
Endothelial Dysfunction in SARS-CoV-2 Infection


Abstract
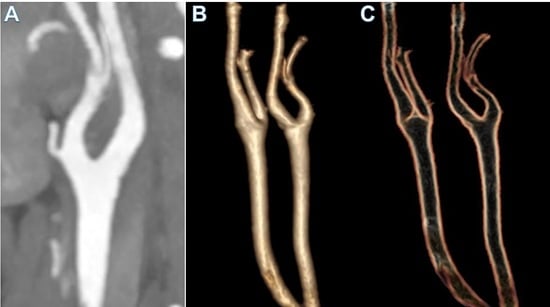
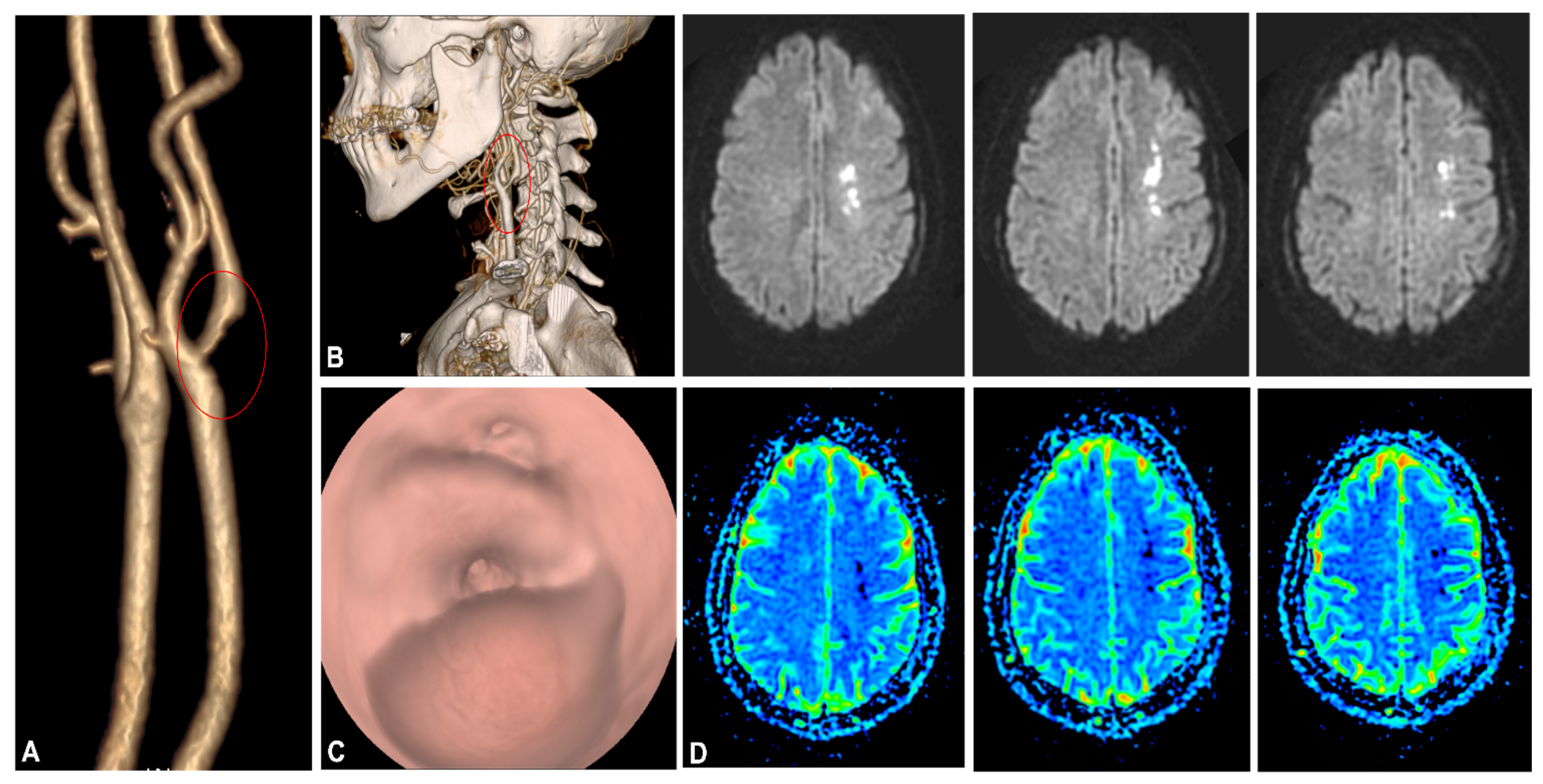
:
1. Introduction
2. The Clinical Problem
3. Pathophysiology
3.1. Coronavirus Infection in Humans
3.2. SARS Cov-2 Host Interaction
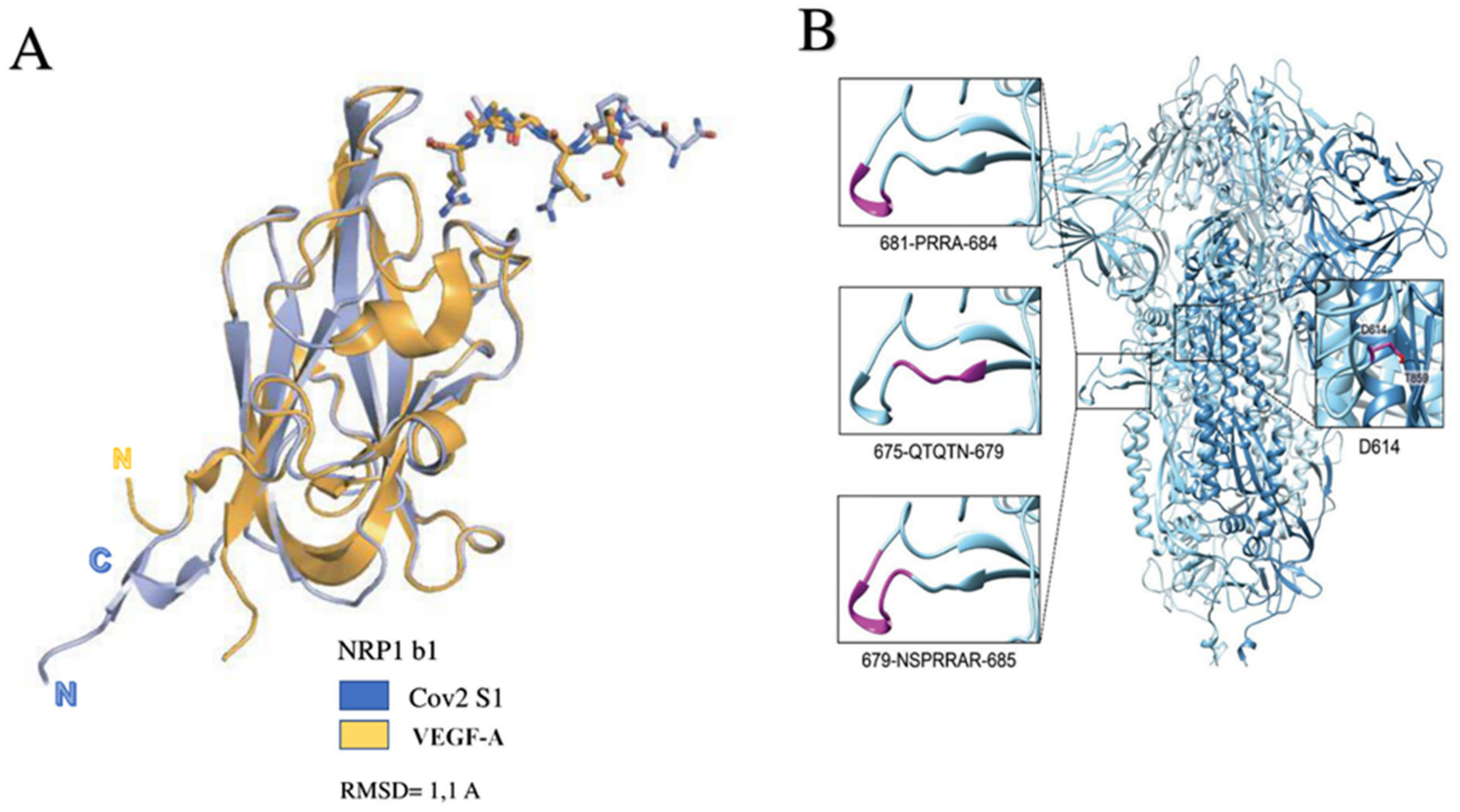
3.3. Functional Characteristics of Glycoprotein S
4. Endothelial Cell Infection and Endotheliitis in SARS-CoV-2 Infection
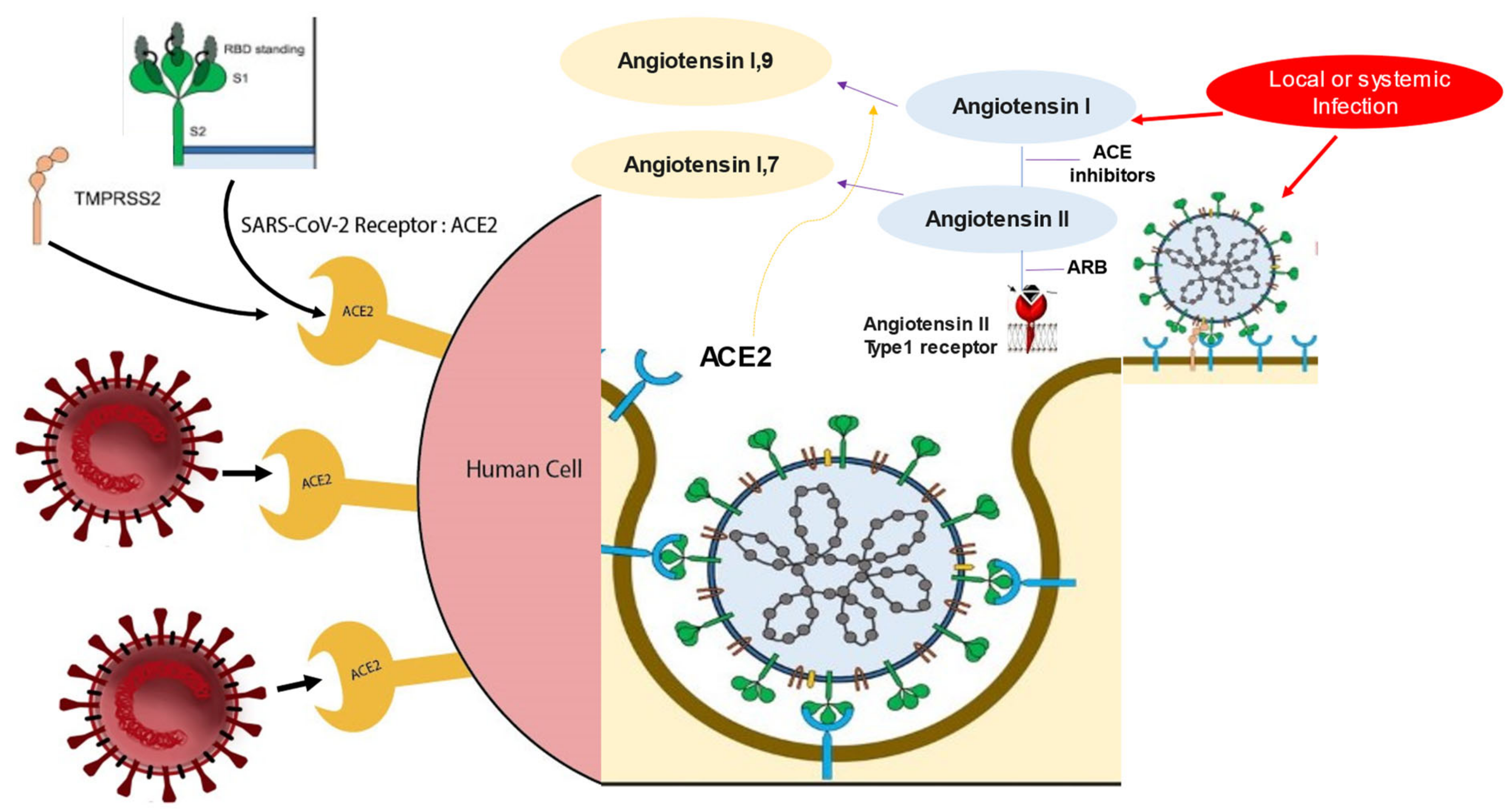
4.1. Interaction of SARS-CoV-2 and ACE2 Receptor: Insight of Influence on Renin–Angiotensin–Aldosterone System Inhibitors
4.2. RAAS Blockers in SARS-CoV-2 Infection—Potential for Benefit or Harm?
4.3. Insights of Angiogenesis and ACE 2 Expression on Endothelial Cells in SARS-CoV-2 Infection
4.4. Other Molecules (Mediators) Affecting Angiogenesis during SARS-CoV-2 Infection
5. The Pathoanatomic Alteration of the Endothelium and SARS-CoV-2 Infection
6. The Role of the Endothelium in Infection: Direct or Vicarious?
7. Comments and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ACA | anterior cerebral artery |
| ACE1 | angiotensin-converting enzyme I |
| ACE2 | angiotensin-converting enzyme 2 |
| Ang-II | angiotensin II |
| Ang-I | angiotensin I |
| Ang 1–7 | angiotensin 1–7 |
| ARDS | severe acute respiratory distress syndrome |
| ARBs | angiotensin II receptor blockers, |
| AT1R | angiotensin type-1 receptor |
| CoVs | coronaviruses |
| CAD | coronary artery disease |
| CVD | cardiovascular disease |
| CatB/L | cathepsin B/L |
| COVID-19 | coronavirus disease 2019 |
| CT | computed tomography |
| DIC | disseminated intravascular coagulation |
| EC | endothelial cell |
| Kb | kilobases |
| ICU | intensive care unit |
| IL | Interleukin |
| MCA | middle cerebral artery |
| MI | myocardial infarction |
| MRI | magnetic resonance |
| NRP | neuropilin |
| PE | pulmonary embolism |
| RBD | receptor-binding domain |
| RMSD | root-mean-square deviation |
| SARS-CoV | severe acute respiratory syndrome coronavirus |
| SARS-CoV-2 | severe acute respiratory syndrome coronavirus 2 |
| S | spike |
| TMPRSS2 | transmembrane serine protease 2 |
| VEGF-A | Vascular Endothelial Growth Factor |
| VTE | venous thromboembolism |
| WHO | World Health Organization |
References
- Cheng, V.C.C.; Lau, S.K.P.; Woo, P.C.Y.; Yuen, K.-Y. Severe Acute Respiratory Syndrome Coronavirus as an Agent of Emerging and Reemerging Infection. Clin. Microbiol. Rev. 2007, 20, 660–694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cowan, L.T.; Lutsey, P.L.; Pankow, J.S.; Matsushita, K.; Ishigami, J.; Lakshminarayan, K. Inpatient and Outpatient Infection as a Trigger of Cardiovascular Disease: The ARIC Study. J. Am. Heart Assoc. 2018, 7, e009683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madjid, M.; Miller, C.C.; Zarubaev, V.V.; Marinich, I.G.; Kiselev, O.I.; Lobzin, Y.V.; Filippov, A.E.; Casscells, S.W. Influenza epidemics and acute respiratory disease activity are associated with a surge in autopsy-confirmed coronary heart disease death: Results from 8 years of autopsies in 34 892 subjects. Eur. Heart J. 2007, 28, 1205–1210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fauci, A.S.; Lane, H.C.; Redfield, R.R. Covid-19—Navigating the Uncharted. N. Engl. J. Med. 2020, 382, 1268–1269. [Google Scholar] [CrossRef]
- Dhainaut, J.-F.; Claessens, Y.-E.; Janes, J.; Nelson, D.R. Underlying Disorders and Their Impact on the Host Response to Infection. Clin. Infect. Dis. 2005, 41, S481–S489. [Google Scholar] [CrossRef] [Green Version]
- Fehr, A.R.; Perlman, S. Coronaviruses: An overview of their replication and pathogenesis. Methods Mol. Biol. 2015, 1282, 1–23. [Google Scholar]
- Zhang, S.-F.; Tuo, J.-L.; Huang, X.-B.; Zhu, X.; Zhang, D.-M.; Zhou, K.; Yuan, L.; Luo, H.-J.; Zheng, B.-J.; Yuen, K.-Y.; et al. Epidemiology characteristics of human coronaviruses in patients with respiratory infection symptoms and phylogenetic analysis of HCoV-OC43 during 2010–2015 in Guangzhou. PLoS ONE 2018, 13, e0191789. [Google Scholar] [CrossRef]
- Team TNCPERE. The epidemiological characteristics of an outbreak of 2019 novel coronavirus diseases (COVID-19)-China, 2020. China CDC Wkly. 2020, 2, 113–122. [Google Scholar] [CrossRef]
- Chen, N.; Zhou, M.; Dong, X.; Qu, J.; Gong, F.; Han, Y.; Qiu, Y.; Wang, J.; Liu, Y.; Wei, Y.; et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: A descriptive study. Lancet 2020, 395, 507–513. [Google Scholar] [CrossRef] [Green Version]
- Su, S.; Wong, G.; Shi, W.; Liu, J.; Lai, A.C.K.; Zhou, J.; Liu, W.; Bi, Y.; Gao, G.F. Epidemiology, Genetic Recombination, and Pathogenesis of Coronaviruses. Trends Microbiol. 2016, 24, 490–502. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Hulswit, R.; Kenney, S.P.; Widjaja, I.; Jung, K.; Alhamo, M.A.; van Dieren, B.; van Kuppeveld, F.J.M.; Saif, L.J.; Bosch, B.-J. Broad receptor engagement of an emerging global coronavirus may potentiate its diverse cross-species transmissibility. Proc. Natl. Acad. Sci. USA 2018, 115, E5135–E5143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Sahly, H.M.; Atmar, R.L.; Glezen, W.P.; Greenberg, S.B. Spectrum of Clinical Illness in Hospitalized Patients with “Common Cold” Virus Infections. Clin. Infect. Dis. 2000, 31, 96–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imai, Y.; Kuba, K.; Rao, S.; Huan, Y.; Guo, F.; Guan, B.; Yang, P.; Sarao, R.; Wada, T.; Leong-Poi, H.; et al. Angiotensin-converting enzyme 2 protects from severe acute lung failure. Nature 2005, 436, 112–116. [Google Scholar] [CrossRef] [PubMed]
- Bonow, R.O.; Fonarow, G.C.; O’Gara, P.T.; Yancy, C.W. Association of Coronavirus Disease 2019 (COVID-19) with Myocardial Injury and Mortality. JAMA Cardiol. 2020, 5, 751–753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Driggin, E.; Madhavan, M.V.; Bikdeli, B.; Chuich, T.; Laracy, J.; Biondi-Zoccai, G.; Brown, T.S.; Der Nigoghossian, C.; Zidar, D.A.; Haythe, J.; et al. Cardiovascular considerations for patients, health care workers, and health systems during the coronavirus disease 2019 (COVID-19) pandemic. J. Am. Coll. Cardiol. 2020, 75, 2352–2371. [Google Scholar] [CrossRef]
- Bikdeli, B.; Madhavan, M.V.; Jimenez, D.; Chuich, T.; Dreyfus, I.; Driggin, E.; Nigoghossian, C.D.; Ageno, W.; Madjid, M.; for the Global COVID-19 Thrombosis Collaborative Group; et al. COVID-19 and thrombotic or thromboembolic disease, implications for prevention, antithrombotic therapy, and follow-up, JACC State-of the- Art Review. J. Am. Coll. Cardiol. 2020, 75, 2950–2973. [Google Scholar] [CrossRef]
- Wang, A.; Mandigo, G.K.; Yim, P.D.; Meyers, P.M.; LaVine, S.D. Stroke and mechanical thrombectomy in patients with COVID-19: Technical observations and patient characteristics. J. NeuroInterv. Surg. 2020, 12, 648–653. [Google Scholar] [CrossRef]
- Viguier, A.; Delamarre, L.; Duplantier, J.; Olivot, J.-M.; Bonneville, F. Acute ischemic stroke complicating common carotid artery thrombosis during a severe COVID-19 infection. J. Neuroradiol. 2020, 47, 393–394. [Google Scholar] [CrossRef]
- Fara, M.G.; Stein, L.K.; Skliut, M.; Morgello, S.; Fifi, J.T.; Dhamoon, M.S. Macrothrombosis and stroke in patients with mild Covid-19 infection. J. Thromb. Haemost. 2020, 18, 2031–2033. [Google Scholar] [CrossRef]
- Román, G.C.; Reis, J.; Spencer, P.S.; Buguet, A.; Öztürk, S.; Wasay, M. World Federation of Neurology Environmental Neurology Specialty Group COVID-19 international neurological registries. Lancet Neurol. 2020, 19, 484–485. [Google Scholar] [CrossRef]
- Tassorelli, C.; Mojoli, F.; Baldanti, F.; Bruno, R.; Benazzo, M. COVID-19: What if the brain had a role in causing the deaths? Eur. J. Neurol. 2020, 27, e41–e42. [Google Scholar] [CrossRef] [PubMed]
- Mao, L.; Jin, H.; Wang, M.; Hu, Y.; Chen, S.; He, Q.; Chang, J.; Hong, C.; Zhou, Y.; Wang, D.; et al. Neurologic Manifestations of Hospitalized Patients With Coronavirus Disease 2019 in Wuhan, China. JAMA Neurol. 2020, 77, 683–690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klok, F.A.; Kruip, M.J.H.A.; Van der Meer, N.J.M.; Arbous, M.S.; Gommers, D.A.M.P.J.; Kant, K.M.; Kaptein, F.H.J.; van Paassen, J.; Stals, M.A.M.; Huisman, M.V.; et al. Incidence of thrombotic complications in critically ill ICU patients with COVID-19. Thromb. Res. 2020, 191, 145–147. [Google Scholar] [CrossRef] [PubMed]
- Tang, N.; Li, D.; Wang, X.; Sun, Z. Abnormal coagulation parameters are associated with poor prognosis in patients with novel coronavirus pneumonia. J. Thromb. Haemost. 2020, 18, 844–847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Llitjos, J.-F.; Leclerc, M.; Chochois, C.; Monsallier, J.-M.; Ramakers, M.; Auvray, M.; Merouani, K. High incidence of venous thromboembolic events in anticoagulated severe COVID-19 patients. J. Thromb. Haemost. 2020, 18, 1743–1746. [Google Scholar] [CrossRef] [PubMed]
- Lodigiani, C.; Iapichino, G.; Carenzo, L.; Cecconi, M.; Ferrazzi, P.; Sebastian, T.; Kucher, N.; Studt, J.-D.; Sacco, C.; for the Humanitas COVID-19 Task Force; et al. Venous and arterial thromboembolic complications in COVID-19 patients admitted to an academic hospital in Milan, Italy. Thromb. Res. 2020, 191, 9–14. [Google Scholar] [CrossRef]
- Marietta, M.; Ageno, W.; Artoni, A.; De Candia, E.; Gresele, P.; Marchetti, M.; Marcucci, R.; Tripodi, A. COVID-19 and haemostasis: A position paper from Italian Society on Thrombosis and Haemostasis (SISET). Blood Transfus. 2020, 18, 167–169. [Google Scholar]
- Ackermann, M.; Verleden, S.E.; Kuehnel, M.; Haverich, A.; Welte, T.; Laenger, F.; Vanstapel, A.; Werlein, C.; Stark, H.; Tzankov, A.; et al. Pulmonary vascular endothelialitis, thrombosis, and angiogenesis in Covid-19. N. Engl. J. Med. 2020, 383, 120–128. [Google Scholar] [CrossRef]
- Wichmann, D.; Sperhake, J.P.; Lütgehetmann, M.; Steurer, S.; Edler, C.; Heinemann, A.; Heinrich, F.; Mushumba, H.; Kniep, I.; Schröder, A.S.; et al. Autopsy findings and venous thromboembolism in patients with CO-VID-19. Ann. Intern. Med. 2020, 173, 268–277. [Google Scholar] [CrossRef]
- Fosse, J.H.; Haraldsen, G.; Falk, K.; Edelmann, R. Endothelial Cells in Emerging Viral Infections. Front. Cardiovasc. Med. 2021, 8, 619690. [Google Scholar] [CrossRef]
- Moores, L.K.; Tritschler, T.; Brosnahan, S.; Carrier, M.; Collen, J.F.; Doerschug, K.; Holley, A.B.; Jimenez, D.; Le Gal, G.; Rali, P.; et al. Prevention, Diagnosis, and Treatment of VTE in Patients with Coronavirus Disease 2019: CHEST guideline and expert panel report. Chest 2020, 158, 1143–1163. [Google Scholar] [CrossRef] [PubMed]
- Spyropoulos, A.C.; Levy, J.H.; Ageno, W.; Connors, J.M.; Hunt, B.J.; Iba, T.; Levi, M.; Samama, C.M.; Giannis, D. Scientific and Standardization Committee communication, clinical guidance on the diagnosis, prevention and treatment of venous thromboembolism in hospitalized patients with COVID-19. J. Thromb. Haemost. 2020, 18, 1859–1865. [Google Scholar] [CrossRef] [PubMed]
- Tang, N.; Bai, H.; Chen, X.; Gong, J.; Li, D.; Sun, Z. Anticoagulant treatment is associated with decreased mortality in severe coro-navirus disease 2019 patients with coagulopathy. J. Thromb. Haemost. 2020, 18, 1094–1099. [Google Scholar] [CrossRef] [PubMed]
- Weiss, S.R.; Navas-Martin, S. Coronavirus Pathogenesis and the Emerging Pathogen Severe Acute Respiratory Syndrome Coronavirus. Microbiol. Mol. Biol. Rev. 2005, 69, 635–664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perlman, S.; Netland, J. Coronaviruses post-SARS: Update on replication and pathogenesis. Nat. Rev. Microbiol. 2009, 7, 439–450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belouzard, S.; Millet, J.K.; Licitra, B.N.; Whittaker, G.R. Mechanisms of Coronavirus Cell Entry Mediated by the Viral Spike Protein. Viruses 2012, 4, 1011–1033. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Guan, X.; Wu, P.; Wang, X.; Zhou, L.; Tong, Y.; Ren, R.; Leung, K.S.M.; Lau, E.H.Y.; Wong, J.Y.; et al. Early Transmission Dynamics in Wuhan, China, of Novel Coronavirus–Infected Pneumonia. N. Engl. J. Med. 2020, 382, 1199–1207. [Google Scholar] [CrossRef]
- Lu, R.; Zhao, X.; Li, J.; Niu, P.; Yang, B.; Wu, H.; Wang, W.; Song, H.; Huang, B.; Zhu, N.; et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: Implications for virus origins and receptor binding. Lancet 2020, 395, 565–574. [Google Scholar] [CrossRef] [Green Version]
- Liang, W.; Zhu, Z.; Guo, J.; Liu, Z.; He, X.; Zhou, W.; Chin, D.P.; Schuchat, A.; for the Beijing Joint SARS Expert Group. Severe Acute Respiratory Syndrome, Beijing, 2003. Emerg. Infect. Dis. 2004, 10, 25–31. [Google Scholar] [CrossRef] [Green Version]
- Aleanizy, F.S.; Mohmed, N.; Alqahtani, F.Y.; El Hadi Mohamed, R.A. Outbreak of Middle East respiratory syndrome coronavirus in Saudi Arabia, a retrospective study. BMC Infect. Dis. 2017, 17, 23. [Google Scholar] [CrossRef] [Green Version]
- Paules, C.I.; Marston, H.D.; Fauci, A.S. Coronavirus Infections—More Than Just the Common Cold. JAMA J. Am. Med. Assoc. 2020, 323, 707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.T.; Leung, K.; Leung, G.M. Nowcasting and forecasting the potential domestic and international spread of the 2019-nCoV outbreak originating inWuhan, China: A modelling study. Lancet 2020, 395, 689–697. [Google Scholar] [CrossRef] [Green Version]
- Cohen, J. Mining coronavirus genomes for clues to the outbreak’s origins. Science 2010, 31, 31. [Google Scholar] [CrossRef]
- del Rio, C.; Malani, P.N. Novel coronavirus—Important information for clinicians. JAMA 2019, 323, 1039–1040. [Google Scholar] [CrossRef]
- Chan, J.F.-W.; Kok, K.-H.; Zhu, Z.; Chu, H.; To, K.K.-W.; Yuan, S.; Yuen, K.-Y. Genomic characterization of the 2019 novel human-pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan. Emerg. Microbes Infect. 2020, 9, 221–236. [Google Scholar] [CrossRef] [Green Version]
- Andersen, K.G.; Rambaut, A.; Lipkin, W.I.; Holmes, E.C.; Garry, R.F. The proximal origin of SARS-CoV-2. Nat. Med. 2020, 26, 450–452. [Google Scholar] [CrossRef] [Green Version]
- Kuba, K.; Imai, Y.; Rao, S.; Jiang, C.; Penninger, J.M. Lessons from SARS: Control of acute lung failure by the SARS receptor ACE2. Klin. Wochenschr. 2006, 84, 814–820. [Google Scholar] [CrossRef]
- Li, F. Receptor Recognition Mechanisms of Coronaviruses: A Decade of Structural Studies. J. Virol. 2014, 89, 1954–1964. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, M.; Kleine-Weber, H.; Pöhlmann, S. A Multibasic Cleavage Site in the Spike Protein of SARS-CoV-2 Is Essential for Infection of Human Lung Cells. Mol. Cell 2020, 78, 779–784.e5. [Google Scholar] [CrossRef]
- Zhou, P.; Yang, X.-L.; Wang, X.-G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.-R.; Zhu, Y.; Li, B.; Huang, C.-L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [Green Version]
- Johnson, M.C.; Lyddon, T.D.; Suarez, R.; Salcedo, B.; LePique, M.; Graham, M.; Ricana, C.; Robinson, C.; Ritter, D.G. Optimized Pseudotyping Conditions for the SARS-CoV-2 Spike Glycoprotein. J. Virol. 2020, 94, e01062-20. [Google Scholar] [CrossRef] [PubMed]
- Laporte, M.; Raeymaekers, V.; Van Berwaer, R.; Vandeput, J.; Marchand-Casas, I.; Thibaut, H.J.; Van Looveren, D.; Martens, K.; Hoffmann, M.; Maes, P.; et al. The SARS-CoV-2 and other human coronavirus spike proteins are fine-tuned towards temperature and proteases of the human airways. PLoS Pathog. 2021, 17, e1009500. [Google Scholar] [CrossRef] [PubMed]
- Daly, J.L.; Simonetti, B.; Klein, K.; Chen, K.E.; Williamson, M.K.; Antón-Plágaro, C.; Shoemark, D.K.; Simón-Gracia, L.; Bauer, M.; Hollandi, R.; et al. Neuropilin-1 is a host factor for SARS-CoV-2 infection. Science 2020, 370, 861–865. [Google Scholar] [CrossRef] [PubMed]
- Teuwen, L.-A.; Geldhof, V.; Pasut, A.; Carmeliet, P. COVID-19: The vasculature unleashed. Nat. Rev. Immunol. 2020, 20, 389–391. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 2020, 181, 271–280.e278. [Google Scholar] [CrossRef]
- Gerhardt, H.; Ruhrberg, C.; Abramsson, A.; Fujisawa, H.; Shima, D.; Betsholtz, C. Neuropilin-1 is required for endothelial tip cell guidance in the developing central nervous system. Dev. Dyn. 2004, 231, 503–509. [Google Scholar] [CrossRef]
- Jones, E.A.V.; Yuan, L.; Breant, C.; Watts, R.J.; Eichmann, A. Separating genetic and hemodynamic defects in neuropilin 1 knockout embryos. Development 2008, 135, 2479–2488. [Google Scholar] [CrossRef] [Green Version]
- Aspalter, I.M.; Gordon, E.; Dubrac, A.; Ragab, A.; Narloch, J.; Vizán, P.; Geudens, I.; Collins, R.T.; Franco, C.A.; Abrahams, C.L.; et al. Alk1 and Alk5 inhibition by Nrp1 controls vascular sprouting down Stream of Notch. Nat. Commun. 2015, 6, 7264. [Google Scholar] [CrossRef] [Green Version]
- Cantuti-Castelvetri, L.; Ojha, R.; Pedro, L.D.; Djannatian, M.; Franz, J.; Kuivanen, S.; van der Meer, F.; Kallio, K.; Kaya, T.; Anastasina, M.; et al. Neuropilin-1 facilitates SARS-CoV-2 cell entry and infectivity. Science 2020, 370, 856–860. [Google Scholar] [CrossRef]
- Zhou, F.; Yu, T.; Du, R.; Fan, G.; Liu, Y.; Liu, Z.; Xiang, J.; Wang, Y.; Song, B.; Gu, X.; et al. Clinical course and risk factors for mortality of adult in patients with COVID-19 in Wuhan, China, a retrospective cohort study. Lancet 2020, 395, 1054–1062. [Google Scholar] [CrossRef]
- Gu, S.X.; Tyagi, T.; Jain, K.; Gu, V.W.; Lee, S.H.; Hwa, J.M.; Kwan, J.M.; Krause, D.S.; Lee, A.I.; Halene, S.; et al. Thrombocytopathy and endotheliopathy: Crucial contributors to COVID-19 thromboinflammation. Nat. Rev. Cardiol. 2020, 18, 194–209. [Google Scholar] [CrossRef] [PubMed]
- Goshua, G.; Pine, A.B.; Meizlish, M.L.; Chang, C.-H.; Zhang, H.; Bahel, P.; Baluha, A.; Bar, N.; Bona, R.D.; Burns, A.J.; et al. Endotheliopathy in COVID-19-associated coagulopathy, evidence from a single-centre, cross-sectional study. Lancet Haematol. 2020, 7, e575–e582. [Google Scholar] [CrossRef]
- Smadja, D.M.; Mentzer, S.J.; Fontenay, M.; Laffan, M.A.; Ackermann, M.; Helms, J.; Jonigk, D.; Chocron, R.; Pier, G.B.; Gendron, N.; et al. COVID-19 is a systemic vascular hemopathy: Insight for mechanistic and clinical aspects. Angiogenesis 2021, 24, 755–788. [Google Scholar] [CrossRef] [PubMed]
- Sardu, C.; Gambardella, J.; Morelli, M.B.; Wang, X.; Marfella, R.; Santulli, G. Hypertension, thrombosis, kidney failure, and diabetes, is COVID-19 an endothelial disease ? A comprehensive evaluation of clinical and basic evidence. J. Clin. Med. 2020, 9, 1417. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Madhavan, M.V.; Sehgal, K.; Nair, N.; Mahajan, S.; Sehrawat, T.S.; Bikdeli, B.; Ahluwalia, N.; Ausiello, J.C.; Wan, E.Y.; et al. Extrapulmonary manifestations of COVID-19. Nat. Med. 2020, 26, 1017–1032. [Google Scholar] [CrossRef]
- Vickers, C.; Hales, P.; Kaushik, V.; Dick, L.; Gavin, J.; Tang, J.; Godbout, K.; Parsons, T.; Baronas, E.; Hsieh, F.; et al. Hydrolysis of biological peptides by human angiotensin-converting enzyme-related car-boxypeptidase. J. Biol. Chem. 2002, 277, 14838–14843. [Google Scholar] [CrossRef] [Green Version]
- Hamming, I.; Timens, W.; Bulthuis, M.L.C.; Lely, A.T.; Navis, G.J.; van Goor, H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus, a first step in understanding SARS pathogenesis. J. Pathol. 2004, 203, 631–637. [Google Scholar] [CrossRef]
- Serfozo, P.; Wysocki, J.; Gulua, G.; Schulze, A.; Ye, M.; Liu, P.; Jin, J.; Bader, M.; Myöhänen, T.; García-Horsman, J.A.; et al. Ang II (angiotensin II) conversion to angiotensin-(1-7) in the circulation is POP (prolylo-ligopeptidase)-dependent and ACE2 (angiotensin-converting enzyme 2) -independent. Hypertension 2020, 75, 173–182. [Google Scholar] [CrossRef]
- Rice, G.I.; Thomas, D.A.; Grant, P.J.; Turner, A.J.; Hooper, N.M. Evaluation of angiotensin-converting enzyme (ACE), its homologue ACE2 and neprilysin in angiotensin peptide metabolism. Biochem. J. 2004, 383, 45–51. [Google Scholar] [CrossRef]
- Ferrario, C.M.; Jessup, J.; Chappell, M.C.; Averill, D.B.; Brosnihan, K.B.; Tallant, E.A.; Diz, D.I.; Gallagher, P.E. Effect of Angiotensin-Converting Enzyme Inhibition and Angiotensin II Receptor Blockers on Cardiac Angiotensin-Converting Enzyme 2. Circulation 2005, 111, 2605–2610. [Google Scholar] [CrossRef] [Green Version]
- Ocaranza, M.P.; Godoy, I.; Jalil, J.E.; Varas, M.; Collantes, P.; Pinto, M.; Roman, M.; Ramirez, C.; Copaja, M.; Diaz-Araya, G.; et al. Enalapril attenuates downregulation of angiotensin-converting enzyme 2 in the late phase of ventricular dysfunction in myocardial infarcted rat. Hypertension 2006, 48, 572–578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vuille-dit-Bille, R.N.; Camargo, S.M.; Emmenegger, L.; Sasse, T.; Kummer, E.; Jando, J.; Hamie, Q.M.; Meier, C.F.; Hunziker, S.; Forras-Kaufmann, Z.; et al. Human intestine luminal ACE2 and amino acid transporter expression increased by ACE-inhibitors. Amino Acids 2015, 47, 693–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sukumaran, V.; Tsuchimochi, H.; Tatsumi, E.; Shirai, M.; Pearson, J.T. Azilsartan ameliorates diabetic cardiomyopathy in young db/ db mice through the modulation of ACE-2/ANG 1-7/Mas receptor cascade. Biochem. Pharmacol. 2017, 144, 90–99. [Google Scholar] [CrossRef] [PubMed]
- Sukumaran, V.; Veeraveedu, P.T.; Lakshmanan, A.P.; Gurusamy, N.; Yamaguchi, K.I.; Ma, M.; Suzuki, K.; Kodama, M.; Watanabe, K. Olmesartan medoxomil treatment potently improves cardiac myo-sininduced dilated cardiomyopathy via the modulation of ACE-2 and ANG 1-7 Mas receptor. Free Radic. Res. 2012, 46, 850–860. [Google Scholar] [CrossRef]
- Sukumaran, V.; Veeraveedu, P.T.; Gurusamy, N.; Lakshmanan, A.P.; Yamaguchi, K.; Ma, M.; Suzuki, K.; Nagata, M.; Takagi, R.; Kodama, M.; et al. Olmesartan attenuates the development of heart failure after experimental autoimmune myocarditis in rats through the modulation of ANG 1–7 mas receptor. Mol. Cell. Endocrinol. 2012, 351, 208–219. [Google Scholar] [CrossRef]
- Lakshmanan, A.P.; Thandavarayan, R.A.; Watanabe, K.; Sari, F.R.; Meilei, H.; Giridharan, V.V.; Sukumaran, V.; Soetikno, V.; Arumugam, S.; Suzuki, K.; et al. Modulation of AT-1R/MAPK cascade by an olmesartan treatment attenuates diabetic nephropathy in streptozotocin-induced diabetic mice. Mol. Cell. Endocrinol. 2012, 348, 104–111. [Google Scholar] [CrossRef]
- Burchill, L.J.; Velkoska, E.; Dean, R.G.; Griggs, K.; Patel, S.K.; Burrell, L.M. Combination renin-angiotensin system blockade and an-giotensin- converting enzyme 2 in experimental myocardial infarction, implications for future therapeutic directions. Clin. Sci. 2012, 123, 649–658. [Google Scholar] [CrossRef] [Green Version]
- Campbell, D.J.; Zeitz, C.J.; Esler, M.D.; Horowitz, J.D. Evidence against a major role for angiotensin converting enzyme-related carboxypeptidase (ACE2) in angiotensin peptide metabolism in the human coronary circulation. J. Hypertens. 2004, 22, 1971–1976. [Google Scholar] [CrossRef]
- Luque, M.; Martin, P.; Martell, N.; Fernandez, C.; Brosnihan, K.B.; Ferrario, C.M. Effects of captopril related to increased levels of prostacyclin and angiotensin-(1-7) in essential hypertension. J. Hypertens. 1996, 14, 799–805. [Google Scholar] [CrossRef]
- Furuhashi, M.; Moniwa, N.; Mita, T.; Fuseya, T.; Ishimura, S.; Ohno, K.; Shibata, S.; Tanaka, M.; Watanabe, Y.; Akasaka, H.; et al. Urinary angiotensinconverting enzyme 2 in hypertensive patients may be increased by olmesartan, an angiotensin II receptor blocker. Am. J. Hypertens. 2015, 28, 15–21. [Google Scholar] [CrossRef] [Green Version]
- Lambert, D.W.; Yarski, M.; Warner, F.J.; Thornhill, P.; Parkin, E.T.; Smith, A.I.; Hooper, N.M.; Turner, A.J. Tumor necrosis factor-α convertase (ADAM17) mediates regulated ectodomain shedding of the severe-acute respiratory syndrome-coronavirus (SARS-CoV) receptor, angiotensin-converting enzyme-2 (ACE2). J. Biol. Chem. 2005, 280, 30113–30119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gurwitz, D. Angiotensin receptor blockers as tentative SARS-CoV-2 therapeutics. Drug Dev. Res. 2020, 81, 537–540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Yang, Y.; Zhang, C.; Huang, F.; Wang, F.; Yuan, J.; Wang, Z.; Li, J.; Li, J.; Feng, C.; et al. Clinical and biochemical indexes from 2019-nCoV infected patients linked to viral loads and lung injury. Sci. China Life Sci. 2020, 63, 364–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sodhi, C.P.; Wohlford-Lenane, C.; Yamaguchi, Y.; Prindle, T.; Fulton, W.B.; Wang, S.; McCray, P.B., Jr.; Chappell, M.; Hackam, D.J.; Jia, H.; et al. Attenuation of pulmonary ACE2 activity impairs inactivation of des-Arg9 bradykinin/BKB1R axis and facilitates LPS-induced neutrophil infiltration. Am. J. Physiol. Lung Cell. Mol. Physiol. 2018, 314, L17–L31. [Google Scholar] [CrossRef] [PubMed]
- Hemnes, A.R.; Rathinasabapathy, A.; Austin, E.A.; Brittain, E.L.; Carrier, E.J.; Chen, X.; Fessel, J.P.; Fike, C.D.; Fong, P.; Fortune, N.; et al. A potential therapeutic role for angiotensin-converting enzyme 2 in human pulmonary arterial hypertension. Eur. Respir. J. 2018, 51, 1702638. [Google Scholar] [CrossRef] [PubMed]
- Tan, W.S.D.; Liao, W.; Zhou, S.; Mei, D.; Wong, W.-S.F. Targeting the renin-angiotensin system as novel therapeutic strategy for pulmonary diseases. Curr. Opin. Pharmacol. 2018, 40, 9–17. [Google Scholar] [CrossRef]
- Shi, S.; Qin, M.; Shen, B.; Cai, Y.; Liu, T.; Yang, F.; Gong, W.; Liu, X.; Liang, J.; Zhao, Q.; et al. Association of cardiac injury with mortality in hospitalized patients with COVID-19 in Wuhan, China. JAMA Cardiol. 2020, 5, 802–810. [Google Scholar] [CrossRef] [Green Version]
- Oudit, G.Y.; Kassiri, Z.; Jiang, C.; Liu, P.P.; Poutanen, S.M.; Penninger, J.M.; Butany, J. SARS-coronavirus modulation of myocardial ACE2 expression and inflammation in patients with SARS. Eur. J. Clin. Investig. 2009, 39, 618–625. [Google Scholar] [CrossRef]
- Basu, R.; Poglitsch, M.; Yogasundaram, H.; Thomas, J.; Rowe, B.H.; Oudit, G.Y. Roles of angiotensin peptides and recombinant human ACE2 in heart failure. J. Am. Coll. Cardiol. 2017, 69, 805–819. [Google Scholar] [CrossRef]
- Nicin, L.; Abplanalp, W.T.; Mellentin, H.; Kattih, B.; Tombor, L.; John, D.; Schmitto, J.D.; Heineke, J.; Emrich, F.; Arsalan, M.; et al. Cell typespecific expression of the putative SARS-CoV-2 receptor ACE2 in human hearts. Eur. Heart J. 2020, 41, 1804–1806. [Google Scholar] [CrossRef] [Green Version]
- Sluimer, J.; Gasc, J.M.; Hamming, I.; van Goor, H.; Michaud, A.; Akker, L.H.V.D.; Jutten, B.; Cleutjens, J.; Bijnens, A.P.J.J.; Corvol, P.; et al. Angiotensinconverting enzyme 2 (ACE2) expression and activity in human carotid atherosclerotic lesions. J. Pathol. 2008, 215, 273–279. [Google Scholar] [CrossRef] [PubMed]
- Hikmet, F.; Mear, L.; Edvinsson, A.; Micke, P.; Uhlen, M.; Lindskog, C. The protein expression profile of ACE2 in human tissues. Mol. Syst. Biol. 2020, 16, e9610. [Google Scholar] [CrossRef] [PubMed]
- Monteil, V.; Kwon, H.; Prado, P.; Hagelkrüys, A.; Wimmer, R.A.; Stahl, M.; Leopoldi, A.; Garreta, E.; Del Pozo, C.H.; Prosper, F.; et al. Inhibition of SARS-CoV-2 infections in engineered human tissues using clini-cal-grade soluble human ACE2. Cell 2020, 18, 905–913. Available online: https://www.cell.com/pbassets/products/coronavirus/CELL_CELL-D-20-00739.pdf (accessed on 17 April 2020). [CrossRef] [PubMed]
- Chen, L.; Li, X.; Chen, M.; Feng, Y.; Xiong, C. The ACE2 expression in human heart indicates new potential mechanism of heart injury among patients infected with SARS-CoV-2. Cardiovasc. Res. 2020, 116, 1097–1100. [Google Scholar] [CrossRef] [Green Version]
- McCracken, I.R.; Saginc, G.; He, L.; Huseynov, A.; Daniels, A.; Fletcher, S.; Peghaire, C.; Kalna, V.; Andaloussi-Mäe, M.; Muhl, L.; et al. Lack of evidence of angiotensin-converting enzyme 2 expression and replicative infection by SARS-CoV-2 in human endothelial cells. Circulation 2021, 143, 865–868. [Google Scholar] [CrossRef]
- Litviňuková, M.; Talavera-López, C.; Maatz, H.; Reichart, D.; Worth, C.L.; Lindberg, E.L.; Kanda, M.; Polanski, K.; Heinig, M.; Lee, M.; et al. Cells of the adult human heart. Nature 2020, 588, 466–472. [Google Scholar] [CrossRef]
- He, L.; Mäe, M.A.; Muhl, L.; Sun, Y.; Pietilä, R.; Nahar, K.; Liébanas, E.V.; Fagerlund, M.J.; Oldner, A.; Liu, J.; et al. Pericyte-specific vascular expression of SARS-CoV-2 receptor ACE2, implications for microvascular inflammation and hypercoagulopathy in COVID-19. BioRxiv 2020. Available online: https://www.biorxiv.org/content/10.1101/2020.05.11.088500v2.full.pdf (accessed on 17 April 2020).
- Rauch, A.; Dupont, A.; Goutay, J.; Caplan, M.; Staessens, S.; Moussa, M.; Jeanpierre, E.; Corseaux, D.; Lille COVID Research Network (LICORNE); Members of the LICORNE Scientific Committee; et al. Endotheliopathy is induced by plasma from critically Ill patients and associated with organ failure in severe COVID-19. Circulation 2020, 142, 1881–1884. [Google Scholar] [CrossRef]
- Aimes, R.T.; Zijlstra, A.; Hooper, J.D.; Ogbourne, S.M.; Sit, M.L.; Fuchs, S.; Gotley, D.C.; Quigley, J.P.; Antalis, T.M. Endothelial cell serine proteases expressed during vascular morphogenesis and angio-genesis. Thromb. Haemost. 2003, 89, 561–572. [Google Scholar] [CrossRef] [Green Version]
- Bryce, C.; Grimes, Z.; Pujadas, E.; Ahuja, S.; Beasley, M.B.; Albrecht, R.; Hernandez, T.; Stock, A.; Zhao, Z.; AlRasheed, M.R.; et al. Pathophysiology of SARS-CoV-2, the Mount Sinai COVID-19 autopsy experience. Mod. Pathol. 2021, 34, 1456–1467. [Google Scholar] [CrossRef]
- Schaefer, I.M.; Padera, R.F.; Solomon, I.H.; Kanjilal, S.; Hammer, M.M.; Hornick, J.L.; Sholl, L.M. In situ detection of SARS-CoV-2 in lungs and airways of patients with COVID-19. Mod. Pathol. 2020, 33, 2104–2114. [Google Scholar] [CrossRef] [PubMed]
- Varga, Z.; Flammer, A.J.; Steiger, P.; Haberecker, M.; Andermatt, R.; Zinkernagel, A.S.; Mehra, M.R.; Schuepbach, R.A.; Ruschitzka, F.; Moch, H. Endothelial cell infection and endotheliitis in COVID-19. Lancet 2020, 395, 1417–1418. [Google Scholar] [CrossRef]
- Delorey, T.M.; Ziegler, C.G.; Heimberg, G.; Normand, R.; Yang, Y.; Segerstolpe, Å.; Abbondanza, D.; Fleming, S.J.; Subramanian, A.; Montoro, D.T.; et al. COVID-19 tissue atlases reveal SARS-CoV-2 pathology and cellular targets. Nature 2021, 595, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Lindner, D.; Fitzek, A.; Bräuninger, H.; Aleshcheva, G.; Edler, C.; Meissner, K.; Scherschel, K.; Kirchhof, P.; Escher, F.; Schultheiss, H.P.; et al. Association of Cardiac Infection With SARS-CoV-2 in Confirmed COVID-19 Autopsy Cases. JAMA Cardiol. 2020, 5, 1281–1285. [Google Scholar] [CrossRef]
- Schimmel, L.; Chew, K.Y.; Stocks, C.J.; Yordanov, T.E.; Essebier, P.; Kulasinghe, A.; Monkman, J.; Dos Santos Miggiolaro, A.F.R.; Cooper, C.; de Noronha, L.; et al. Endothelial cells are not productively infected by SARS-CoV-2. Clin. Transl. Immunol. 2021, 10, e1350. [Google Scholar] [CrossRef]
- Magro, C.; Mulvey, J.J.; Berlin, D.; Nuovo, G.; Salvatore, S.; Harp, J.; Baxter-Stoltzfus, A.; Laurence, J. Complement associated microvascular injury and thrombosis in the pathogenesis of severe COVID-19 infection, a report of five cases. Transl. Res. 2020, 220, 1–13. [Google Scholar] [CrossRef]
- O’Donnell, J.S.; Sharif, K.; Emery, P.; Bridgewood, C.; McGonagle, D. Why the immune mechanisms of pulmonary intravascular coagulopathy in COVID-19 pneumonia are distinct from macrophage activation syndrome with disseminated intravascular coagulation. Lancet Rheumatol. 2020, 2, e437–e445. [Google Scholar]
- Mentzer, S.J.; Konerding, M.A. Intussusceptive angiogenesis, expansion and remodeling of microvascular networks. Angiogenesis 2014, 17, 499–509. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Xiao, M.; Zhang, S.; Xia, P.; Cao, W.; Jiang, W.; Chen, H.; Ding, X.; Zhao, H.; Zhang, H.; et al. Coagulopathy and Antiphospholipid Antibodies in Patients with Covid-19. N. Engl. J. Med. 2020, 382, e38. [Google Scholar] [CrossRef]
- Madjid, M.; Safavi-Naeini, P.; Solomon, S.D.; Vardeny, O. Potential effects of coronaviruses on the cardiovascular system: A review. JAMA Cardiol. 2020, 5, 831–840. [Google Scholar] [CrossRef] [Green Version]
- Yan, R.; Zhang, Y.; Li, Y.; Xia, L.; Guo, Y.; Zhou, Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science 2020, 367, 1444–1448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giacinto, O.; Satriano, U.; Nenna, A.; Spadaccio, C.; Lusini, M.; Mastroianni, C.; Nappi, F.; Chello, M. Inflammatory Response and Endothelial Dysfunction Following Cardiopulmonary Bypass: Pathophysiology and Pharmacological Targets. Recent Pat. Inflamm. Allergy Drug Discov. 2019, 13, 158–173. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| First Author/Year Ref | Type of Study | Number of Patients | Mean Age (Yrs) | Autopsy (n) | Findings |
|---|---|---|---|---|---|
| Bryce et al., 2021 [100] | OS | 100 | 29 to 94 years (Median 68) | Lung 99 Heart 97 Spleen 86 Lymphnodes 60 Kidney 94 | 82 cases DAD; Hemophagocytosis Higher cytokines IL-6, IL-8, and TNFα. |
| Ackerman et al., 2020 [28] | OS | 14 SARS-CoV-2 7 H1N1 7 | 68 ± 9.2 years (female) 80 ± 11.5 years (male) | Lung 14 | Alveolar capillary microthrombi 9 times more in SARS-CoV-2 Higher CD3, CD4 and CD-8 positive T cells in SARS-CoV-2 Lower neutrophils (CD15) |
| Schaefer et al., 2020 [101] | OS | 7 | 50 to 77 (Median 66) Male 16 Female 23 | Lung 7 | 5 cases diffuse DAD; 2 cases alveolar injuries. SARS-CoV-2 infection involving epithelial lung cell in acute phase No endothelial cell infection |
| Varga et al., 2020 [102] | OS | 3 | 58 to 61 years (Median 63) | Kidney 2 Lung 2 Heart 1 Liver 1 Intestin 2 | Lymphocytic endotheliitis in lung, heart, kidney, and liver. Apoptotic bodies in the heart Mononuclear cells in lung |
| Delorey et al., 2021 [103] | OS | 32 | 30 to 89 years Male 20 Female 12 | Kidney 16 Lung 24 Heart 19 Liver 16 | Higher viral RNAs in phagocytic mononuclear and endothelial lung cells. Transcriptional alterations in multiple cell types in the heart tissue. |
| Lindner et al., 2020 [104] | Prospective | 39 | 78 to 89 years (Median 68) Male 16 Female 23 | Heart 39 | SARS-CoV-2 infects directly the myocardium Absence of inflammatory cell infiltrates in patient with SARS-CoV-2 infection. Higher cytokine response |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nappi, F.; Avtaar Singh, S.S. Endothelial Dysfunction in SARS-CoV-2 Infection. Biomedicines 2022, 10, 654. https://doi.org/10.3390/biomedicines10030654
Nappi F, Avtaar Singh SS. Endothelial Dysfunction in SARS-CoV-2 Infection. Biomedicines. 2022; 10(3):654. https://doi.org/10.3390/biomedicines10030654
Chicago/Turabian StyleNappi, Francesco, and Sanjeet Singh Avtaar Singh. 2022. "Endothelial Dysfunction in SARS-CoV-2 Infection" Biomedicines 10, no. 3: 654. https://doi.org/10.3390/biomedicines10030654
APA StyleNappi, F., & Avtaar Singh, S. S. (2022). Endothelial Dysfunction in SARS-CoV-2 Infection. Biomedicines, 10(3), 654. https://doi.org/10.3390/biomedicines10030654