
The Mitochondrial Genome in Aging and Disease and the Future of Mitochondrial Therapeutics

 , ,
, ,
Abstract
:1. Introduction
2. mtDNA and Its Role in Mitochondrial Function
Importance of mtDNA Maintenance in Aging
3. Mitochondrial Therapeutics
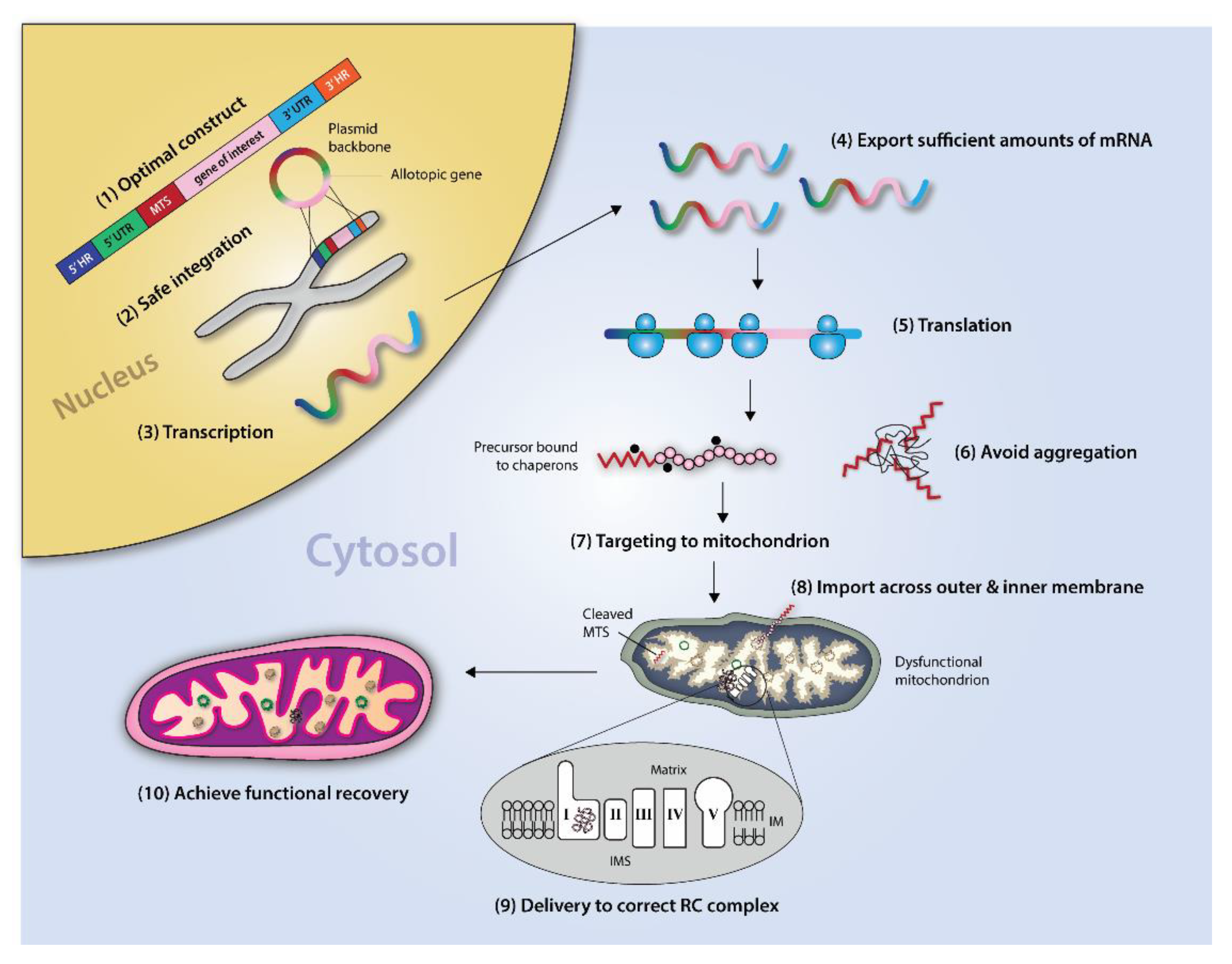
4. Allotopic Expression
Allotopic Expression of mtDNA Genes to Correct Underlying mtDNA Damage
5. Circumventing Biological Roadblocks
5.1. Mitochondrial Targeting
5.2. Probing the Hydrophobicity Threshold
5.3. Coupling for Co-Translational Import
5.4. Piecewise Import
6. Genetic and Molecular Characteristics of mtDNA-Encoded Proteins Present Inherent Challenges for Successful AE
Paucity of Animal Models to Validate Allotopic Expression
7. Allotopic Expression Has Been Demonstrated In Vivo
A Safe Harbor Expression System for Allotopic Genes
8. Allotopic Expression Gene Therapy in Human Clinical Trials
9. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AE | Allotopic expression |
| AHSCT | Allogeneic hematopoietic stem cell transplantation |
| ALS | Amyotrophic lateral sclerosis |
| ATP | Adenosine triphosphate |
| ETC | Electron transport chain |
| HR | Homologous region |
| HSP | Hereditary spastic paraplegias |
| IMS | Intermembrane space |
| iPSCs | Induced pluripotent stem cells |
| AAV/AAV2 | Adeno-associated virus (vector) |
| NADH | Nicotinamide adenine dinucleotide dehydrogenase |
| LHON | Leber’s hereditary optic neuropathy |
| NARP | Neuropathy, ataxia, and retinitis pigmentosa |
| MELAS | Mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes |
| MERRF | Myoclonic epilepsy with ragged-red fibers |
| MIDD | Maternally inherited diabetes and deafness |
| MILS | Maternally inherited Leigh syndrome |
| MIM/IM | Mitochondrial inner membrane |
| MNGIE | Mitochondrial neurogastrointestinal encephalomyopathy |
| mtDNA | Mitochondrial DNA |
| MTS | Mitochondrial targeting sequence |
| OXPHOS | Oxidative phosphorylation |
| RC | Respiratory complex |
| RNS | Reactive nitrogen species |
| ROS | Reactive oxygen species |
| SCA | Spinocerebellar ataxias |
| TALENS | Transcription activator-like effector nucleases |
| TM | Transmembrane domain |
| UTR | Untranslated regions |
References
- Chan, D.C. Mitochondria: Dynamic Organelles in Disease, Aging, and Development. Cell 2006, 125, 1241–1252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baker, M.J.; Frazier, A.E.; Gulbis, J.M.; Ryan, M.T. Mitochondrial protein-import machinery: Correlating structure with function. Trends Cell Biol. 2007, 17, 456–464. [Google Scholar] [CrossRef]
- Jornayvaz, F.R.; Shulman, G.I. Regulation of mitochondrial biogenesis. Essays Biochem. 2010, 47, 69–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Twig, G.; Shirihai, O.S. The interplay between mitochondrial dynamics and mitophagy. Antioxid. Redox Signal. 2011, 14, 1939–1951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westermann, B. Merging mitochondria matters. EMBO Rep. 2002, 3, 527–531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomes, L.C.; Scorrano, L. High levels of Fis1, a pro-fission mitochondrial protein, trigger autophagy. Biochim. Biophys. Acta-Bioenerg. 2008, 1777, 860–866. [Google Scholar] [CrossRef] [PubMed]
- Malena, A.; Loro, E.; Di Re, M.; Holt, I.J.; Vergani, L. Inhibition of mitochondrial fission favours mutant over wild-type mitochondrial DNA. Hum. Mol. Genet. 2009, 18, 3407–3416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, L.K.; Tiwari, M.; Rai, N.K.; Bai, Y. Mitophagy activation repairs Leber’s hereditary optic neuropathy-associated mitochondrial dysfunction and improves cell survival. Hum. Mol. Genet. 2019, 28, 422–433. [Google Scholar] [CrossRef]
- Ashar, F.N.; Zhang, Y.; Longchamps, R.J.; Lane, J.; Moes, A.; Grove, M.L.; Mychaleckyj, J.C.; Taylor, K.D.; Coresh, J.; Rotter, J.I.; et al. Association of mitochondrial DNA copy number with cardiovascular disease. JAMA Cardiol. 2017, 2, 1247–1255. [Google Scholar] [CrossRef]
- Herbers, E.; Kekäläinen, N.J.; Hangas, A.; Pohjoismäki, J.L.; Goffart, S. Tissue specific differences in mitochondrial DNA maintenance and expression. Mitochondrion 2019, 44, 85–92. [Google Scholar] [CrossRef]
- Zsurka, G.; Peeva, V.; Kotlyar, A.; Kunz, W.S. Is there still any role for oxidative stress in mitochondrial DNA-dependent aging? Genes 2018, 9, 175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, S.; Bankier, A.T.; Barrell, B.G.; De Bruijn, M.H.L.; Coulson, A.R.; Drouin, J.; Eperon, I.C.; Nierlich, D.P.; Roe, B.A.; Sanger, F.; et al. Sequence and organization of the human mitochondrial genome. Nature 1981, 290, 457–465. [Google Scholar] [CrossRef] [PubMed]
- Howell, N.; Kubacka, I.; Mackey, D.A. How rapidly does the human mitochondrial genome evolve? Am. J. Hum. Genet. 1996, 59, 501–509. [Google Scholar]
- Jazin, E.; Soodyall, H.; Jalonen, P.; Lindholm, E.; Stoneking, M.; Gyllensten, U. Mitochondrial mutation rate revisited: Hot spots and polymorphism. Nat. Genet. 1998, 18, 109–110. [Google Scholar] [CrossRef]
- Raha, S.; Robinson, B.H. Mitochondria, oxygen free radicals, disease and ageing. Trends Biochem. Sci. 2000, 25, 502–508. [Google Scholar] [CrossRef]
- Yu-Wai-Man, P.; Griffiths, P.G.; Chinnery, P.F. Mitochondrial optic neuropathies-Disease mechanisms and therapeutic strategies. Prog. Retin. Eye Res. 2011, 30, 81–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, K.; Lu, J.; Ma, F.; Keinan, A.; Gu, Z. Extensive pathogenicity of mitochondrial heteroplasmy in healthy human individuals. Proc. Natl. Acad. Sci. USA 2014, 111, 10654–10659. [Google Scholar] [CrossRef] [Green Version]
- Chinnery, P.F.; Hudson, G. Mitochondrial genetics. Br. Med. Bull. 2013, 106, 135–159. [Google Scholar] [CrossRef] [Green Version]
- Khrapko, K.; Turnbull, D. Mitochondrial DNA mutations in aging. In Progress in Molecular Biology and Translational Science; Academic Press: San Diego, CA, USA, 2014; Volume 127, pp. 29–62. ISBN 9780123946256. [Google Scholar]
- Rossignol, R.; Faustin, B.; Rocher, C.; Malgat, M.; Mazat, J.P.; Letellier, T. Mitochondrial threshold effects. Biochem. J. 2003, 370, 751–762. [Google Scholar] [CrossRef] [Green Version]
- Reinecke, F.; Smeitink, J.A.M.; van der Westhuizen, F.H. OXPHOS gene expression and control in mitochondrial disorders. Biochim. Biophys. Acta-Mol. Basis Dis. 2009, 1792, 1113–1121. [Google Scholar] [CrossRef] [Green Version]
- Schon, E.A.; Dimauro, S.; Hirano, M. Human mitochondrial DNA: Roles of inherited and somatic mutations. Nat. Rev. Genet. 2012, 13, 878–890. [Google Scholar] [CrossRef]
- Kaufmann, P.; Engelstad, K.; Wei, Y.; Kulikova, R.; Oskoui, M.; Sproule, D.M.; Battista, V.; Koenigsberger, D.Y.; Pascual, J.M.; Shanske, S.; et al. Natural history of MELAS associated with mitochondrial DNA m.3243A>G genotype. Neurology 2011, 77, 1965–1971. [Google Scholar] [CrossRef] [Green Version]
- Tick Chong, P.S.; Vucic, S.; Hedley-Whyte, E.T.; Dreyer, M.; Cros, D. Multiple symmetric lipomatosis (Madelung’s disease) caused by the MERRF (A8344G)’ mutation: A report of two cases and review of the literature. J. Clin. Neuromuscul. Dis. 2003, 5, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Goto, Y.I.; Nonaka, I.; Horai, S. A mutation in the tRNALeu(UUR) gene associated with the MELAS subgroup of mitochondrial encephalomyopathies. Nature 1990, 348, 651–653. [Google Scholar] [CrossRef] [PubMed]
- Kirby, D.M.; McFarland, R.; Ohtake, A.; Dunning, C.; Ryan, M.T.; Wilson, C.; Ketteridge, D.; Turnbull, D.M.; Thorburn, D.R.; Taylor, R.W. Mutations of the mitochondrial ND1 gene as a cause of MELAS. J. Med. Genet. 2004, 41, 784–789. [Google Scholar] [CrossRef] [Green Version]
- Santorelli, F.M.; Tanji, K.; Kulikova, R.; Shanske, S.; Vilarinho, L.; Hays, A.P.; DiMauro, S. Identification of a novel mutation in the mtDNA ND5 gene associated with MELAS. Biochem. Biophys. Res. Commun. 1997, 238, 326–328. [Google Scholar] [CrossRef]
- Lin, Y.; Xu, X.; Zhao, D.; Liu, F.; Luo, Y.; Du, J.; Wang, D.; Ji, K.; Zhao, Y.; Yan, C. A novel m.11406 T > A mutation in mitochondrial ND4 gene causes MELAS syndrome. Mitochondrion 2020, 54, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Shoffner, J.M.; Lott, M.T.; Lezza, A.M.S.; Seibel, P.; Ballinger, S.W.; Wallace, D.C. Myoclonic epilepsy and ragged-red fiber disease (MERRF) is associated with a mitochondrial DNA tRNALys mutation. Cell 1990, 61, 931–937. [Google Scholar] [CrossRef]
- Howell, N. Leber hereditary optic neuropathy: Respiratory chain dysfunction and degeneration of the optic nerve. Vis. Res. 1998, 38, 1495–1504. [Google Scholar] [CrossRef] [Green Version]
- Howell, N.; Bindoff, L.A.; McCullough, D.A.; Kubacka, I.; Poulton, J.; Mackey, D.; Taylor, L.; Turnbull, D.M. Leber hereditary optic neuropathy: Identification of the same mitochondrial ND1 mutation in six pedigrees. Am. J. Hum. Genet. 1991, 49, 939. [Google Scholar] [PubMed]
- Huoponen, K.; Vilkki, J.; Aula, P.; Nikoskelainen, E.K.; Savontaus, M.L. A new mtDNA mutation associated with Leber hereditary optic neuroretinopathy. Am. J. Hum. Genet. 1991, 48, 1147. [Google Scholar] [PubMed]
- Wallace, D.C.; Singh, G.; Lott, M.T.; Hodge, J.A.; Schurr, T.G.; Lezza, A.M.S.; Elsas, L.J.; Nikoskelainen, E.K. Mitochondrial DNA mutation associated with Leber’s hereditary optic neuropathy. Science 1988, 242, 1427–1430. [Google Scholar] [CrossRef]
- Johns, D.R.; Neufeld, M.J.; Park, R.D. An ND-6 mitochondrial DNA mutation associated with Leber hereditary optic neuropathy. Biochem. Biophys. Res. Commun. 1992, 187, 1551–1557. [Google Scholar] [CrossRef]
- Sarzi, E.; Brown, M.D.; Lebon, S.; Chretien, D.; Munnich, A.; Rotig, A.; Procaccio, V. A novel recurrent mitochondrial DNA mutation inND3 gene is associated with isolated complex I deficiency causing Leigh syndrome and dystonia. Am. J. Med. Genet. Part A 2007, 143A, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Moslemi, A.R.; Darin, N.; Tulinius, M.; Oldfors, A.; Holme, E. Two new mutations in the MTATP6 gene associated with Leigh syndrome. Neuropediatrics 2005, 36, 314–318. [Google Scholar] [CrossRef]
- Kytövuori, L.; Lipponen, J.; Rusanen, H.; Komulainen, T.; Martikainen, M.H.; Majamaa, K. A novel mutation m.8561C>G in MT-ATP6/8 causing a mitochondrial syndrome with ataxia, peripheral neuropathy, diabetes mellitus, and hypergonadotropic hypogonadism. J. Neurol. 2016, 263, 2188–2195. [Google Scholar] [CrossRef]
- Sgarbi, G.; Baracca, A.; Lenaz, G.; Valentino, L.M.; Carelli, V.; Solaini, G. Inefficient coupling between proton transport and ATP synthesis may be the pathogenic mechanism for NARP and Leigh syndrome resulting from the T8993G mutation in mtDNA. Biochem. J. 2006, 395, 493–500. [Google Scholar] [CrossRef] [Green Version]
- Holt, I.J.; Harding, A.E.; Petty, R.K.; Morgan-Hughes, J.A. A new mitochondrial disease associated with mitochondrial DNA heteroplasmy. Am. J. Hum. Genet. 1990, 46, 428–433. [Google Scholar]
- Ganetzky, R.D.; Stendel, C.; McCormick, E.M.; Zolkipli-Cunningham, Z.; Goldstein, A.C.; Klopstock, T.; Falk, M.J. MT-ATP6 mitochondrial disease variants: Phenotypic and biochemical features analysis in 218 published cases and cohort of 14 new cases. Hum. Mutat. 2019, 40, 499–515. [Google Scholar] [CrossRef]
- Khan, N.A.; Govindaraj, P.; Meena, A.K.; Thangaraj, K. Mitochondrial disorders: Challenges in diagnosis & treatment. Indian J. Med. Res. Suppl. 2015, 141, 13–26. [Google Scholar] [CrossRef]
- Boggan, R.M.; Lim, A.; Taylor, R.W.; McFarland, R.; Pickett, S.J. Resolving complexity in mitochondrial disease: Towards precision medicine. Mol. Genet. Metab. 2019, 128, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Contreras, M.; Sweetwyne, M.T.; Kohrn, B.F.; Tsantilas, K.A.; Hipp, M.J.; Schmidt, E.K.; Fredrickson, J.; Whitson, J.A.; Campbell, M.D.; Rabinovitch, P.S.; et al. A replication-linked mutational gradient drives somatic mutation accumulation and influences germline polymorphisms and genome composition in mitochondrial DNA. Nucleic Acids Res. 2021, 49, 11103–11118. [Google Scholar] [CrossRef]
- Kowald, A.; Kirkwood, T.B.L. Resolving the enigma of the clonal expansion of mtDNA deletions. Genes 2018, 9, 126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chinnery, P.F.; Samuels, D.C.; Elson, J.; Turnbull, D.M. Accumulation of mitochondrial DNA mutations in ageing, cancer, and mitochondrial disease: Is there a common mechanism? Lancet 2002, 360, 1323–1325. [Google Scholar] [CrossRef]
- Trifunovic, A.; Hansson, A.; Wredenberg, A.; Rovio, A.T.; Dufour, E.; Khvorostov, I.; Spelbrink, J.N.; Wibom, R.; Jacobs, H.T.; Larsson, N.G. Somatic mtDNA mutations cause aging phenotypes without affecting reactive oxygen species production. Proc. Natl. Acad. Sci. USA 2005, 102, 17993–17998. [Google Scholar] [CrossRef] [Green Version]
- Herbst, A.; Lee, C.C.; Vandiver, A.R.; Aiken, J.M.; McKenzie, D.; Hoang, A.; Allison, D.; Liu, N.; Wanagat, J. Mitochondrial DNA deletion mutations increase exponentially with age in human skeletal muscle. Aging Clin. Exp. Res. 2021, 33, 1811–1820. [Google Scholar] [CrossRef] [PubMed]
- Arbeithuber, B.; Hester, J.; Cremona, M.A.; Stoler, N.; Zaidi, A.; Higgins, B.; Anthony, K.; Chiaromonte, F.; Diaz, F.J.; Makova, K.D. Age-related accumulation of de novo mitochondrial mutations in mammalian oocytes and somatic tissues. PLoS Biol. 2020, 18, e3000745. [Google Scholar] [CrossRef] [PubMed]
- Kowald, A.; Kirkwood, T.B.L. Transcription could be the key to the selection advantage of mitochondrial deletion mutants in aging. Proc. Natl. Acad. Sci. USA 2014, 111, 2972–2977. [Google Scholar] [CrossRef] [Green Version]
- De Grey, A.D.N.J. A proposed refinement of the mitochondrial free radical theory of aging. BioEssays 1997, 19, 161–166. [Google Scholar] [CrossRef]
- Carelli, V.; Maresca, A.; Caporali, L.; Trifunov, S.; Zanna, C.; Rugolo, M. Mitochondria: Biogenesis and mitophagy balance in segregation and clonal expansion of mitochondrial DNA mutations. Int. J. Biochem. Cell Biol. 2015, 63, 21–24. [Google Scholar] [CrossRef]
- Lin, M.T.; Cantuti-Castelvetri, I.; Zheng, K.; Jackson, K.E.; Tan, Y.B.; Arzberger, T.; Lees, A.J.; Betensky, R.A.; Beal, M.F.; Simon, D.K. Somatic mitochondrial DNA mutations in early Parkinson and incidental lewy body disease. Ann. Neurol. 2012, 71, 850–854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoekstra, J.G.; Hipp, M.J.; Montine, T.J.; Kennedy, S.R. Mitochondrial DNA mutations increase in early stage Alzheimer disease and are inconsistent with oxidative damage. Ann. Neurol. 2016, 80, 301–306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bua, E.; Johnson, J.; Herbst, A.; Delong, B.; McKenzie, D.; Salamat, S.; Aiken, J.M. Mitochondrial DNA-deletion mutations accumulate intracellularly to detrimental levels in aged human skeletal muscle fibers. Am. J. Hum. Genet. 2006, 79, 469–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schon, E.A.; Przedborski, S. Mitochondria: The Next (Neurode)Generation. Neuron 2011, 70, 1033–1053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Coo, I.F.M.; Renier, W.O.; Ruitenbeek, W.; Ter Laak, H.J.; Bakker, M.; Schägger, H.; Van Oost, B.A.; Smeets, H.J.M. A 4–base pair deletion in the mitochondrial cytochrome b gene associated with parkinsonism/MELAS overlap syndrome. Ann. Neurol. 1999, 45, 130–133. [Google Scholar] [CrossRef]
- Silvestri, G.; Mongini, T.; Odoardi, F.; Modoni, A.; DeRosa, G.; Doriguzzi, C.; Palmucci, L.; Tonali, P.; Servidei, S. A new mtDNA mutation associated with a progressive encephalopathy and cytochrome c oxidase deficiency. Neurology 2000, 54, 1693–1696. [Google Scholar] [CrossRef] [PubMed]
- Conley, K.E.; Jubrias, S.A.; Esselman, P.C. Oxidative capacity and ageing in human muscle. J. Physiol. 2000, 526, 203–210. [Google Scholar] [CrossRef]
- Short, K.R.; Bigelow, M.L.; Kahl, J.; Singh, R.; Coenen-Schimke, J.; Raghavakaimal, S.; Nair, K.S. Decline in skeletal muscle mitochondrial function with aging in humans. Proc. Natl. Acad. Sci. USA 2005, 102, 5618–5623. [Google Scholar] [CrossRef] [Green Version]
- Nicolson, G.L. Mitochondrial dysfunction and chronic disease: Treatment with natural supplements. Integr. Med. 2014, 13, 35–43. [Google Scholar]
- Kwong, J.Q.; Henning, M.S.; Starkov, A.A.; Manfredi, G. The mitochondrial respiratory chain is a modulator of apoptosis. J. Cell Biol. 2007, 179, 1163–1177. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Sharma, L.K.; Bai, Y. Implications of mitochondrial DNA mutations and mitochondrial dysfunction in tumorigenesis. Cell Res. 2009, 19, 802–815. [Google Scholar] [CrossRef] [PubMed]
- Tasdogan, A.; McFadden, D.G.; Mishra, P. Mitochondrial DNA Haplotypes as Genetic Modifiers of Cancer. Trends Cancer 2020, 6, 1044–1058. [Google Scholar] [CrossRef]
- Lin, Y.-H.; Lim, S.; Chen, C.; Chi, H.; Yeh, C.; Lin, W. Functional Role of Mitochondrial DNA in Cancer Progression. Int. J. Mol. Sci. 2022, 23, 1659. [Google Scholar] [CrossRef]
- Gopal, R.K.; Kübler, K.; Calvo, S.E.; Polak, P.; Livitz, D.; Rosebrock, D.; Sadow, P.M.; Campbell, B.; Donovan, S.E.; Amin, S.; et al. Widespread Chromosomal Losses and Mitochondrial DNA Alterations as Genetic Drivers in Hürthle Cell Carcinoma. Cancer Cell 2018, 34, 242–255.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kopinski, P.K.; Singh, L.N.; Zhang, S.; Lott, M.T.; Wallace, D.C. Mitochondrial DNA variation and cancer. Nat. Rev. Cancer 2021, 21, 431–445. [Google Scholar] [CrossRef]
- Lanza, I.R.; Nair, K.S. Mitochondrial function as a determinant of life span. Pflugers Arch. Eur. J. Physiol. 2010, 459, 277–289. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.; Wang, Y.; Ye, K.; Picard, M.; Gu, Z. Independent impacts of aging on mitochondrial DNA quantity and quality in humans. BMC Genom. 2017, 18, 890. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Zhang, Y.; Lu, L.; Yang, H. Therapeutic Effects of Idebenone on Leber Hereditary Optic Neuropathy. Curr. Eye Res. 2020, 45, 1315–1323. [Google Scholar] [CrossRef] [PubMed]
- Enns, G.M.; Cohen, B.H. Clinical Trials in Mitochondrial Disease: An Update on EPI-734 and RP103. J. Inborn Errors Metab. Screen. 2017, 5, 232640981773301. [Google Scholar] [CrossRef]
- Singh, A.; Faccenda, D.; Campanella, M. Pharmacological advances in mitochondrial therapy. EBioMedicine 2021, 65, 103244. [Google Scholar] [CrossRef] [PubMed]
- Iyer, S.; Thomas, R.R.; Portell, F.R.; Dunham, L.D.; Quigley, C.K.; Bennett, J.P. Recombinant mitochondrial transcription factor A with N-terminal mitochondrial transduction domain increases respiration and mitochondrial gene expression. Mitochondrion 2009, 9, 196–203. [Google Scholar] [CrossRef] [Green Version]
- Dai, Y.; Zheng, K.; Clark, J.; Swerdlow, R.H.; Pulst, S.M.; Sutton, J.P.; Shinobu, L.A.; Simon, D.K. Rapamycin drives selection against a pathogenic heteroplasmic mitochondrial DNA mutation. Hum. Mol. Genet. 2014, 23, 637–647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, X.; Lewin, A.S.; Sun, L.; Hauswirth, W.W.; Guy, J. SOD2 gene transfer protects against optic neuropathy induced by deficiency of complex I. Ann. Neurol. 2004, 56, 182–191. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.; Sun, L.; Hauswirth, W.W.; Lewin, A.S.; Guy, J. Use of mitochondrial antioxidant defenses for rescue of cells with a leber hereditary optic neuropathy-causing mutation. Arch. Ophthalmol. 2007, 125, 268–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bax, B.E.; Levene, M.; Bain, M.D.; Fairbanks, L.D.; Filosto, M.; Uçar, S.K.; Klopstock, T.; Kornblum, C.; Mandel, H.; Rahman, S.; et al. Erythrocyte encapsulated thymidine phosphorylase for the treatment of patients with mitochondrial neurogastrointestinal encephalomyopathy: Study protocol for a multi-centre, multiple dose, open label trial. J. Clin. Med. 2019, 8, 1096. [Google Scholar] [CrossRef] [Green Version]
- Hirano, M.; Carelli, V.; De Giorgio, R.; Pironi, L.; Accarino, A.; Cenacchi, G.; D’Alessandro, R.; Filosto, M.; Martí, R.; Nonino, F.; et al. Mitochondrial neurogastrointestinal encephalomyopathy (MNGIE): Position paper on diagnosis, prognosis, and treatment by the MNGIE International Network. J. Inherit. Metab. Dis. 2021, 44, 376–387. [Google Scholar] [CrossRef]
- Sendra, L.; García-Mares, A.; Herrero, M.J.; Aliño, S.F. Mitochondrial dna replacement techniques to prevent human mitochondrial diseases. Int. J. Mol. Sci. 2021, 22, 551. [Google Scholar] [CrossRef]
- Tang, J.-X.; Pyle, A.; Taylor, R.W.; Oláhová, M. Interrogating Mitochondrial Biology and Disease Using CRISPR/Cas9 Gene Editing. Genes 2021, 12, 1604. [Google Scholar] [CrossRef]
- Yang, X.; Jiang, J.; Li, Z.; Liang, J.; Xiang, Y. Strategies for mitochondrial gene editing. Comput. Struct. Biotechnol. J. 2021, 19, 3319–3329. [Google Scholar] [CrossRef]
- Nissanka, N.; Moraes, C.T. Mitochondrial DNA heteroplasmy in disease and targeted nuclease-based therapeutic approaches. EMBO Rep. 2020, 21, e49612. [Google Scholar] [CrossRef]
- Tonin, Y.; Heckel, A.M.; Vysokikh, M.; Dovydenko, I.; Meschaninova, M.; Rötig, A.; Munnich, A.; Venyaminova, A.; Tarassov, I.; Entelis, N. Modeling of antigenomic therapy of mitochondrial diseases by mitochondrially addressed RNA targeting a pathogenic point mutation in mitochondrial DNA. J. Biol. Chem. 2014, 289, 13323–13334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Comte, C.; Tonin, Y.; Heckel-Mager, A.M.; Boucheham, A.; Smirnov, A.; Auré, K.; Lombès, A.; Martin, R.P.; Entelis, N.; Tarassov, I. Mitochondrial targeting of recombinant RNAs modulates the level of a heteroplasmic mutation in human mitochondrial DNA associated with Kearns Sayre Syndrome. Nucleic Acids Res. 2013, 41, 418–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bayona-Bafaluy, M.P.; Blits, B.; Battersby, B.J.; Shoubridge, E.A.; Moraes, C.T. Rapid directional shift of mitochondrial DNA heteroplasmy in animal tissues by a mitochondrially targeted restriction endonuclease. Proc. Natl. Acad. Sci. USA 2005, 102, 14392–14397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reddy, P.; Ocampo, A.; Suzuki, K.; Luo, J.; Bacman, S.R.; Williams, S.L.; Sugawara, A.; Okamura, D.; Tsunekawa, Y.; Wu, J.; et al. Selective elimination of mitochondrial mutations in the germline by genome editing. Cell 2015, 161, 459–469. [Google Scholar] [CrossRef] [Green Version]
- Bacman, S.R.; Williams, S.L.; Garcia, S.; Moraes, C.T. Organ-specific shifts in mtDNA heteroplasmy following systemic delivery of a mitochondria-targeted restriction endonuclease. Gene Ther. 2010, 17, 713–720. [Google Scholar] [CrossRef]
- Mok, B.Y.; de Moraes, M.H.; Zeng, J.; Bosch, D.E.; Kotrys, A.V.; Raguram, A.; Hsu, F.S.; Radey, M.C.; Peterson, S.B.; Mootha, V.K.; et al. A bacterial cytidine deaminase toxin enables CRISPR-free mitochondrial base editing. Nature 2020, 583, 631–637. [Google Scholar] [CrossRef]
- Pereira, C.V.; Moraes, C.T. Current strategies towards therapeutic manipulation of mtDNA heteroplasmy. Front. Biosci.-Landmark 2017, 22, 991–1010. [Google Scholar] [CrossRef]
- Jackson, C.B.; Turnbull, D.M.; Minczuk, M.; Gammage, P.A. Therapeutic Manipulation of mtDNA Heteroplasmy: A Shifting Perspective. Trends Mol. Med. 2020, 26, 698–709. [Google Scholar] [CrossRef] [Green Version]
- Rai, P.K.; Craven, L.; Hoogewijs, K.; Russell, O.M.; Lightowlers, R.N. Advances in methods for reducing mitochondrial DNA disease by replacing or manipulating the mitochondrial genome. Essays Biochem. 2018, 62, 455–465. [Google Scholar] [CrossRef]
- Bian, W.-P.; Chen, Y.-L.; Luo, J.-J.; Wang, C.; Xie, S.-L.; Pei, D.-S. Knock-In Strategy for Editing Human and Zebrafish Mitochondrial DNA Using Mito-CRISPR/Cas9 System. ACS Synth. Biol. 2019, 8, 621–632. [Google Scholar] [CrossRef]
- Yoo, B.-C.; Yadav, N.S.; Orozco, E.M.; Sakai, H. Cas9/gRNA-mediated genome editing of yeast mitochondria and Chlamydomonas chloroplasts. PeerJ 2020, 8, e8362. [Google Scholar] [CrossRef] [Green Version]
- Antón, Z.; Mullally, G.; Ford, H.C.; van der Kamp, M.W.; Szczelkun, M.D.; Lane, J.D. Mitochondrial import, health and mtDNA copy number variability seen when using type II and type V CRISPR effectors. J. Cell Sci. 2021, 133, 1–32. [Google Scholar] [CrossRef]
- Hussain, S.R.A.; Yalvac, M.E.; Khoo, B.; Eckardt, S.; McLaughlin, K.J. Adapting CRISPR/Cas9 System for Targeting Mitochondrial Genome. Front. Genet. 2021, 12, 627050. [Google Scholar] [CrossRef]
- Jo, A.; Ham, S.; Lee, G.H.; Lee, Y.I.; Kim, S.; Lee, Y.S.; Shin, J.H.; Lee, Y. Efficient mitochondrial genome editing by CRISPR/Cas9. BioMed Res. Int. 2015, 2015, 305716. [Google Scholar] [CrossRef] [Green Version]
- Gearing, D.P.; McMullen, G.L.; Nagley, P. Chemical synthesis of a mitochondrial gene designed for expression in the yeast nucleus. Biochem. Int. 1985, 10, 907–915. [Google Scholar] [PubMed]
- Supekova, L.; Supek, F.; Greer, J.E.; Schultz, P.G. A single mutation in the first transmembrane domain of yeast COX2 enables its allotopic expression. Proc. Natl. Acad. Sci. USA 2010, 107, 5047–5052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gearing, D.P.; Nagley, P. Yeast mitochondrial ATPase subunit 8, normally a mitochondrial gene product, expressed in vitro and imported back into the organelle. EMBO J. 1986, 5, 3651–3655. [Google Scholar] [CrossRef]
- Oca-Cossio, J.; Kenyon, L.; Hao, H.; Moraes, C.T. Limitations of Allotopic Expression of Mitochondrial Genes in Mammalian Cells. Genetics 2003, 165, 707–720. [Google Scholar] [CrossRef]
- Perales-Clemente, E.; Fernández-Silva, P.; Acín-Pérez, R.; Pérez-Martos, A.; Enríquez, J.A. Allotopic expression of mitochondrial-encoded genes in mammals: Achieved goal, undemonstrated mechanism or impossible task? Nucleic Acids Res. 2011, 39, 225–234. [Google Scholar] [CrossRef] [Green Version]
- Kunze, M.; Berger, J. The similarity between N-terminal targeting signals for protein import into different organelles and its evolutionary relevance. Front. Physiol. 2015, 6, 259. [Google Scholar] [CrossRef] [Green Version]
- Manfredi, G.; Fu, J.; Ojaimi, J.; Sadlock, J.E.; Kwong, J.Q.; Guy, J.; Schon, E.A. Rescue of a deficiency in ATP synthesis by transfer of MTATP6, a mitochondrial DNA-encoded gene, to the nucleus. Nat. Genet. 2002, 30, 394–399. [Google Scholar] [CrossRef] [PubMed]
- Calvo, S.E.; Julien, O.; Clauser, K.R.; Shen, H.; Kamer, K.J.; Wells, J.A.; Mootha, V.K. Comparative Analysis of Mitochondrial N-Termini from Mouse, Human, and Yeast. Mol. Cell. Proteom. 2017, 16, 512–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chin, R.M.; Panavas, T.; Brown, J.M.; Johnson, K.K. Optimized Mitochondrial Targeting of Proteins Encoded by Modified mRNAs Rescues Cells Harboring Mutations in mtATP6. Cell Rep. 2018, 22, 2818–2826. [Google Scholar] [CrossRef] [Green Version]
- Guy, J.; Qi, X.; Pallotti, F.; Schon, E.A.; Manfredi, G.; Carelli, V.; Martinuzzi, A.; Hauswirth, W.W.; Lewin, A.S. Rescue of a mitochondrial deficiency causing leber hereditary optic neuropathy. Ann. Neurol. 2002, 52, 534–542. [Google Scholar] [CrossRef]
- Farrell, L.B.; Gearing, D.P.; Nagley, P. Reprogrammed expression of subunit 9 of the mitochondrial ATPase complex of Saccharomyces cerevisiae Expression in vitro from a chemically synthesized gene and import into isolated mitochondria. Eur. J. Biochem. 1988, 173, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Law, R.H.P.; Farrell, L.B.; Nero, D.; Devenish, R.J.; Nagley, P. Studies on the import into mitochondria of yeast ATP synthase subunits 8 and 9 encoded by artificial nuclear genes. FEBS Lett. 1988, 236, 501–505. [Google Scholar] [CrossRef] [Green Version]
- Galanis, M.; Devenish, R.J.; Nagley, P. Duplication of leader sequence for protein targeting to mitochondria leads to increased import efficiency. FEBS Lett. 1991, 282, 425–430. [Google Scholar] [CrossRef] [Green Version]
- Bietenhader, M.; Martos, A.; Tetaud, E.; Aiyar, R.S.; Sellem, C.H.; Kucharczyk, R.; Clauder-Münster, S.; Giraud, M.F.; Godard, F.; Salin, B.; et al. Experimental Relocation of the Mitochondrial ATP9 Gene to the Nucleus Reveals Forces Underlying Mitochondrial Genome Evolution. PLoS Genet. 2012, 8, e1002876. [Google Scholar] [CrossRef]
- Zullo, S.J.; Parks, W.T.; Chloupkova, M.; Wei, B.; Weiner, H.; Fenton, W.A.; Eisenstadt, J.M.; Merril, C.R. Stable transformation of CHO cells and human NARP cybrids confers oligomycin resistance (olir) following transfer of a mitochondrial DNA-encoded olir ATPase6 gene to the nuclear genome: A model system for mtDNA gene therapy. Rejuvenation Res. 2005, 8, 18–28. [Google Scholar] [CrossRef]
- Ojaimi, J.; Pan, J.; Santra, S.; Snell, W.J.; Schon, E.A. An algal nucleus-encoded subunit of mitochondrial ATP synthase rescues a defect in the analogous human mitochondrial-encoded subunit. Mol. Biol. Cell 2002, 13, 3836–3844. [Google Scholar] [CrossRef] [Green Version]
- Bokori-Brown, M.; Holt, I.J. Expression of algal nuclear ATP synthase subunit 6 in human cells results in protein targeting to mitochondria but no assembly into ATP synthase. Rejuvenation Res. 2006, 9, 455–469. [Google Scholar] [CrossRef]
- Kaltimbacher, V.; Bonnet, C.; Lecoeuvre, G.; Forster, V.; Sahel, J.A.; Corral-Debrinski, M. mRNA localization to the mitochondrial surface allows the efficient translocation inside the organelle of a nuclear recoded ATP6 protein. RNA 2006, 12, 1408–1417. [Google Scholar] [CrossRef] [Green Version]
- Bonnet, C.; Kaltimbacher, V.; Ellouze, S.; Augustin, S.; Bénit, P.; Forster, V.; Rustin, P.; Sahel, J.A.; Corral-Debrinski, M. Allotopic mRNA localization to the mitochondrial surface rescues respiratory chain defects in fibroblasts harboring mitochondrial DNA mutations affecting complex I or V subunits. Rejuvenation Res. 2007, 10, 127–143. [Google Scholar] [CrossRef] [PubMed]
- Figueroa-Martínez, F.; Vázquez-Acevedo, M.; Cortés-Hernández, P.; García-Trejo, J.J.; Davidson, E.; King, M.P.; González-Halphen, D. What limits the allotopic expression of nucleus-encoded mitochondrial genes? The case of the chimeric Cox3 and Atp6 genes. Mitochondrion 2011, 11, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Boominathan, A.; Vanhoozer, S.; Basisty, N.; Powers, K.; Crampton, A.L.; Wang, X.; Friedricks, N.; Schilling, B.; Brand, M.D.; O’Connor, M.S. Stable nuclear expression of ATP8 and ATP6 genes rescues a mtDNA Complex v null mutant. Nucleic Acids Res. 2016, 44, 9342–9357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunn, D.A.; Pinkert, C.A. Nuclear expression of a mitochondrial DNA gene: Mitochondrial targeting of allotopically expressed mutant ATP6 in transgenic mice. J. Biomed. Biotechnol. 2012, 2012, 541245. [Google Scholar] [CrossRef] [PubMed]
- Law, R.H.P.; Devenish, R.J.; Nagley, P. Assembly of imported subunit 8 into the ATP synthase complex of isolated yeast mitochondria. Eur. J. Biochem. 1990, 188, 421–429. [Google Scholar] [CrossRef]
- Nagley, P.; Farrell, L.B.; Gearing, D.P.; Nero, D.; Meltzer, S.; Devenish, R.J. Assembly of functional proton-translocating ATPase complex in yeast mitochondria with cytoplasmically synthesized subunit 8, a polypeptide normally encoded within the organelle. Proc. Natl. Acad. Sci. USA 1988, 85, 2091–2095. [Google Scholar] [CrossRef] [Green Version]
- Claros, M.G.; Perea, J.; Shu, Y.; Samatey, F.A.; Popot, J.-L.; Jacq, C. Limitations to in vivo Import of Hydrophobic Proteins into Yeast Mitochondria: The Case of a Cytoplasmically Synthesized Apocytochrome b. Eur. J. Biochem. 1995, 228, 762–771. [Google Scholar] [CrossRef]
- Bonnet, C.; Augustin, S.; Ellouze, S.; Bénit, P.; Bouaita, A.; Rustin, P.; Sahel, J.A.; Corral-Debrinski, M. The optimized allotopic expression of ND1 or ND4 genes restores respiratory chain complex I activity in fibroblasts harboring mutations in these genes. Biochim. Biophys. Acta-Mol. Cell Res. 2008, 1783, 1707–1717. [Google Scholar] [CrossRef] [Green Version]
- Calabrese, C.; Iommarini, L.; Kurelac, I.; Calvaruso, M.A.; Capristo, M.; Lollini, P.-L.; Nanni, P.; Bergamini, C.; Nicoletti, G.; De Giovanni, C.; et al. Respiratory complex I is essential to induce a Warburg profile in mitochondria-defective tumor cells. Cancer Metab. 2013, 1, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iommarini, L.; Ghelli, A.; Tropeano, C.V.; Kurelac, I.; Leone, G.; Vidoni, S.; Lombes, A.; Zeviani, M.; Gasparre, G.; Porcelli, A.M. Unravelling the effects of the mutation m.3571insC/MT-ND1 on respiratory complexes structural organization. Int. J. Mol. Sci. 2018, 19, 764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
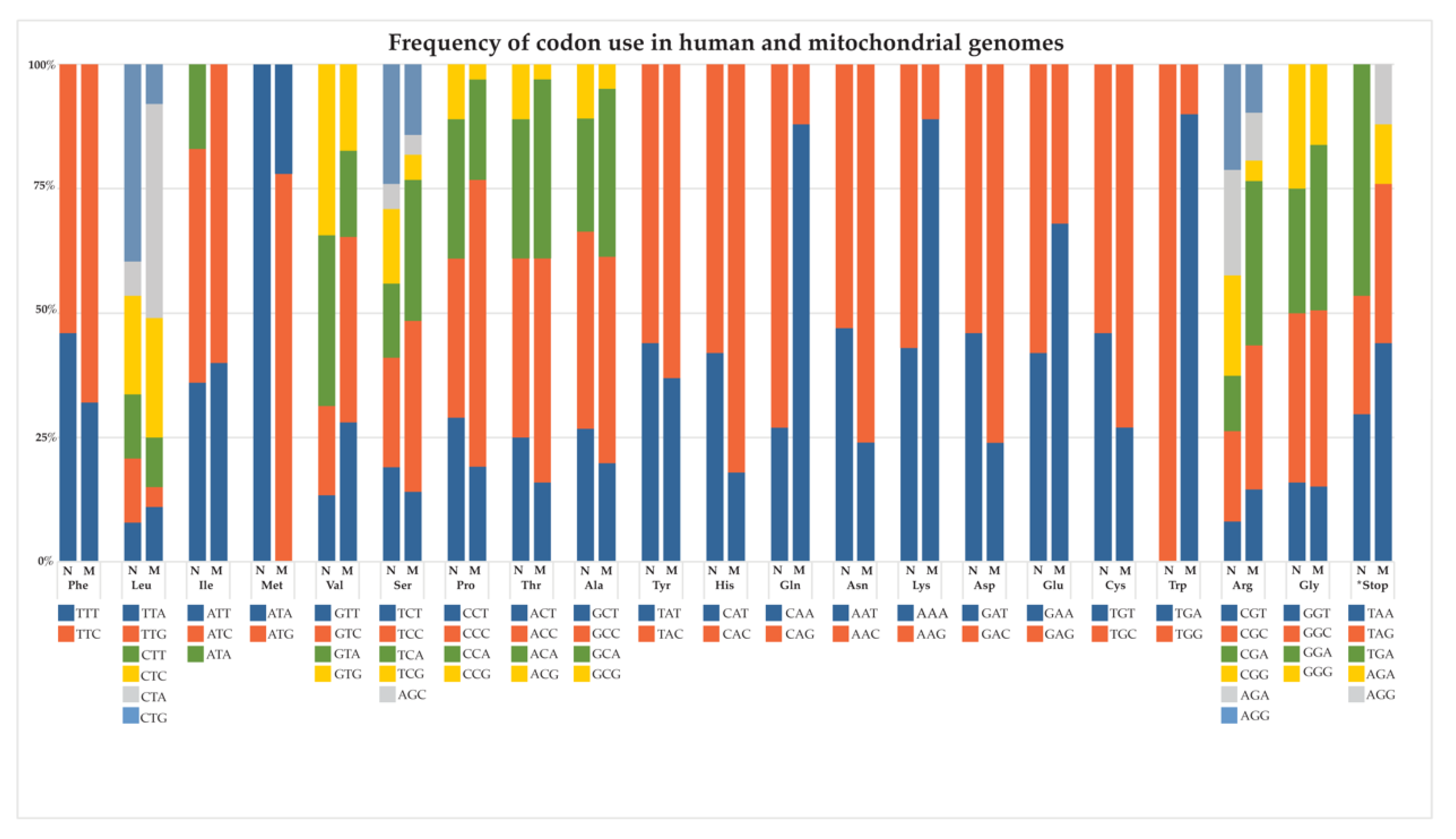
- Lewis, C.J.; Dixit, B.; Batiuk, E.; Hall, C.J.; O’Connor, M.S.; Boominathan, A. Codon optimization is an essential parameter for the efficient allotopic expression of mtDNA genes. Redox Biol. 2020, 30, 101429. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.; Sun, L.; Lewin, A.S.; Hauswirth, W.W.; Guy, J. The mutant human ND4 subunit of complex I induces optic neuropathy in the mouse. Investig. Ophthalmol. Vis. Sci. 2007, 48, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Koilkonda, R.D.; Chou, T.H.; Porciatti, V.; Hauswirth, W.W.; Guy, J. Induction of rapid and highly efficient expression of the human ND4 complex I subunit in the mouse visual system by self-complementary adeno-associated virus. Arch. Ophthalmol. 2010, 128, 876–883. [Google Scholar] [CrossRef] [Green Version]
- Ellouze, S.; Augustin, S.; Bouaita, A.; Bonnet, C.; Simonutti, M.; Forster, V.; Picaud, S.; Sahel, J.A.; Corral-Debrinski, M. Optimized Allotopic Expression of the Human Mitochondrial ND4 Prevents Blindness in a Rat Model of Mitochondrial Dysfunction. Am. J. Hum. Genet. 2008, 83, 373–387. [Google Scholar] [CrossRef] [Green Version]
- Cwerman-Thibault, H.; Augustin, S.; Lechauve, C.; Ayache, J.; Ellouze, S.; Sahel, J.A.; Corral-Debrinski, M. Nuclear expression of mitochondrial ND4 leads to the protein assembling in complex I and prevents optic atrophy and visual loss. Mol. Ther.-Methods Clin. Dev. 2015, 2, 15003. [Google Scholar] [CrossRef]
- Singh, R.K.; Saini, S.K.; Prakasam, G.; Kalairasan, P.; Bamezai, R.N.K. Role of ectopically expressed mtDNA encoded cytochrome c oxidase subunit I (MT-COI) in tumorigenesis. Mitochondrion 2019, 49, 56–65. [Google Scholar] [CrossRef]
- Tsukihara, T.; Shimokata, K.; Katayama, Y.; Shimada, H.; Muramoto, K.; Aoyama, H.; Mochizuki, M.; Shinzawa-Itoh, K.; Yamashita, E.; Yao, M.; et al. The low-spin heme of cytochrome c oxidase as the driving element of the proton-pumping process. Proc. Natl. Acad. Sci. USA 2003, 100, 15304–15309. [Google Scholar] [CrossRef] [Green Version]
- Shimokata, K.; Katayama, Y.; Murayama, H.; Suematsu, M.; Tsukihara, T.; Muramoto, K.; Aoyama, H.; Yoshikawa, S.; Shimada, H. The proton pumping pathway of bovine heart cytochrome c oxidase. Proc. Natl. Acad. Sci. USA 2007, 104, 4200–4205. [Google Scholar] [CrossRef] [Green Version]
- Rubalcava-Gracia, D.; García-Rincón, J.; Pérez-Montfort, R.; Hamel, P.P.; González-Halphen, D. Key within-membrane residues and precursor dosage impact the allotopic expression of yeast subunit II of cytochrome c oxidase. Mol. Biol. Cell 2019, 30, 2358–2366. [Google Scholar] [CrossRef] [PubMed]
- Rubalcava-Gracia, D.; Vázquez-Acevedo, M.; Funes, S.; Pérez-Martínez, X.; González-Halphen, D. Mitochondrial versus nuclear gene expression and membrane protein assembly: The case of subunit 2 of yeast cytochrome c oxidase. Mol. Biol. Cell 2018, 29, 820–833. [Google Scholar] [CrossRef] [Green Version]
- Cruz-Torres, V.; Vázquez-Acevedo, M.; García-Villegas, R.; Pérez-Martínez, X.; Mendoza-Hernández, G.; González-Halphen, D. The cytosol-synthesized subunit II (Cox2) precursor with the point mutation W56R is correctly processed in yeast mitochondria to rescue cytochrome oxidase. Biochim. Biophys. Acta-Bioenerg. 2012, 1817, 2128–2139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Markantone, D.M.; Towheed, A.; Crain, A.T.; Collins, J.M.; Celotto, A.M.; Palladino, M.J. Protein coding mitochondrial-targeted RNAs rescue mitochondrial disease in vivo. Neurobiol. Dis. 2018, 117, 203–210. [Google Scholar] [CrossRef]
- Gadir, N.; Haim-Vilmovsky, L.; Kraut-Cohen, J.; Gerst, J.E. Localization of mRNAs coding for mitochondrial proteins in the yeast Saccharomyces cerevisiae. RNA 2011, 17, 1551–1565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Margeot, A.; Blugeon, C.; Sylvestre, J.; Vialette, S.; Jacq, C.; Corral-Debrinski, M. In Saccharomyces cerevisiae, ATP2 mRNA sorting to the vicinity of mitochondria is essential for respiratory function. EMBO J. 2002, 21, 6893–6904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.M.; Liu, D.R. Discovery of a mRNA mitochondrial localization element in Saccharomyces cerevisiae by nonhomologous random recombination and in vivo selection. Nucleic Acids Res. 2007, 35, 6750–6761. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Sun, X.; Wee, J.; Guo, X.; Gu, Z. Novel insights into global translational regulation through Pumilio family RNA-binding protein Puf3p revealed by ribosomal profiling. Curr. Genet. 2019, 65, 201–212. [Google Scholar] [CrossRef]
- Sen, A.; Cox, R.T. Clueless is a conserved ribonucleoprotein that binds the ribosome at the mitochondrial outer membrane. Biol. Open 2016, 5, 195–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mauro, V.P.; Chappell, S.A. A critical analysis of codon optimization in human therapeutics. Trends Mol. Med. 2014, 20, 604–613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fåhraeus, R.; Marin, M.; Olivares-Illana, V. Whisper mutations: Cryptic messages within the genetic code. Oncogene 2016, 35, 3753–3759. [Google Scholar] [CrossRef] [PubMed]
- Firth, A.E. Mapping overlapping functional elements embedded within the protein-coding regions of RNA viruses. Nucleic Acids Res. 2014, 42, 12425–12439. [Google Scholar] [CrossRef] [PubMed]
- Mauro, V.P. Codon Optimization in the Production of Recombinant Biotherapeutics: Potential Risks and Considerations. BioDrugs 2018, 32, 69–81. [Google Scholar] [CrossRef]
- Scarpulla, R.C. Nucleus-encoded regulators of mitochondrial function: Integration of respiratory chain expression, nutrient sensing and metabolic stress. Biochim. Biophys. Acta-Gene Regul. Mech. 2012, 1819, 1088–1097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gureev, A.P.; Shaforostova, E.A.; Popov, V.N. Regulation of mitochondrial biogenesis as a way for active longevity: Interaction between the Nrf2 and PGC-1α signaling pathways. Front. Genet. 2019, 10, 435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, W.; Waymire, K.G.; Narula, N.; Li, P.; Rocher, C.; Coskun, P.E.; Vannan, M.A.; Narula, J.; MacGregor, G.R.; Wallace, D.C. A mouse model of mitochondrial disease reveals germline selection against severe mtDNA mutations. Science 2008, 319, 958–962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inoue, K.; Nakada, K.; Ogura, A.; Isobe, K.; Goto, Y.I.; Nonaka, I.; Hayashi, J.I. Generation of mice with mitochondrial dysfunction by introducing mouse mtDNA carrying a deletion into zygotes. Nat. Genet. 2000, 26, 176–181. [Google Scholar] [CrossRef]
- Weiss, H.; Wester-Rosenloef, L.; Koch, C.; Koch, F.; Baltrusch, S.; Tiedge, M.; Ibrahim, S. The mitochondrial Atp8 mutation induces mitochondrial ROS generation, secretory dysfunction, and β-cell mass adaptation in conplastic B6-mt FVB mice. Endocrinology 2012, 153, 4666–4676. [Google Scholar] [CrossRef]
- Lin, C.S.; Sharpley, M.S.; Fan, W.; Waymire, K.G.; Sadun, A.A.; Carelli, V.; Ross-Cisneros, F.N.; Baciu, P.; Sung, E.; McManus, M.J.; et al. Mouse mtDNA mutant model of Leber hereditary optic neuropathy. Proc. Natl. Acad. Sci. USA 2012, 109, 20065–20070. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Ozdemir, S.S.; Koilkonda, R.D.; Chou, T.H.; Porciatti, V.; Chiodo, V.; Boye, S.L.; Hauswirth, W.W.; Lewin, A.S.; Guy, J. Mutant NADH dehydrogenase subunit 4 gene delivery to mitochondria by targeting sequence-modified adeno-associated virus induces visual loss and optic atrophy in mice. Mol. Vis. 2012, 18, 1668–1683. [Google Scholar] [PubMed]
- Larsson, N.G.; Wang, J.; Wilhelmsson, H.; Oldfors, A.; Rustin, P.; Lewandoski, M.; Barsh, G.S.; Clayton, D.A. Mitochondrial transcription factor A is necessary for mtDNA maintenance and embryogenesis in mice. Nat. Genet. 1998, 18, 231–236. [Google Scholar] [CrossRef]
- Williams, S.L.; Huang, J.; Edwards, Y.J.K.; Ulloa, R.H.; Dillon, L.M.; Prolla, T.A.; Vance, J.M.; Moraes, C.T.; Züchner, S. The mtDNA mutation spectrum of the progeroid polg mutator mouse includes abundant control region multimers. Cell Metab. 2010, 12, 675–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tyynismaa, H.; Mjosund, K.P.; Wanrooij, S.; Lappalainen, I.; Ylikallio, E.; Jalanko, A.; Spelbrink, J.N.; Paetau, A.; Suomalainen, A. Mutant mitochondrial helicase Twinkle causes multiple mtDNA deletions and a late-onset mitochondrial disease in mice. Proc. Natl. Acad. Sci. USA 2005, 102, 17687–17692. [Google Scholar] [CrossRef] [Green Version]
- Song, L.; Shan, Y.; Kent Lloyd, K.C.; Cortopassi, G.A. Mutant twinkle increases dopaminergic neurodegeneration, mtDNA deletions and modulates Parkin expression. Hum. Mol. Genet. 2012, 21, 5147–5158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milenkovic, D.; Matic, S.; Kühl, I.; Ruzzenente, B.; Freyer, C.; Jemt, E.; Park, C.B.; Falkenberg, M.; Larsson, N.G. Twinkle is an essential mitochondrial helicase required for synthesis of nascent D-loop strands and complete mtDNA replication. Hum. Mol. Genet. 2013, 22, 1983–1993. [Google Scholar] [CrossRef] [PubMed]
- Tiranti, V.; Hoertnagel, K.; Carrozzo, R.; Calimberti, C.; Munaro, M.; Granatiero, M.; Zelante, L.; Gasparini, P.; Marzella, R.; Rocchi, M.; et al. Mutations of SURF-1 in Leigh disease associated with cytochrome C oxidase deficiency. Am. J. Hum. Genet. 1998, 63, 1609–1621. [Google Scholar] [CrossRef] [Green Version]
- Vempati, U.D.; Torraco, A.; Moraes, C.T. Mouse models of oxidative phosphorylation dysfunction and disease. Methods 2008, 46, 241–247. [Google Scholar] [CrossRef] [Green Version]
- Ruzzenente, B.; Rötig, A.; Metodiev, M.D. Mouse models for mitochondrial diseases. Hum. Mol. Genet. 2016, 25, R115–R122. [Google Scholar] [CrossRef] [Green Version]
- Singh, P.; Bahr, T.; Zhao, X.; Hu, P.; Daadi, M.; Huang, T.; Bai, Y. Creating Cell Model 2.0 Using Patient Samples Carrying a Pathogenic Mitochondrial DNA Mutation: iPSC Approach for LHON. Methods Mol. Biol. 2021, 1–13. [Google Scholar] [CrossRef]
- Meshrkey, F.; Cabrera Ayuso, A.; Rao, R.R.; Iyer, S. Quantitative analysis of mitochondrial morphologies in human induced pluripotent stem cells for Leigh syndrome. Stem Cell Res. 2021, 57, 102572. [Google Scholar] [CrossRef] [PubMed]
- McKnight, C.L.; Low, Y.C.; Elliott, D.A.; Thorburn, D.R.; Frazier, A.E. Modelling mitochondrial disease in human pluripotent stem cells: What have we learned? Int. J. Mol. Sci. 2021, 22, 7730. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Gaffney, D.J.; Chinnery, P.F. Cell reprogramming shapes the mitochondrial DNA landscape. Nat. Commun. 2021, 12, 5241. [Google Scholar] [CrossRef] [PubMed]
- Kosanke, M.; Davenport, C.; Szepes, M.; Wiehlmann, L.; Kohrn, T.; Dorda, M.; Gruber, J.; Menge, K.; Sievert, M.; Melchert, A.; et al. iPSC culture expansion selects against putatively actionable mutations in the mitochondrial genome. Stem Cell Rep. 2021, 16, 2488–2502. [Google Scholar] [CrossRef]
- Koilkonda, R.; Yu, H.; Talla, V.; Porciatti, V.; Feuer, W.J.; Hauswirth, W.W.; Chiodo, V.; Erger, K.E.; Boye, S.L.; Lewin, A.S.; et al. LHON gene therapy vector prevents visual loss and optic neuropathy induced by G11778A mutant mitochondrial DNA: Biodistribution and toxicology profile. Investig. Ophthalmol. Vis. Sci. 2014, 55, 7739–7753. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Koilkonda, R.D.; Chou, T.H.; Porciatti, V.; Ozdemir, S.S.; Chiodo, V.; Boye, S.L.; Boye, S.E.; Hauswirth, W.W.; Lewin, A.S.; et al. Gene delivery to mitochondria by targeting modified adenoassociated virus suppresses Leber’s hereditary optic neuropathy in a mouse model. Proc. Natl. Acad. Sci. USA 2012, 109, E1238–E1247. [Google Scholar] [CrossRef] [Green Version]
- Dunn, D.A.; Pinkert, C.A. Allotopic expression of ATP6 in the mouse as a transgenic model of mitochondrial disease. In Methods in Molecular Biology; Humana Press: New York, NY, USA, 2015; Volume 1265, pp. 255–269. ISBN 9781493922888. [Google Scholar]
- Yu, H.; Sant, D.W.; Wang, G.; Guy, J. Mitochondrial transfer of the Mutant human ND6T14484C gene causes visual loss and optic neuropathy. Transl. Vis. Sci. Technol. 2020, 9, 1–12. [Google Scholar] [CrossRef]
- Zhao, C.; Farruggio, A.P.; Bjornson, C.R.R.; Chavez, C.L.; Geisinger, J.M.; Neal, T.L.; Karow, M.; Calos, M.P. Recombinase-mediated reprogramming and dystrophin gene addition in mdx mouse induced pluripotent stem cells. PLoS ONE 2014, 9, e96279. [Google Scholar] [CrossRef]
- Ou, L.; Przybilla, M.J.; Tăbăran, A.F.; Overn, P.; O’Sullivan, M.G.; Jiang, X.; Sidhu, R.; Kell, P.J.; Ory, D.S.; Whitley, C.B. A novel gene editing system to treat both Tay–Sachs and Sandhoff diseases. Gene Ther. 2020, 27, 226–236. [Google Scholar] [CrossRef]
- Li, G.; Zhang, X.; Wang, H.; Mo, J.; Zhong, C.; Shi, J.; Zhou, R.; Li, Z.; Yang, H.; Wu, Z.; et al. CRISPR/Cas9-mediated integration of large transgene into pig CEP112 locus. G3 Genes Genomes Genet. 2020, 10, 467–473. [Google Scholar] [CrossRef] [Green Version]
- Kazuki, Y.; Oshimura, M. Human artificial chromosomes for gene delivery and the development of animal models. Mol. Ther. 2011, 19, 1591–1601. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.; Yang, H.; Colosi, P. Effect of Genome Size on AAV Vector Packaging. Mol. Ther. 2010, 18, 80. [Google Scholar] [CrossRef]
- Matkarimov, B.T.; Saparbaev, M.K. DNA Repair and Mutagenesis in Vertebrate Mitochondria: Evidence for Asymmetric DNA Strand Inheritance. Adv. Exp. Med. Biol. 2020, 1241, 77–100. [Google Scholar] [CrossRef]
- Zhao, X.; Cui, L.; Xiao, Y.; Mao, Q.; Aishanjiang, M.; Kong, W.; Liu, Y.; Chen, H.; Hong, F.; Jia, Z.; et al. Hypertension-associated mitochondrial DNA 4401A>G mutation caused the aberrant processing of tRNAMet, all 8 tRNAs and ND6 mRNA in the light-strand transcript. Nucleic Acids Res. 2019, 47, 10340–10356. [Google Scholar] [CrossRef]
- Wan, X.; Pei, H.; Zhao, M.J.; Yang, S.; Hu, W.K.; He, H.; Ma, S.Q.; Zhang, G.; Dong, X.Y.; Chen, C.; et al. Efficacy and Safety of rAAV2-ND4 Treatment for Leberâ (tm) s Hereditary Optic Neuropathy. Sci. Rep. 2016, 6, 21587. [Google Scholar] [CrossRef]
- Biousse, V.; Newman, N.J.; Yu-Wai-Man, P.; Carelli, V.; Moster, M.L.; Vignal-Clermont, C.; Klopstock, T.; Sadun, A.A.; Sergott, R.C.; Hage, R.; et al. Long-Term Follow-Up After Unilateral Intravitreal Gene Therapy for Leber Hereditary Optic Neuropathy: The RESTORE Study. J. Neuroophthalmol. 2021, 41, 309–315. [Google Scholar] [CrossRef]
- Birk, A.V.; Liu, S.; Soong, Y.; Mills, W.; Singh, P.; Warren, J.D.; Seshan, S.V.; Pardee, J.D.; Szeto, H.H. The mitochondrial-targeted compound SS-31 re-energizes ischemic mitochondria by interacting with cardiolipin. J. Am. Soc. Nephrol. 2013, 24, 1250–1261. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Expressed mtDNA Gene | Expression System | Gene Origin | Strategy Features | References | ||
|---|---|---|---|---|---|---|
| MTS | Gene | Other | ||||
| ATP6 | S. cerevisiae | S. cerevisiae | + * | Optimized | --- | [106,107] |
| ATP6 | S. cerevisiae | S. cerevisiae | ++ * | Optimized | --- | [108] |
| ATP6 | S. cerevisiae | P. anserine | +, + * | Recoded | --- | [109] |
| ATP6 | Oli-sensitive CHO line (11-11); NARP cybrids (T8993G) | C. griseus | + | Recoded | --- | [110] |
| ATP6 | HEK293, COS7 cell lines; NARP and MILS cybrids (JCP213, JCP261) | C. reinhardtii | --- | Unchanged | --- | [111] |
| ATP6 | HEK293, 143B WT cell lines; NARP cybrids JCP261 (206.8993E (T8993G)) | H. sapiens, C. reinhardtii | + * | Recoded | --- | [112] |
| ATP6 | NARP cybrids JCP261 (T8993G) | H. sapiens | + | Recoded | --- | [102] |
| ATP6 | HeLa | H. sapiens | + | Recoded | 3′UTR | [113] |
| ATP6 | NARP cybrids (T8993G) | H. sapiens | + | Recoded | 3′UTR | [114] |
| ATP6 | CHO; NARP cybrids (T8933G) | H. sapiens | + * | Recoded | Multiple residue substitutions to reduce TM hydrophobicity | [115] |
| ATP6, ATP8 | A8/A6 mutant cybrids (G8529A) | H. sapiens | + | Recoded or optimized | coexpression of ATP6 and ATP8 | [116] |
| ATP6 | Transgenic ATP6 WT or NARP/MILS mutant (L156R in ATP6) mice | M. musculus | + | Recoded or mutant recoded ATP6 | --- | [117] |
| ATP8 | S. cerevisiae | S. cerevisiae | +, + * | Optimized | --- | [98,107,118,119] |
| ATP8 | S. cerevisiae | S. cerevisiae | + * | Optimized | --- | [108] |
| ATP8 | HeLa, COS-7 | H. sapiens | +, ++, + *+ | Recoded | --- | [99] |
| CYB | S. cerevisiae | S. cerevisiae | + * | Recoded | Piecewise import as TM bundles | [120] |
| CYB | HeLa, COS-7 | H. sapiens | +, ++, + *+ | Recoded | --- | [99] |
| ND1 | LHON ND1 cybrids (G3460A) | H. sapiens | + | 3′UTR | [121] | |
| ND1 | Heteroplasmic ND1 KO cybrid line (mt3571insC) | H. sapiens | + | Recoded | 5′UTR and 3′UTR | [122,123] |
| ND1 | HEK293 and 143B WT lines; homoplasmic ND1 KO cybrid line (mt3571insC) | H. sapiens | + | Recoded, optimized | --- | [124] |
| ND4 | LHON cybrids (G11778A) | H. sapiens | + | Recoded | --- | [105] |
| ND4 | M. musculus (DBA/1J) | H. sapiens | + | Recoded or mutant recoded ND4 (R340H) | In vivo | [125] |
| ND4 | M. Musculus (DBA/1J) | H. sapiens | + | Recoded | In vivo | [126] |
| ND4 | HeLa, COS-7 | H. sapiens | +, ++, + *+ | Recoded | --- | [99] |
| ND4 | LHON ND4 cybrids (G11778A) | H. sapiens | + | Recoded | 3′UTR | [114,121] |
| ND4 | In vivo in rat retina induced LHON model (G11778A) | H. sapiens | + | Recoded, optimized, or mutant recoded ND4 (G11778A) | 3′UTR; IRES, β globin intron introduced into gene construct | [127,128] |
| ND6 | Mouse NIH3T3 ND6 KO mutant line (del13887) | M. musculus | + | Recoded | --- | [100] |
| COX1 | HeLa, HEK293T, MCF-7, MDA-MB231 | H. sapiens | + | Optimized | --- | [129] |
| COX1 | HeLa | Bos taurus | + | Recoded or mutant recoded COX1 (D51N) | --- | [130,131] |
| COX2 | S. cerevisiae | S. cerevisiae | + | Recoded | Single, double, or triple residue substitutions to reduce TM hydrophobicity | [132] |
| COX2 | S. cerevisiae | S. cerevisiae | + | Recoded | W56R mutation to reduce TM hydrophobicity | [133,134] |
| COX2 | S. cerevisiae | S. cerevisiae | +, + * | Recoded | 3′UTR; W56R mutation to reduce TM hydrophobicity | [97] |
| COX3 | CHO; COX3 15bp deletion cell line CSP112.5 | H. sapiens | + * | Recoded | multiple residue substitutions to reduce TM hydrophobicity | [115] |
| ATP6, ATP8, ND1, ND2, ND3, ND4, ND4L, ND5, ND6, COX1, COX2, COX3, CYB | HEK293 and 143B WT cell lines | H. sapiens | + | Recoded and optimized | --- | [124] |
| mtATP6 (mRNA) | Drosophila model for mitochondrial encephalomyopathies (MEs) | D. melanogaster | --- | Drosophila mtATP6 mRNA | mRNA targeted to mitochondrial matrix for expression (mtTRESPro) | [135] |
| mtND1, mtND3, mtND4, mtND6, mtCOX2, mtCOX3, mtATP6, mtATP8 (mRNAs) | HeLa | H. sapiens | MTS Panel | Human mRNAs | 3′UTR Panel | [104] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saravanan, S.; Lewis, C.J.; Dixit, B.; O’Connor, M.S.; Stolzing, A.; Boominathan, A. The Mitochondrial Genome in Aging and Disease and the Future of Mitochondrial Therapeutics. Biomedicines 2022, 10, 490. https://doi.org/10.3390/biomedicines10020490
Saravanan S, Lewis CJ, Dixit B, O’Connor MS, Stolzing A, Boominathan A. The Mitochondrial Genome in Aging and Disease and the Future of Mitochondrial Therapeutics. Biomedicines. 2022; 10(2):490. https://doi.org/10.3390/biomedicines10020490
Chicago/Turabian StyleSaravanan, Sanjana, Caitlin J. Lewis, Bhavna Dixit, Matthew S. O’Connor, Alexandra Stolzing, and Amutha Boominathan. 2022. "The Mitochondrial Genome in Aging and Disease and the Future of Mitochondrial Therapeutics" Biomedicines 10, no. 2: 490. https://doi.org/10.3390/biomedicines10020490
APA StyleSaravanan, S., Lewis, C. J., Dixit, B., O’Connor, M. S., Stolzing, A., & Boominathan, A. (2022). The Mitochondrial Genome in Aging and Disease and the Future of Mitochondrial Therapeutics. Biomedicines, 10(2), 490. https://doi.org/10.3390/biomedicines10020490