
«One Small Step for Mouse»: High CO2 Inhalation as a New Therapeutic Strategy for Parkinson’s Disease

Abstract
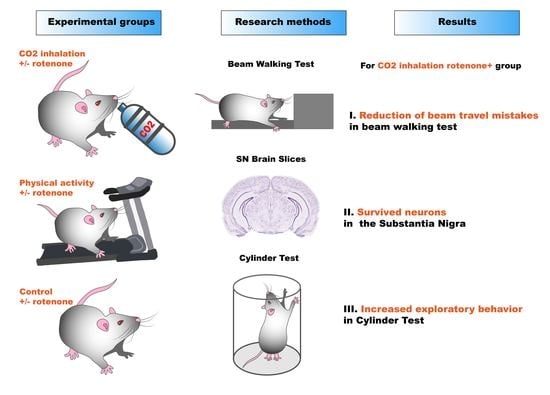
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Experimental Design and Induction of Parkinsonism
- Control mice (I) (received only vehicle) were divided randomly in 3 groups:
- I-0 negative control (n = 3)
- I-a moderate physical activity treatment (n = 3)
- I-b CO2 treatment (n = 3).
- Rotenone treated (II) mice were randomly divided into 3 groups:
- II-0 negative control (n = 9)
- II-a moderate physical activity treatment (n = 9)
- II-b CO2 treatment (n = 8).
2.3. Experimental Therapy Treatments
2.3.1. Forced Moderate Physical Activity
2.3.2. CO2 Inhalation
2.4. Behavioral Evaluation
2.4.1. Beam Walking Test
2.4.2. Cylinder Test
2.5. Histology Preparations
2.6. Primary Rat Cortical Neuroglial Culture
Cell Viability
2.7. Statistical Analysis
3. Results
3.1. Action of Sodium Lacatate on Rotenone Exposed Neurons
3.2. Development of PD-like Symptoms in Rotenone Exposed Mice
3.3. Moderate Physical Activity Treatment
3.4. High CO2 Treatment
3.5. Survival Analysis
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rocca, W.A. The future burden of Parkinson’s disease. Mov. Disord. 2018, 33, 8–9. [Google Scholar] [CrossRef] [PubMed]
- Dauer, W.; Przedborski, S. Parkinson’s disease: Mechanisms and models. Neuron 2003, 39, 889–909. [Google Scholar] [CrossRef] [Green Version]
- Oertel, W.; Schulz, J.B. Current and experimental treatments of Parkinson disease: A guide for neuroscientists. J. Neurochem. 2016, 139, 325–337. [Google Scholar] [CrossRef] [PubMed]
- Kramer, M.L.; Schulz-Schaeffer, W.J. Presynaptic α-Synuclein aggregates, not Lewy bodies, cause neurodegeneration in de-mentia with Lewy bodies. J. Neurosci. 2007, 27, 1405–1410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winklhofer, K.F.; Haass, C. Mitochondrial dysfunction in Parkinson’s disease. Biochim. Biophys. Acta—Mol. Basis Dis. 2010, 1802, 29–44. [Google Scholar] [CrossRef] [PubMed]
- Venderova, K.; Park, D. Programmed cell death in Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2012, 2, a009365. [Google Scholar] [CrossRef] [Green Version]
- Lynch-Day, M.A.; Mao, K.; Wang, K.; Zhao, M.; Klionsky, D.J. The role of autophagy in Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2012, 2, a009357. [Google Scholar] [CrossRef] [Green Version]
- Mercado, G.; Valdés, P.; Hetz, C. An ERcentric view of Parkinson’s disease. Trends Mol. Med. 2013, 19, 165–175. [Google Scholar] [CrossRef]
- Zaichick, S.V.; McGrath, K.M.; Caraveo, G. The role of Ca2+ signaling in Parkinson’s disease. Dis. Model. Mech. 2017, 10, 519–535. [Google Scholar] [CrossRef] [Green Version]
- McCormack, A.L.; Thiruchelvamb, M.; Manning-Bog, A.B.; Thiffaulta, C.; Langstona, J.W.; Cory-Slechta, D.A.; Di Monte, D.A. Environmental risk factors and Parkinson’s disease: Selective degeneration of nigral dopaminergic neurons caused by the herbicide paraquat. Neurobiol. Dis. 2002, 10, 119–127. [Google Scholar] [CrossRef]
- Konnova, E.A.; Swanberg, M. Animal Models of Parkinson’s Disease. In Parkinson’s Disease: Pathogenesis and Clinical Aspects; Stoker, T.B., Greenland, J.C., Eds.; Codon Publications: Brisbane, Australia, 2018. [Google Scholar] [CrossRef]
- Tieu, K. A guide to neurotoxic animal models of Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2011, 1, a009316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Betarbet, R.; Sherer, T.B.; MacKenzie, G.; Garcia-Osuna, M.; Panov, A.V.; Greenamyre, J.T. Chronic systemic pesticide exposure reproduces features of Parkinson’s disease. Nat. Neurosci. 2000, 3, 1301–1306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sherer, T.B.; Betarbet, R.; Stout, A.K.; Lund, S.; Baptista, M.; Panov, A.V.; Cookson, M.R.; Greenamyre, J.T. An in vitro model of Parkinson’s disease: Linking mitochondrial impairment to altered α-synuclein metabolism and oxidative damage. J. Neurosci. 2002, 22, 7006–7015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spatola, M.; Wider, C. Genetics of Parkinson’s disease: The yield. Park. Relat. Disord. 2014, 20, S35–S38. [Google Scholar] [CrossRef]
- Siddiqui, I.J.; Pervaiz, N.; Abbasi, A.A. The Parkinson Disease gene SNCA: Evolutionary and structural insights with pathological implication. Sci. Rep. 2016, 6, 24475. [Google Scholar] [CrossRef]
- Exner, N.; Lutz, A.K.; Haass, C.; Winklhofer, K.F. Mitochondrial dysfunction in Parkinson’s disease: Molecular mechanisms and pathophysiological consequences. EMBO J. 2012, 31, 3038–3062. [Google Scholar] [CrossRef] [Green Version]
- Williams, E.T.; Chen, X.; Moore, D.J. VPS35, the retromer complex and Parkinson’s disease. J. Park. Dis. 2017, 7, 219–233. [Google Scholar] [CrossRef] [Green Version]
- Mehta, Z.B.; Fine, N.; Pullen, T.J.; Cane, M.C.; Hu, M.; Chabosseau, P.; Meur, G.; Velayos-Baeza, A.; Monaco, A.P.; Marselli, L.; et al. Changes in the expression of the type 2 diabetes-associated gene VPS13C in the β-cell are associated with glucose intolerance in humans and mice. Am. J. Physiol. Endocrinol. Metab. 2016, 311, E488–E507. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Liu, W.; Li, R.; Yang, H. Mitophagy in Parkinson’s disease: From pathogenesis to treatment. Cells 2019, 8, 712. [Google Scholar] [CrossRef]
- Youle, R.J.; Narendra, D.P. Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol. 2011, 12, 9–14. [Google Scholar] [CrossRef]
- Berezhnov, A.V.; Soutar, M.P.; Fedotova, E.I.; Frolova, M.S.; Plun-Favreau, H.; Zinchenko, V.P.; Abramov, A.Y. Intracellular pH Modulates Autophagy and Mitophagy. J. Biol. Chem. 2016, 291, 8701–8708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fedotova, E.I.; Dolgacheva, L.P.; Abramov, A.Y.; Berezhnov, A.V. Lactate and Pyruvate Activate Autophagy and Mitophagy that Protect Cells in Toxic Model of Parkinson’s Disease. Mol. Neurobiol. 2022, 59, 177–190. [Google Scholar] [CrossRef] [PubMed]
- Messias, L.H.D.; Gobatto, C.A.; Beck, W.R.; Manchado-Gobatto, F.B. The lactate minimum test: Concept, methodological aspects and insights for future investigations in human and animal models. Front. Physiol. 2017, 8, 389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Åstrand, P.-O.; Hallbäck, I.; Hedman, R.; Saltin, B. Blood lactates after prolonged severe exercise. J. Appl. Physiol. 1963, 18, 619–622. [Google Scholar] [CrossRef]
- Du, J.; Price, M.P.; Taugher, R.J.; Grigsby, D.; Ash, J.J.; Stark, A.C.; Saad, Z.H.; Singh, K.; Mandal, J.; Wemmie, A.J.; et al. Transient acidosis while retrieving a fear-related memory enhances its lability. eLife 2017, 6, e22564. [Google Scholar] [CrossRef] [PubMed]
- Magnotta, V.A.; Heo, H.-Y.; Dlouhy, B.J.; Dahdaleh, N.S.; Follmer, R.L.; Thedens, D.R.; Welsh, M.J.; Wemmie, J.A. Detecting activity-evoked pH changes in human brain. Proc. Natl. Acad. Sci. USA 2012, 109, 8270–8273. [Google Scholar] [CrossRef] [Green Version]
- Fleming, S.M.; Ekhator, O.R.; Ghisays, V. Assessment of sensorimotor function in mouse models of Parkinson’s disease. J. Vis. Exp. 2013, 76, e50303. [Google Scholar] [CrossRef] [Green Version]
- Dias, V.; Junn, E.; Mouradian, M.M. The role of oxidative stress in Parkinson’s disease. J. Parkinsons Dis. 2013, 3, 461–491. [Google Scholar] [CrossRef] [Green Version]
- Schapira, A.H.; Cooper, J.M.; Dexter, D.; Clark, J.B.; Jenner, P.; Marsden, C.D. Mitochondrial complex I deficiency in Parkin-son’s disease. J. Neurochem. 1990, 54, 823–827. [Google Scholar] [CrossRef]
- Innos, J.; Hickey, M.A. Using Rotenone to Model Parkinson’s Disease in Mice: A Review of the Role of Pharmacokinetics. Chem. Res. Toxicol. 2021, 34, 1223–1239. [Google Scholar] [CrossRef]
- Bastioli, G.; Arnold, J.C.; Mancini, M.; Mar, A.C.; Gamallo–Lana, B.; Saadipour, K.; Chao, M.V.; Rice, M.E. Voluntary Exercise Boosts Striatal Dopamine Release: Evidence for the Necessary and Sufficient Role of BDNF. J. Neurosci. 2022, 42, 4725–4736. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Saavedra, M.; De Repentigny, Y.; Yang, D.; O’Meara, R.W.; Yan, K.; Hashem, L.E.; Racacho, L.; Ioshikhes, I.; Bulman, D.E.; Parks, R.J.; et al. Voluntary Running Triggers VGF-Mediated Oligodendrogenesis to Prolong the Lifespan of Snf2h-Null Ataxic Mice. Cell Rep. 2016, 17, 862–875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komilova, N.R.; Angelova, P.R.; Berezhnov, A.V.; Stelmashchuk, O.A.; Mirkhodjaev, U.Z.; Houlden, H.; Gourine, A.V.; Esteras, N.; Abramov, A.Y. Metabolically induced intracellular pH changes activate mitophagy, autophagy, and cell protection in familial forms of Parkinson’s disease. FEBS J. 2022, 289, 699–711. [Google Scholar] [CrossRef] [PubMed]
- Cannon, J.R.; Tapias, V.; Na, H.M.; Honick, A.S.; Drolet, R.E.; Greenamyre, J.T. A highly reproducible rotenone model of Parkinson’s disease. Neurobiol. Dis. 2009, 34, 279–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, T.; Li, C.; Han, B.; Wang, Z.; Meng, X.; Zhang, L.; He, J.; Fu, F. Neuroprotective effects of Danshensu on rote-none-induced Parkinson’s disease models in vitro and in vivo. BMC Complement. Med. Ther. 2020, 20, 20. [Google Scholar] [CrossRef] [Green Version]
- Park, J.-S.; Koentjoro, B.; Sue, C.M. Commentary: Nix restores mitophagy and mitochondrial function to protect against PINK1/Parkin-related Parkinson’s disease. Front. Mol. Neurosci. 2017, 10, 297. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nadeev, A.D.; Kritskaya, K.A.; Fedotova, E.I.; Berezhnov, A.V. «One Small Step for Mouse»: High CO2 Inhalation as a New Therapeutic Strategy for Parkinson’s Disease. Biomedicines 2022, 10, 2832. https://doi.org/10.3390/biomedicines10112832
Nadeev AD, Kritskaya KA, Fedotova EI, Berezhnov AV. «One Small Step for Mouse»: High CO2 Inhalation as a New Therapeutic Strategy for Parkinson’s Disease. Biomedicines. 2022; 10(11):2832. https://doi.org/10.3390/biomedicines10112832
Chicago/Turabian StyleNadeev, Alexander D., Kristina A. Kritskaya, Evgeniya I. Fedotova, and Alexey V. Berezhnov. 2022. "«One Small Step for Mouse»: High CO2 Inhalation as a New Therapeutic Strategy for Parkinson’s Disease" Biomedicines 10, no. 11: 2832. https://doi.org/10.3390/biomedicines10112832
APA StyleNadeev, A. D., Kritskaya, K. A., Fedotova, E. I., & Berezhnov, A. V. (2022). «One Small Step for Mouse»: High CO2 Inhalation as a New Therapeutic Strategy for Parkinson’s Disease. Biomedicines, 10(11), 2832. https://doi.org/10.3390/biomedicines10112832