
Effect of Renal Ischemia Reperfusion on Brain Neuroinflammation
 , , , , , , ,
, , , , , , ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Experimental Animals
2.2. Animal Model of Kidney Ischemia Reperfusion
2.3. In Vivo BBB Permeability Measurements
2.4. Immunohistochemistry (IHC)
2.5. Immunofluorescence
2.6. Terminal Deoxynucleotidyl Transferase dUTP Nick-End Labelling (TUNEL) Assay
2.7. Western Blot Analysis
2.8. Reverse Transcription and Quantitative PCR (qPCR) Analysis
2.9. Single Microglia Separate Using CD1b+ Beads Kit
2.10. Immunofluorescence (IF) Analysis
2.11. Statistical Analysis
3. Results
3.1. Kidney IR Injury Increases the Permeability of BBB
3.2. Kidney IR Injury Induced Microglia and A1 Astrocyte Activation in Striatum and VM
3.3. Kidney IR Injury Induced Microglia and A1 Astrocyte Activation in the VM
3.4. Microglia and A1 Astrocytes Were Activated by Kidney IR Injury in the Hippocampus and Cortex
3.5. Kidney IR Injury Induced Microglia Activation and A1 Astrocyte Formation in the Cortex
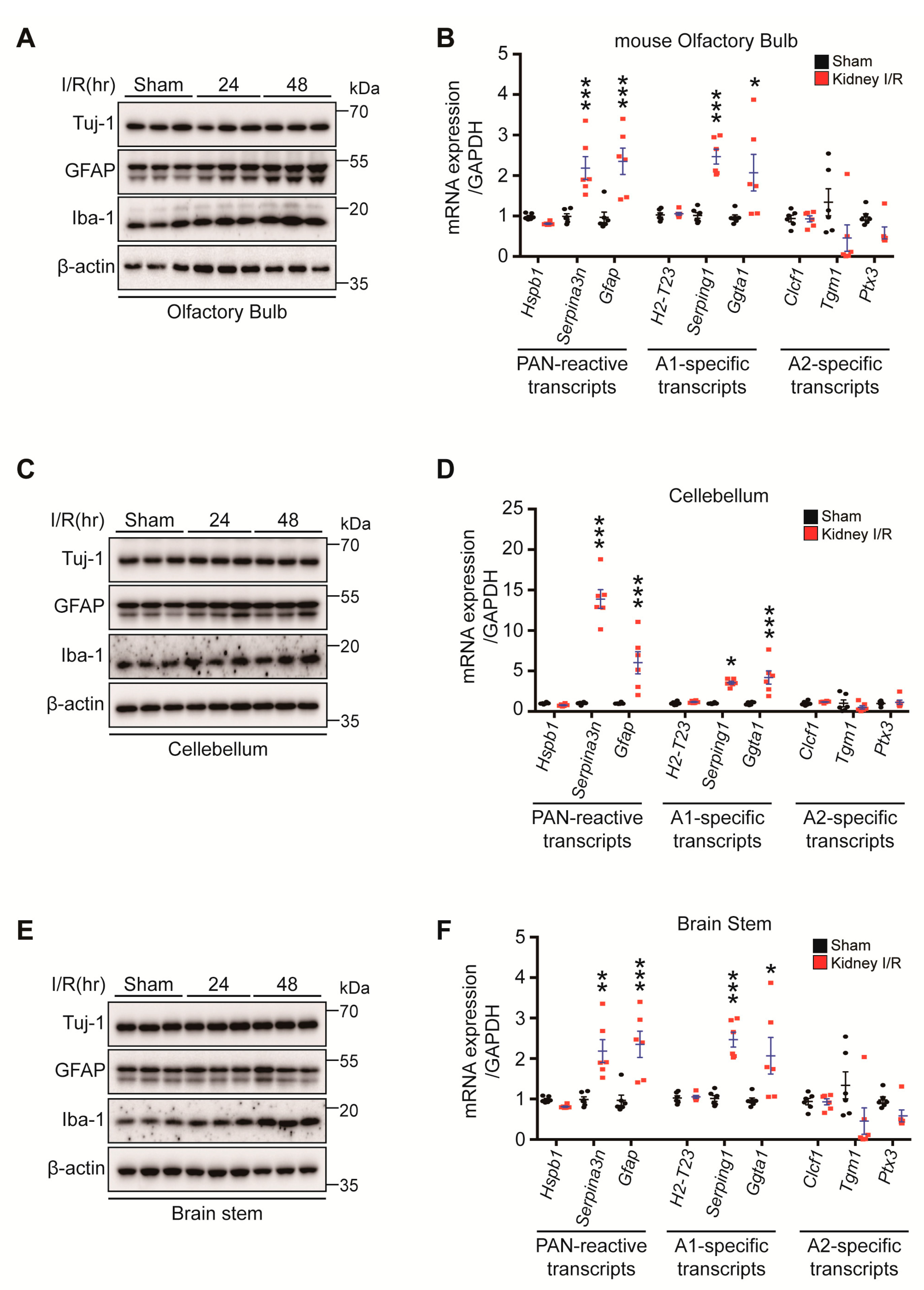
3.6. Kidney IR Induced Microglia Activation and A1 Astrocyte Formation in the Olfactory Bulb, Cerebellum, and Brain Stem
3.7. Colocalization of Iba-1 and Cytokines, Neutrophils and Cytokines in STR, VM, HIP and CTX
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sharfuddin, A.A.; Molitoris, B.A. Pathophysiology of ischemic acute kidney injury. Nat. Rev. Nephrol. 2011, 7, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Leither, M.D.; Murphy, D.P.; Bicknese, L.; Reule, S.; Vock, D.M.; Ishani, A.; Foley, R.N.; Drawz, P.E. The impact of outpatient acute kidney injury on mortality and chronic kidney disease: A retrospective cohort study. Nephrol. Dial. Transplant. 2019, 34, 493–501. [Google Scholar] [CrossRef] [PubMed]
- Meersch, M.; Kullmar, M.; Schmidt, C.; Gerss, J.; Weinhage, T.; Margraf, A.; Ermert, T.; Kellum, J.A.; Zarbock, A. Long-Term Clinical Outcomes after Early Initiation of RRT in Critically Ill Patients with AKI. J. Am. Soc. Nephrol. 2018, 29, 1011–1019. [Google Scholar] [CrossRef] [Green Version]
- Sawhney, S.; Beaulieu, M.; Black, C.; Djurdjev, O.; Espino-Hernandez, G.; Marks, A.; McLernon, D.J.; Sheriff, Z.; Levin, A. Predicting kidney failure risk after acute kidney injury among people receiving nephrology clinic care. Nephrol. Dial. Transplant. 2018, 35, 836–845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- See, E.J.; Jayasinghe, K.; Glassford, N.; Bailey, M.; Johnson, D.W.; Polkinghorne, K.R.; Toussaint, N.D.; Bellomo, R. Long-term risk of adverse outcomes after acute kidney injury: A systematic review and meta-analysis of cohort studies using consensus definitions of exposure. Kidney Int. 2019, 95, 160–172. [Google Scholar] [CrossRef]
- Go, A.S.; Hsu, C.Y.; Yang, J.; Tan, T.C.; Zheng, S.; Ordonez, J.D.; Liu, K.D. Acute Kidney Injury and Risk of Heart Failure and Atherosclerotic Events. Clin. J. Am. Soc. Nephrol. 2018, 13, 833–841. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Park, S.; Kang, M.W.; Yoo, H.W.; Han, K.; Kim, Y.; Lee, J.P.; Joo, K.W.; Lim, C.S.; Kim, Y.S.; et al. Postdischarge long-term cardiovascular outcomes of intensive care unit survivors who developed dialysis-requiring acute kidney injury after cardiac surgery. J. Crit. Care 2019, 50, 92–98. [Google Scholar] [CrossRef]
- Faubel, S. Pulmonary complications after acute kidney injury. Adv. Chronic. Kidney Dis. 2008, 15, 284–296. [Google Scholar] [CrossRef]
- Park, S.W.; Chen, S.W.; Kim, M.; Brown, K.M.; Kolls, J.K.; D’Agati, V.D.; Lee, H.T. Cytokines induce small intestine and liver injury after renal ischemia or nephrectomy. Lab. Investig. 2011, 91, 63–84. [Google Scholar] [CrossRef] [Green Version]
- Odutayo, A.; Wong, C.X.; Farkouh, M.; Altman, D.G.; Hopewell, S.; Emdin, C.A.; Hunn, B.H. AKI and Long-Term Risk for Cardiovascular Events and Mortality. J. Am. Soc. Nephrol. 2017, 28, 377–387. [Google Scholar] [CrossRef] [Green Version]
- Nongnuch, A.; Panorchan, K.; Davenport, A. Brain-kidney crosstalk. Crit. Care 2014, 18, 225. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Liang, Y.; Chigurupati, S.; Lathia, J.D.; Pletnikov, M.; Sun, Z.; Crow, M.; Ross, C.A.; Mattson, M.P.; Rabb, H. Acute kidney injury leads to inflammation and functional changes in the brain. J. Am. Soc. Nephrol. 2008, 19, 1360–1370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parpura, V.; Heneka, M.T.; Montana, V.; Oliet, S.H.R.; Schousboe, A.; Haydon, P.G.; Stout Jr, R.F.; Spray, D.C.; Reichenbach, A.; Pannicke, T.; et al. Glial cells in (patho)physiology. J. Neurochem. 2012, 121, 4–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colonna, M.; Butovsky, O. Microglia Function in the Central Nervous System During Health and Neurodegeneration. Annu. Rev. Immunol. 2017, 35, 441–468. [Google Scholar] [CrossRef] [PubMed]
- Tsodyks, M.V.; Markram, H. The neural code between neocortical pyramidal neurons depends on neurotransmitter release’s probability. Proc. Natl. Acad. Sci. USA 1997, 94, 719–723. [Google Scholar] [CrossRef] [Green Version]
- Mosley, R.L.; Benner, E.J.; Kadiu, I.; Thomas, M.; Boska, M.D.; Hasan, K.; Laurie, C.; Gendelman, H.E. Neuroinflammation, Oxidative Stress and the Pathogenesis of Parkinson’s Disease. Clin. Neurosci. Res. 2006, 6, 261–281. [Google Scholar] [CrossRef] [Green Version]
- Glass, C.K.; Saijo, K.; Winner, B.; Marchetto, M.C.; Gage, F.H. Mechanisms underlying inflammation in neurodegeneration. Cell 2010, 140, 918–934. [Google Scholar] [CrossRef] [Green Version]
- Block, M.L.; Hong, J.S. Microglia and inflammation-mediated neurodegeneration: Multiple triggers with a common mechanism. Prog. Neurobiol. 2005, 76, 77–98. [Google Scholar] [CrossRef]
- Hirsch, E.C.; Hunot, S. Neuroinflammation in Parkinson’s disease: A target for neuroprotection? Lancet Neurol. 2009, 8, 382–397. [Google Scholar] [CrossRef]
- Vezzani, A.; Aronica, E.; Mazarati, A.; Pittman, Q.J. Epilepsy and brain inflammation. Exp. Neurol. 2013, 244, 11–21. [Google Scholar] [CrossRef]
- Park, E.J.; Dusabimana, T.; Je, J.; Jeong, K.; Yun, S.P.; Kim, H.J.; Kim, H.; Park, S.W. Honokiol Protects the Kidney from Renal Ischemia and Reperfusion Injury by Upregulating the Glutathione Biosynthetic Enzymes. Biomedicines 2020, 8, 352. [Google Scholar] [CrossRef]
- Yepes, M.; Sandkvist, M.; Moore, E.G.; Bugge, T.H.; Strickland, D.K.; Lawrence, D.A. Tissue-type plasminogen activator induces opening of the blood-brain barrier via the LDL receptor-related protein. J. Clin. Investig. 2003, 112, 1533–1540. [Google Scholar] [CrossRef] [PubMed]
- Carroll, J.A.; Race, B.; Williams, K.; Striebel, J.; Chesebro, B. RNA-seq and network analysis reveal unique glial gene expression signatures during prion infection. Mol. Brain 2020, 13, 71. [Google Scholar] [CrossRef] [PubMed]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Munch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellomo, R.; Kellum, J.A.; Ronco, C. Acute kidney injury. Lancet 2012, 380, 756–766. [Google Scholar] [CrossRef]
- Kim, D.J.; Park, S.H.; Sheen, M.R.; Jeon, U.S.; Kim, S.W.; Koh, E.S.; Woo, S.K. Comparison of experimental lung injury from acute renal failure with injury due to sepsis. Respiration 2006, 73, 815–824. [Google Scholar] [CrossRef]
- Jorres, A.; Gahl, G.M.; Dobis, C.; Polenakovic, M.H.; Cakalaroski, K.; Rutkowski, B.; Kisielnicka, E.; Krieter, D.H.; Rumpf, K.W.; Guenther, C.; et al. Haemodialysis-membrane biocompatibility and mortality of patients with dialysis-dependent acute renal failure: A prospective randomised multicentre trial. International Multicentre Study Group. Lancet 1999, 354, 1337–1341. [Google Scholar] [CrossRef]
- Francoz, C.; Glotz, D.; Moreau, R.; Durand, F. The evaluation of renal function and disease in patients with cirrhosis. J. Hepatol. 2010, 52, 605–613. [Google Scholar] [CrossRef] [Green Version]
- Burn, D.J.; Bates, D. Neurology and the kidney. J. Neurol. Neurosurg. Psychiatry 1998, 65, 810–821. [Google Scholar] [CrossRef] [Green Version]
- Brouns, R.; De Deyn, P.P. Neurological complications in renal failure: A review. Clin. Neurol. Neurosurg. 2004, 107, 1–16. [Google Scholar] [CrossRef]
- Hoke, T.S.; Douglas, I.S.; Klein, C.L.; He, Z.; Fang, W.; Thurman, J.M.; Tao, Y.; Dursun, B.; Voelkel, N.F.; Edelstein, C.L.; et al. Acute renal failure after bilateral nephrectomy is associated with cytokine-mediated pulmonary injury. J. Am. Soc. Nephrol. 2007, 18, 155–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ronco, C.; Haapio, M.; House, A.A.; Anavekar, N.; Bellomo, R. Cardiorenal syndrome. J. Am. Coll. Cardiol. 2008, 52, 1527–1539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyazawa, S.; Watanabe, H.; Miyaji, C.; Hotta, O.; Abo, T. Leukocyte accumulation and changes in extra-renal organs during renal ischemia reperfusion in mice. J. Lab. Clin. Med. 2002, 139, 269–278. [Google Scholar] [CrossRef] [PubMed]
- Golab, F.; Kadkhodaee, M.; Zahmatkesh, M.; Hedayati, M.; Arab, H.; Schuster, R.; Zahedi, K.; Lentsch, A.B.; Soleimani, M. Ischemic and non-ischemic acute kidney injury cause hepatic damage. Kidney Int. 2009, 75, 783–792. [Google Scholar] [CrossRef] [Green Version]
- Kniesel, U.; Wolburg, H. Tight junctions of the blood-brain barrier. Cell Mol. Neurobiol. 2000, 20, 57–76. [Google Scholar] [CrossRef]
- Rothwell, N.J. Annual review prize lecture cytokines—Killers in the brain? J. Physiol. 1999, 514 Pt 1, 3–17. [Google Scholar] [CrossRef] [PubMed]
- Vitkovic, L.; Konsman, J.P.; Bockaert, J.; Dantzer, R.; Homburger, V.; Jacque, C. Cytokine signals propagate through the brain. Mol. Psychiatry 2000, 5, 604–615. [Google Scholar] [CrossRef] [Green Version]
- Aron, A.R.; Schlaghecken, F.; Fletcher, P.C.; Bullmore, E.T.; Eimer, M.; Barker, R.; Sahakian, B.J.; Robbins, T.W. Inhibition of subliminally primed responses is mediated by the caudate and thalamus: Evidence from functional MRI and Huntington’s disease. Brain 2003, 126, 713–723. [Google Scholar] [CrossRef] [Green Version]
- Band, G.P.; van Boxtel, G.J. Inhibitory motor control in stop paradigms: Review and reinterpretation of neural mechanisms. Acta Psychol. 1999, 101, 179–211. [Google Scholar] [CrossRef]
- Kaji, R. Basal ganglia as a sensory gating devise for motor control. J. Med. Investig. 2001, 48, 142–146. [Google Scholar]
- Mink, J.W. The basal ganglia: Focused selection and inhibition of competing motor programs. Prog. Neurobiol. 1996, 50, 381–425. [Google Scholar] [CrossRef]
- Dubois, B.; Defontaines, B.; Deweer, B.; Malapani, C.; Pillon, B. Cognitive and behavioral changes in patients with focal lesions of the basal ganglia. Adv. Neurol. 1995, 65, 29–41. [Google Scholar] [PubMed]
- Saint-Cyr, J.A.; Taylor, A.E.; Nicholson, K. Behavior and the basal ganglia. Adv. Neurol. 1995, 65, 1–28. [Google Scholar]
- Korotkova, T.M.; Ponomarenko, A.A.; Brown, R.E.; Haas, H.L. Functional diversity of ventral midbrain dopamine and GABAergic neurons. Mol. Neurobiol. 2004, 29, 243–259. [Google Scholar] [CrossRef]
- Neuhoff, H.; Neu, A.; Liss, B.; Roeper, J. I(h) channels contribute to the different functional properties of identified dopaminergic subpopulations in the midbrain. J. Neurosci. 2002, 22, 1290–1302. [Google Scholar] [CrossRef] [Green Version]
- Blumenstock, S.; Dudanova, I. Cortical and Striatal Circuits in Huntington’s Disease. Front. Neurosci. 2020, 14, 82. [Google Scholar] [CrossRef]
- Ásgrímsdóttir, E.S.; Arenas, E. Midbrain Dopaminergic Neuron Development at the Single Cell Level: In vivo and in Stem Cells. Front. Cell Dev. Biol. 2020, 8, 463. [Google Scholar] [CrossRef]
- Fahn, S. Description of Parkinson’s disease as a clinical syndrome. Ann. N. Y. Acad. Sci. 2003, 991, 1–14. [Google Scholar] [CrossRef]
- Marr, D. Simple memory: A theory for archicortex. Philos. Trans. R. Soc. Lond B Biol. Sci. 1971, 262, 23–81. [Google Scholar] [CrossRef]
- Small, S.A.; Schobel, S.A.; Buxton, R.B.; Witter, M.P.; Barnes, C.A. A pathophysiological framework of hippocampal dysfunction in ageing and disease. Nat. Rev. Neurosci. 2011, 12, 585–601. [Google Scholar] [CrossRef] [Green Version]
- Shipp, S. Structure and function of the cerebral cortex. Curr. Biol. 2007, 17, R443–R449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Servick, K. Deep brain stimulation takes new aim at depression. Science 2021, 374, 1182–1183. [Google Scholar] [CrossRef] [PubMed]
- Arnold, S.E.; Hyman, B.T.; Flory, J.; Damasio, A.R.; Van Hoesen, G.W. The topographical and neuroanatomical distribution of neurofibrillary tangles and neuritic plaques in the cerebral cortex of patients with Alzheimer’s disease. Cereb. Cortex 1991, 1, 103–116. [Google Scholar] [CrossRef] [PubMed]
- Duyckaerts, C.; Colle, M.A.; Dessi, F.; Piette, F.; Hauw, J.J. Progression of Alzheimer histopathological changes. Acta Neurol. Belg. 1998, 98, 180–185. [Google Scholar]
- Braak, H.; Bohl, J.R.; Muller, C.M.; Rub, U.; de Vos, R.A.; Del Tredici, K. Stanley Fahn Lecture 2005: The staging procedure for the inclusion body pathology associated with sporadic Parkinson’s disease reconsidered. Mov. Disord. 2006, 21, 2042–2051. [Google Scholar] [CrossRef]
- Brettschneider, J.; Del Tredici, K.; Lee, V.M.; Trojanowski, J.Q. Spreading of pathology in neurodegenerative diseases: A focus on human studies. Nat. Rev. Neurosci. 2015, 16, 109–120. [Google Scholar] [CrossRef] [Green Version]
- Braak, H.; Del Tredici, K. Spreading of Tau Pathology in Sporadic Alzheimer’s Disease Along Cortico-cortical Top-Down Connections. Cereb. Cortex 2018, 28, 3372–3384. [Google Scholar] [CrossRef] [Green Version]
- Jin, M.; Shepardson, N.; Yang, T.; Chen, G.; Walsh, D.; Selkoe, D.J. Soluble amyloid beta-protein dimers isolated from Alzheimer cortex directly induce Tau hyperphosphorylation and neuritic degeneration. Proc. Natl. Acad. Sci. USA 2011, 108, 5819–5824. [Google Scholar] [CrossRef] [Green Version]
- Moreno-Jiménez, E.P.; Flor-García, M.; Terreros-Roncal, J.; Rábano, A.; Cafini, F.; Pallas-Bazarra, N.; Ávila, J.; Llorens-Martín, M. Adult hippocampal neurogenesis is abundant in neurologically healthy subjects and drops sharply in patients with Alzheimer’s disease. Nat. Med. 2019, 25, 554–560. [Google Scholar] [CrossRef]
- Sabuncu, M.R.; Desikan, R.S.; Sepulcre, J.; Yeo, B.T.; Liu, H.; Schmansky, N.J.; Reuter, M.; Weiner, M.W.; Buckner, R.L.; Sperling, R.A.; et al. The dynamics of cortical and hippocampal atrophy in Alzheimer disease. Arch. Neurol. 2011, 68, 1040–1048. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Yamada, K.; Liddelow, S.A.; Smith, S.T.; Zhao, L.; Luo, W.; Tsai, R.M.; Spina, S.; Grinberg, L.T.; Rojas, J.C.; et al. ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature 2017, 549, 523–527. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Zhang, N.; Zhang, Q.; Li, C.; Sandhu, A.F.; Iii, G.W.; Lin, S.; Lv, P.; Liu, Y.; Wu, Q.; et al. Inflammatory factors and amyloid β-induced microglial polarization promote inflammatory crosstalk with astrocytes. Aging 2020, 12, 22538–22549. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.W.; Chang, N.P.; Krishnagiri, M.; Patel, A.P.; Lindman, M.; Angel, J.P.; Kung, P.L.; Atkins, C.; Daniels, B.P. Fibrillar α-synuclein induces neurotoxic astrocyte activation via RIP kinase signaling and NF-κB. Cell Death Dis. 2021, 12, 756. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genes | Forward Primer | Reverse Primer |
|---|---|---|
| C1q | TCTGCACTGTACCCGGCTA | CCCTGGTAAATGTGACCCTTTT |
| Il1a | GCACCTTACACCTACCAGAGT | AAACTTCTGCCTGACGAGCTT |
| Il1β | GCAACTGTTCCTGAACTCAACT | ATCTTTTGGGGTCCGTCAACT |
| Tnfa | CCCTCACACTCAGATCATCTTCT | GCTACGACGTGGGCTACAG |
| Il6 | TAGTCCTTCCTACCCCAATTTCC | TTGGTCCTTAGCCACTCCTTC |
| C3 | CCAGCTCCCCATTAGCTCTG | GCACTTGCCTCTTTAGGAAGTC |
| Steap4 | CCCGAATCGTGTCTTTCCTA | GGCCTGAGTAATGGTTGCAT |
| S1pr3 | AAGCCTAGCGGGAGAGAAAC | TCAGGGAACAATTGGGAGAG |
| Hspb1 | GACATGAGCAGTCGGATTGA | GGATGGGGTGTAGGGGTACT |
| Cxcl10 | CCCACGTGTTGAGATCATTG | CACTGGGTAAAGGGGAGTGA |
| Osmr | GTGAAGGACCCAAAGCATGT | GCCTAATACCTGGTGCGTGT |
| Cp | TGTGATGGGAATGGGCAATGA | AGTGTATAGAGGATGTTCCAGGTCA |
| Serpinga 3n | CCTGGAGGATGTCCTTTCAA | TTATCAGGAAAGGCCGGATTG |
| Aspg | GCTGCTGGCCATTTACACTG | GTGGGCCTGTGCATACTCTT |
| Timp1 | AGTGATTTCCCCGCCAACTC | GGGGCCATCATCATGGTATCTGC |
| Cd44 | ACCTTGGCCACCACTCCTAA | GCAGTAGGCTGAAGGGTTGT |
| Gfap | AGAAAGGTTGAATCGCTGGA | CGGCGATAGTCGTTAGCTTC |
| H2-T23 | GGACCCGCGAATGACATAGC | GCACCTCAGGGTGACTTCAT |
| Serping 1 | ACAGCCCCCTCTGAATTCTT | GGATGCTCTCCAAGTTGCTC |
| H2-D1 | TCCGAGATTGTAAAGCGTGAAGA | ACAGGGCAGTGCAGGGATAG |
| Ggta1 | GTGAACAGCATGAGGGGTTT | GTTTTGTTGCCTCTGGGTGT |
| Ligp1 | GGGGCAATAGCTCATTGGTA | ACCTCGAAGACATCCCCTTT |
| Gbp2 | GGGGTCACTGTCTCTGACCACT | GGGAAACCTGGGATGAGATT |
| Fbln5 | CTTCAGATGCAAGCAACAA | AGGCAGTGTCAGAGGCCTTA |
| Ugt1a | CCTATGGGTCACTTGCCACT | AAAACCATGTTGGGCATGAT |
| Fkbp5 | TATGCTTATGGCTCGGCTGG | CAGCCTTCCAGGTGGACTTT |
| Psmb8 | CAGTCCTGAAGAGGCCTACG | CACTTTCACCCAACCGTCTT |
| Srgn | GCAAGGTTATCCTGCTCGGA | TGGGAGGGCCGATGTTATTG |
| Clcf1 | CTTCAATCCTCCTCCTCGACTGG | TACGTCGGAGTTCAGCTGTG |
| Ptx3 | AACAAGCTCTGTTGCCCATT | TCCCAAATGGAACATTGGAT |
| S100a10 | CCTCTGGCTGTGGACAAAAT | CTGCTCACAAGAAGCAGTGG |
| Cd109 | CACAGTCGGGAGCCCTAAAG | GCAGCGATTTCGATGTCCAC |
| Ptgs2 | GCTGTACAAGCAGTGGCAAA | CCCCAAAGATAGCATCTGGA |
| Emp1 | GAGACACTGGCCAGAAAAGC | TAAAAGGCAAGGGAATGCAC |
| Slc10a6 | GCTTCGGTGGTATGATGCTT | CCACAGGCTTTTCTGGTGAT |
| Tm4sf1 | GCCCAAGCATATTGTGGAGT | AGGGTAGGATGTGGCACAAG |
| Cd14 | GGACTGATCTCAGCCCTCTG | GCTTCAGCCCAGTGAAAGAC |
| Tgm1 | CTGTTGGTCCCGTCCCAAA | GGACCTTCCATTGTGCCTGG |
| Sphk1 | GATGCATGAGGTGGTGAATG | TGCTCGTACCCAGCATAGTG |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, B.; Ines, I.; Je, J.; Park, E.J.; Seong, H.; Jo, M.G.; Kim, H.; Kim, S.-H.; Kim, S.J.; Kim, H.J.; et al. Effect of Renal Ischemia Reperfusion on Brain Neuroinflammation. Biomedicines 2022, 10, 2993. https://doi.org/10.3390/biomedicines10112993
Lee B, Ines I, Je J, Park EJ, Seong H, Jo MG, Kim H, Kim S-H, Kim SJ, Kim HJ, et al. Effect of Renal Ischemia Reperfusion on Brain Neuroinflammation. Biomedicines. 2022; 10(11):2993. https://doi.org/10.3390/biomedicines10112993
Chicago/Turabian StyleLee, Bina, Ingabire Ines, Jihyun Je, Eun Jung Park, Hyemin Seong, Min Gi Jo, Hwajin Kim, Seon-Hee Kim, Seong Jae Kim, Hye Jung Kim, and et al. 2022. "Effect of Renal Ischemia Reperfusion on Brain Neuroinflammation" Biomedicines 10, no. 11: 2993. https://doi.org/10.3390/biomedicines10112993
APA StyleLee, B., Ines, I., Je, J., Park, E. J., Seong, H., Jo, M. G., Kim, H., Kim, S. -H., Kim, S. J., Kim, H. J., Kim, M., Park, S. W., & Yun, S. P. (2022). Effect of Renal Ischemia Reperfusion on Brain Neuroinflammation. Biomedicines, 10(11), 2993. https://doi.org/10.3390/biomedicines10112993