 3012509. [Google Scholar] [CrossRef] [Green Version]
- Larsson, E.; Tremaroli, V.; Lee, Y.S.; Koren, O.; Nookaew, I.; Fricker, A.; Nielsen, J.; Ley, R.E.; Bäckhed, F. Analysis of gut microbial regulation of host gene expression along the length of the gut and regulation of gut microbial ecology through MyD88. Gut 2012, 61, 1124–1131. [Google Scholar] [CrossRef]
- Brown, J.M.; Hazen, S.L. The gut microbial endocrine organ: Bacterially derived signals driving cardiometabolic diseases. Annu. Rev. Med. 2015, 66, 343–359. [Google Scholar] [CrossRef] [Green Version]
- McMillan, A.; Hazen, S.L. Gut Microbiota Involvement in Ventricular Remodeling Post-Myocardial Infarction. Circulation 2019, 139, 660–662. [Google Scholar] [CrossRef] [PubMed]
- Drapkina, O.M.; Yafarova, A.A.; Kaburova, A.N. Targeting Gut Microbiota as a Novel Strategy for Prevention and Treatment of Hypertension, Atrial Fibrillation and Heart Failure: Current Knowledge and Future Perspectives. Biomedicines 2022, 10, 2019. [Google Scholar] [CrossRef] [PubMed]
- Costanza, A.C.; Moscavitch, S.D.; Faria Neto, H.C.; Mesquita, E.T. Probiotic therapy with Saccharomyces boulardii for heart failure patients: A randomized, double-blind, placebo-controlled pilot trial. Int. J. Cardiol. 2015, 179, 348–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerisano, G.; Buonamici, P.; Valenti, R.; Sciagrà, R.; Raspanti, S.; Santini, A.; Carrabba, N.; Dovellini, E.V.; Romito, R.; Pupi, A.; et al. Early short-term doxycycline therapy in patients with acute myocardial infarction and left ventricular dysfunction to prevent the ominous progression to adverse remodelling: The TIPTOP trial. Eur. Heart J. 2014, 35, 184–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Von Haehling, S.; Schefold, J.C.; Jankowska, E.A.; Springer, J.; Vazir, A.; Kalra, P.R.; Sandek, A.; Fauler, G.; Stojakovic, T.; Trauner, M.; et al. Ursodeoxycholic acid in patients with chronic heart failure: A double-blind, randomized, placebo-controlled, crossover trial. J. Am. Coll. Cardiol. 2012, 59, 585–592. [Google Scholar] [CrossRef]


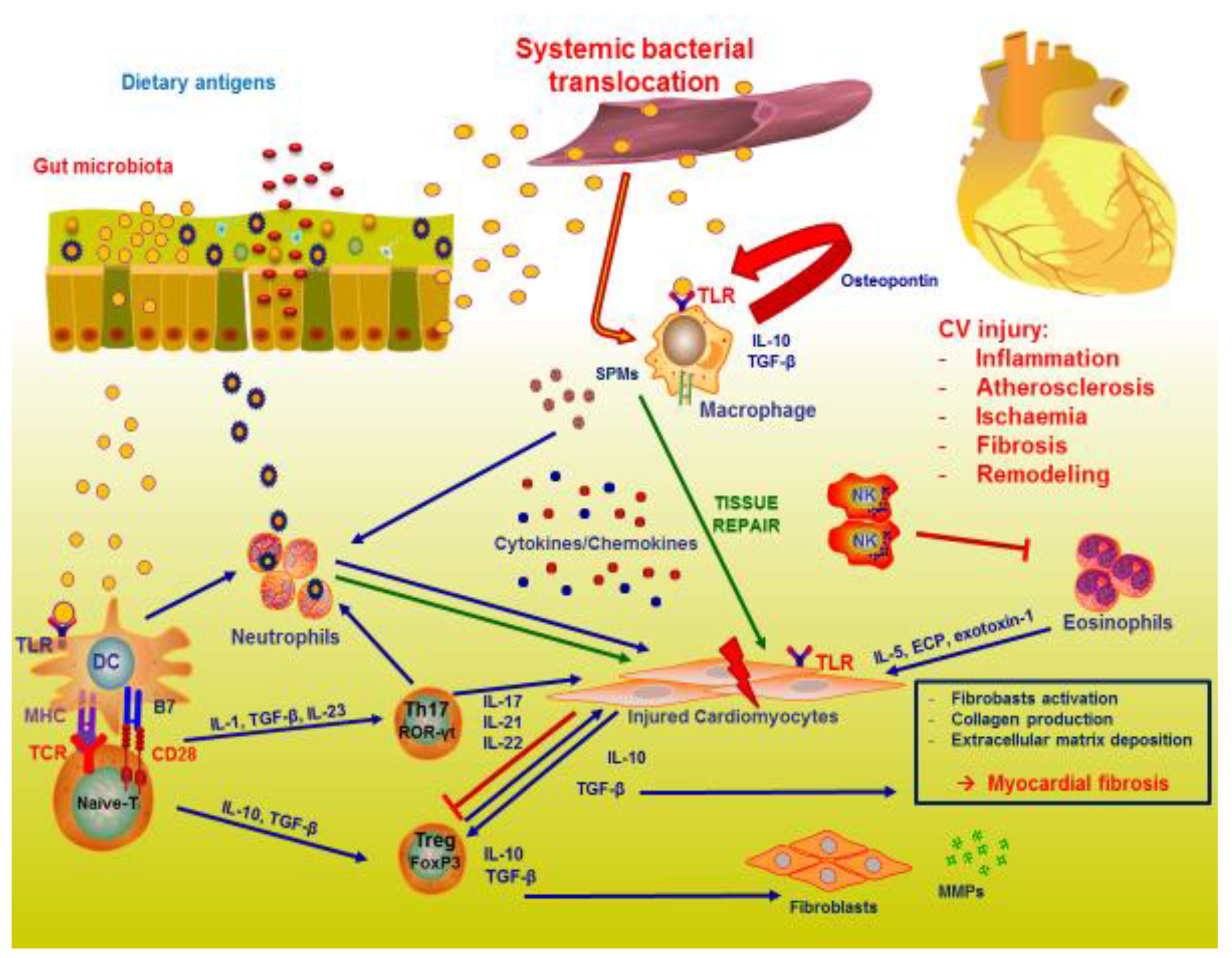{kind=link}
| Cytokines | Effects | Productive Cells | |
|---|---|---|---|
| Pro-Inflammatory Cytokines | TNF-α IL-1β |
| Neutrophils, Dendritic cells, Macrophages, Mononuclear cells |
| IL-6 IL-8 IL-13 MIP-1α ICAM-1, VCAM-1 CCR2, CCL2 TLRs |
| Neutrophils, Dendritic cells, Macrophages, Mononuclear cells | |
| IL-6 | Neutrophils, Dendritic cells, Macrophages, Mononuclear cells | ||
| Anti-Inflammatory Cytokines | IL-10 |
| Macrophages, Mononuclear cells, Tregs |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cianci, R.; Franza, L.; Borriello, R.; Pagliari, D.; Gasbarrini, A.; Gambassi, G. The Role of Gut Microbiota in Heart Failure: When Friends Become Enemies. Biomedicines 2022, 10, 2712. https://doi.org/10.3390/biomedicines10112712
Cianci R, Franza L, Borriello R, Pagliari D, Gasbarrini A, Gambassi G. The Role of Gut Microbiota in Heart Failure: When Friends Become Enemies. Biomedicines. 2022; 10(11):2712. https://doi.org/10.3390/biomedicines10112712
Chicago/Turabian StyleCianci, Rossella, Laura Franza, Raffaele Borriello, Danilo Pagliari, Antonio Gasbarrini, and Giovanni Gambassi. 2022. "The Role of Gut Microbiota in Heart Failure: When Friends Become Enemies" Biomedicines 10, no. 11: 2712. https://doi.org/10.3390/biomedicines10112712
APA StyleCianci, R., Franza, L., Borriello, R., Pagliari, D., Gasbarrini, A., & Gambassi, G. (2022). The Role of Gut Microbiota in Heart Failure: When Friends Become Enemies. Biomedicines, 10(11), 2712. https://doi.org/10.3390/biomedicines10112712