
ER-phagy in the Occurrence and Development of Cancer

{kind=link}
{kind=link}
Abstract
:1. Background
2. ER-phagy-Related Receptors and Their Protumor or Antitumor Role
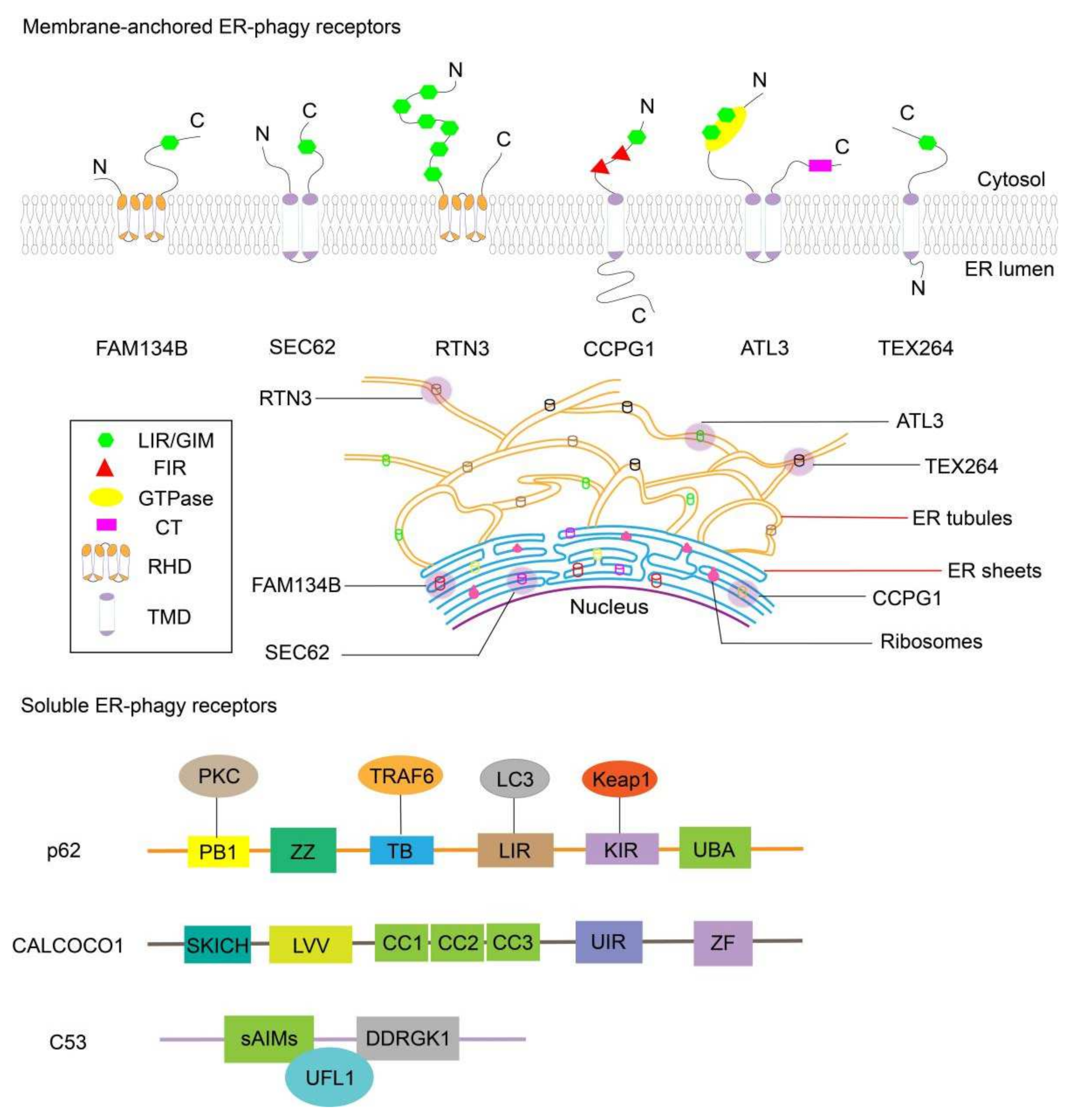
2.1. FAM134B
2.2. SEC62
2.3. RTN3
2.4. CCPG1
2.5. ATL3
2.6. TEX264
2.7. CALCOCO1
2.8. C53
2.9. SQSTM1/p62
3. The Procancer or Anticancer Roles and Molecular Pathways of ER-phagy
3.1. The Procancer Role of ER-phagy
3.1.1. ER-phagy Is Advantageous to the Invasion and Metastasis of Tumor Cells
3.1.2. ER-phagy Mediates Drug Resistance in Tumor Cells
3.1.3. ER-phagy Facilitates Cancer Cell Survival by Promoting Angiogenesis
3.1.4. ER-phagy Protects Tumor Cells by Immunosuppression
3.2. The Role of ER-phagy in Inhibiting Tumorigenesis
3.2.1. Excessive ER-phagy Contributes to Cancer Cell Death
3.2.2. ER-phagy Inhibits Tumor Cell Migration
3.2.3. ER-phagy Adversely Affects the Stemness of Tumor Cells
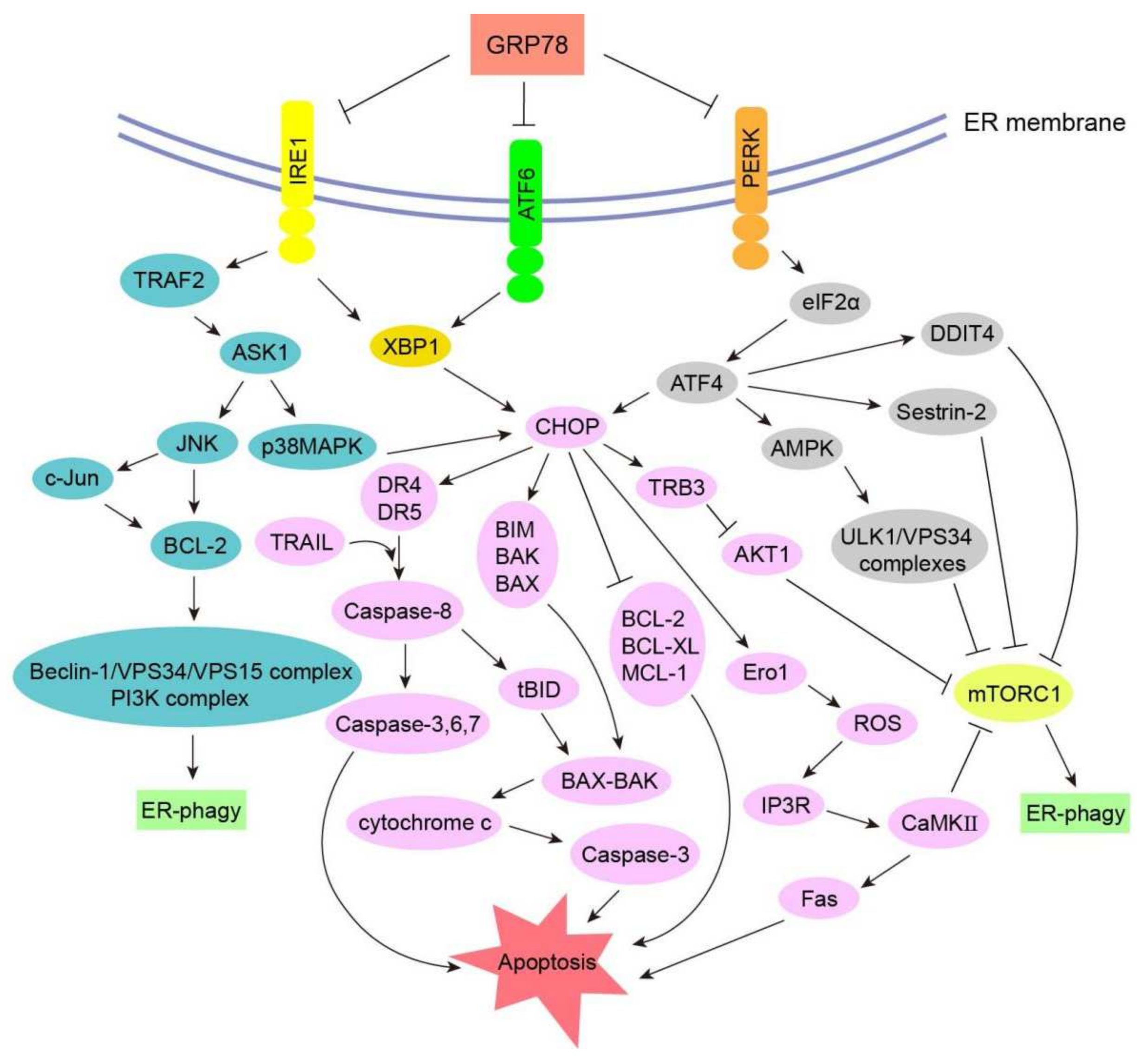
3.3. Signal Transduction Pathways of ER-phagy in Cancers
3.3.1. CHOP–BCL-2 Protein Family
3.3.2. Caspase Signaling Pathway
3.3.3. Ero1 Signaling Pathway
3.3.4. GADD34 Signaling Pathway
3.3.5. ASK1-p38 MAPK/ASK1-JNK Signaling Pathway
4. ER-phagy as a Potential Target for Cancer Therapy
4.1. Activating ER-phagy to Treat Cancer
4.2. Inhibition of ER-phagy to Treat Cancer
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ER | Endoplasmic reticulum |
| HSC70 | Heat shock cognate protein of 70 kDa |
| LAMP2A | Lysosome-associated membrane protein type 2A |
| FKB | Flavokawain B |
| MAP1LC3 | Microtubule-associated protein light chain 3 |
| GABARAP | Gamma-aminobutyric acid receptor-associated protein |
| FAM134B | Family with sequence similarity 134 member B |
| SEC62 | Translocation protein SEC62 |
| RTN3 | Reticulon 3 |
| CCPG1 | Cell-cycle progression gene 1 |
| ATL3 | Atlastin GTPase 3 |
| TEX264 | Testis-expressed protein 264 |
| p62/SQSTM1 | Sequestosome 1 |
| RHD | Reticulon-homology domain |
| LIR | LC3-interacting region |
| HCC | Hepatocellular carcinoma |
| Chk2 | Checkpoint kinase 2 |
| FIR | FIP200-interacting region |
| ATL | Atlastin |
| 3HB | Three-helix bundle |
| CT | C-terminal tail |
| GIMs | GABARAP-interacting motifs |
| GyrI | Gyrase inhibitor |
| CALCOCO1 | Calcium-binding and coiled-coil domain-containing protein 1 |
| C53 | CDK5 regulatory subunit-associated protein 3 |
| UFL1 | Ubiquitin-fold modifier 1-specific ligase 1 |
| DDRGK1 | DDRGK domain-containing protein 1 |
| PB1 | Phox-BEM1 |
| PKC | Protein kinase C |
| TB | TRAF6-binding |
| KIR | Keap1-binding region |
| TRIM13 | Tripartite motif containing 13 |
| IRE1 | Inositol-requiring enzyme 1 |
| ATF6 | Activating transcription factor 6 |
| PERK | Protein kinase R-like ER kinase |
| GRP78/BiP | 78 kDa glucose-regulated protein/binding immunoglobulin protein |
| LD | Luminal domain |
| SBD | Substrate binding domain |
| RNase | Endoribonuclease |
| XBP1s | Spliced X-box binding protein 1 |
| TNF | Tumor necrosis factor |
| TRAF2 | TNF receptor-associated factor 2 |
| ASK1 | Apoptosis signal-regulating kinase 1 |
| JNK | JUN N-terminal kinase |
| BCL-2 | B cell lymphoma 2 |
| PI3K | Phosphatidylinositol 3-kinase |
| ERp72 | ER protein 72 |
| eIF2α | Eukaryotic initiation factor 2α |
| ATF4 | Activating transcription factor 4 |
| CHOP | C/EBP-homologous protein |
| AMPK | Adenosine monophosphate-activated protein kinase |
| TRB3 | Tribbles homologue 3 |
| mTORC1 | Mammalian target of rapamycin complex 1 |
| DDIT4 | DNA damage-inducible transcript 4 |
| ULK1 | Uncoordinated-51-like kinase 1 |
| AMBRA1 | Activating molecule in Beclin-1-regulated autophagy protein 1 |
| PI3P | Phosphatidyl-inositol-3-phosphate |
| PI | Phosphatidylinositol |
| WIPIs | WD repeat domain phosphoinositide-interacting proteins |
| PE | Phosphatidylethanolamine |
| LAMP3 | Lysosomal-associated membrane protein 3 |
| VEGF | Vascular endothelial growth factor |
| TRAIL | TNF-related apoptosis-inducing ligand |
| Ero1 | ER oxidoreductin 1 |
| CaMK | Calcium-/calmodulin-dependent protein kinase |
| GADD34 | Growth arrest and DNA damage-inducible protein 34 |
| PP1 | Phosphatase 1 |
| p38MAPK | p38 mitogen-activated protein kinase |
| CSC | Cancer stem cells |
| DSF | Disulfiram |
| CQ | Chloroquine |
| HCQ | Hydroxychloroquine |
| EGCG | Epigallocatechin gallate |
| Atg5 | Autophagy related protein 5 |
| FIP200 | Focal adhesion kinase family interacting protein of 200 kDa |
| C/EBPβ | CCAAT/enhancer-binding protein beta |
| EB1 | End-binding protein 1 |
| ESCRT-III | Endosomal sorting complex required for transport-III |
| UPR | Unfolded protein response |
| VEGFA | Vascular endothelial growth factor A |
| ROS | Reactive oxygen species |
| MHC | Major histocompatibility complex |
| LGR5 | Leucine-rich repeat-containing G protein-coupled receptor 5 |
| OLFM4 | Olfactomedin 4 |
| BCL-XL | B cell lymphoma-extra large |
| MCL-1 | Myeloid cell leukemia-1 |
| BAK | BCL-2 antagonist killer |
| BAX | BCL-2-associated X protein |
| DR4 | Death receptor 4 |
| DR5 | Death receptor 5 |
| BID | BH3-interacting domain death agonist |
References
- Jiang, P.; Mizushima, N. Autophagy and human diseases. Cell Res. 2014, 24, 69–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, K.H.; Lee, M.S. Autophagy—A key player in cellular and body metabolism. Nat. Rev. Endocrinol. 2014, 10, 322–337. [Google Scholar] [CrossRef] [PubMed]
- Yoshii, S.R.; Mizushima, N. Monitoring and Measuring Autophagy. Int. J. Mol. Sci. 2017, 18, 1865. [Google Scholar] [CrossRef] [PubMed]
- Mehrbod, P.; Ande, S.R.; Alizadeh, J. The roles of apoptosis, autophagy and unfolded protein response in arbovirus, influenza virus, and HIV infections. Virulence 2019, 10, 376–413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaushik, S.; Cuervo, A.M. The coming of age of chaperone-mediated autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 365–381. [Google Scholar] [CrossRef] [PubMed]
- Parzych, K.R.; Klionsky, D.J. An overview of autophagy Morphology, mechanism, and regulation. Antioxid. Redox. Signal 2014, 20, 460–473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, R.; Pattison, J.S. Macroautophagy and Chaperone-Mediated Autophagy in Heart Failure The Known and the Unknown. Oxidative Med. Cell. Longev. 2018, 2018, 8602041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolender, R.P.; Weibel, E.R. A morphometric study of the removal of phenobarbital-induced membranes from hepatocytes after cessation of threatment. J. Cell Biol. 1973, 56, 746–761. [Google Scholar] [CrossRef]
- Bernales, S.; McDonald, K.L.; Walter, P. Autophagy counterbalances endoplasmic reticulum expansion during the unfolded protein response. PLoS Biol. 2006, 4, e423. [Google Scholar] [CrossRef] [Green Version]
- Wilkinson, S. ER-phagy Shaping up and destressing the endoplasmic reticulum. FEBS J. 2019, 286, 2645–2663. [Google Scholar] [CrossRef] [Green Version]
- Molinari, M. ER-phagy responses in yeast, plants, and mammalian cells and their crosstalk with UPR and ERAD. Dev. Cell 2021, 56, 949–966. [Google Scholar] [CrossRef]
- Eldeeb, M.A.; Zorca, C.E.; Ragheb, M.A.; Rashidi, F.B.; Salah El-Din, D.S. Fine-tuning ER-phagy by post-translational modifications. BioEssays News Rev. Mol. Cell. Dev. Biol. 2021, 43, e2000212. [Google Scholar] [CrossRef]
- Mowers, E.E.; Sharifi, M.N.; Macleod, K.F. Functions of autophagy in the tumor microenvironment and cancer metastasis. FEBS J. 2018, 285, 1751–1766. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.J.; Lei, Y.H.; Yao, N.; Wang, C.R.; Hu, N.; Ye, W.C.; Zhang, D.M.; Chen, Z.S. Autophagy and multidrug resistance in cancer. Chin. J. Cancer 2017, 36, 52. [Google Scholar] [CrossRef]
- Levine, B.; Kroemer, G. Biological Functions of Autophagy Genes A Disease Perspective. Cell 2019, 176, 11–42. [Google Scholar] [CrossRef] [Green Version]
- Antunes, F.; Erustes, A.G.; Costa, A.J.; Nascimento, A.C.; Bincoletto, C.; Ureshino, R.P.; Pereira, G.J.S.; Smaili, S.S. Autophagy and intermittent fasting The connection for cancer therapy? Clinics 2018, 73, e814s. [Google Scholar] [CrossRef]
- Huang, T.; Song, X.; Yang, Y.; Wan, X.; Alvarez, A.A.; Sastry, N.; Feng, H.; Hu, B.; Cheng, S.Y. Autophagy and Hallmarks of Cancer. Crit. Rev. Oncog. 2018, 23, 247–267. [Google Scholar] [CrossRef]
- Amaravadi, R.; Kimmelman, A.C.; White, E. Recent insights into the function of autophagy in cancer. Genes Dev. 2016, 30, 1913–1930. [Google Scholar] [CrossRef]
- Mizushima, N. A Dual Binding Receptor for ER-phagy. Dev. Cell 2018, 44, 133–135. [Google Scholar] [CrossRef] [Green Version]
- Smith, M.; Wilkinson, S. ER homeostasis and autophagy. Essays Biochem. 2017, 61, 625–635. [Google Scholar] [CrossRef] [Green Version]
- D’Eletto, M.; Oliverio, S.; Di Sano, F. Reticulon Homology Domain-Containing Proteins and ER-Phagy. Front. Cell Dev. Biol. 2020, 8, 90. [Google Scholar] [CrossRef] [PubMed]
- Khaminets, A.; Heinrich, T.; Mari, M.; Grumati, P.; Huebner, A.K.; Akutsu, M.; Liebmann, L.; Stolz, A.; Nietzsche, S.; Koch, N.; et al. Regulation of endoplasmic reticulum turnover by selective autophagy. Nature 2015, 522, 354–358. [Google Scholar] [CrossRef] [PubMed]
- Loi, M.; Fregno, I.; Guerra, C.; Molinari, M. Eat it right ER-phagy and recovER-phagy. Biochem. Soc. Trans. 2018, 46, 699–706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhaskara, R.M.; Grumati, P.; Garcia-Pardo, J.; Kalayil, S.; Covarrubias-Pinto, A.; Chen, W.; Kudryashev, M.; Dikic, I.; Hummer, G. Curvature induction and membrane remodeling by FAM134B reticulon homology domain assist selective ER-phagy. Nat. Commun. 2019, 10, 2370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohno, S.; Shiozaki, Y.; Keenan, A.L.; Miyazaki-Anzai, S.; Miyazaki, M. An N-terminal-truncated isoform of FAM134B (FAM134B-2) regulates starvation-induced hepatic selective ER-phagy. Life Sci. Alliance 2019, 2, e201900340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Islam, F.; Chaousis, S.; Wahab, R.; Gopalan, V.; Lam, A.K. Protein interactions of FAM134B with EB1 and APC/beta-catenin in vitro in colon carcinoma. Mol. Carcinog. 2018, 57, 1480–1491. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.Q.; Chen, J.; Huang, W.Q.; Ning, D.; Liu, Q.M.; Wang, C.; Zhang, L.; Ren, L.; Chu, L.; Liang, H.F.; et al. FAM134B induces tumorigenesis and epithelial-to-mesenchymal transition via Akt signaling in hepatocellular carcinoma. Mol. Oncol. 2019, 13, 792–810. [Google Scholar] [CrossRef] [Green Version]
- Mo, J.; Chen, J.; Zhang, B. Critical roles of FAM134B in ER-phagy and diseases. Cell Death Dis. 2020, 11, 983. [Google Scholar] [CrossRef] [PubMed]
- Linxweiler, M.; Schick, B.; Zimmermann, R. Let’s talk about Secs Sec61, Sec62 and Sec63 in signal transduction, oncology and personalized medicine. Signal Transduct. Target Ther. 2017, 2, 17002. [Google Scholar] [CrossRef] [PubMed]
- Fumagalli, F.; Noack, J.; Bergmann, T.J.; Cebollero, E.; Pisoni, G.B.; Fasana, E.; Fregno, I.; Galli, C.; Loi, M.; Solda, T.; et al. Translocon component Sec62 acts in endoplasmic reticulum turnover during stress recovery. Nat. Cell Biol. 2016, 18, 1173–1184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loi, M.; Raimondi, A.; Morone, D.; Molinari, M. ESCRT-III-driven piecemeal micro-ER-phagy remodels the ER during recovery from ER stress. Nat. Commun. 2019, 10, 5058. [Google Scholar] [CrossRef] [Green Version]
- Loi, M.; Molinari, M. Mechanistic insights in recov-ER-phagy Micro-ER-phagy to recover from stress. Autophagy 2020, 16, 385–386. [Google Scholar] [CrossRef]
- Bergmann, T.J.; Fumagalli, F.; Loi, M.; Molinari, M. Role of SEC62 in ER maintenance A link with ER stress tolerance in SEC62-overexpressing tumors? Mol. Cell Oncol. 2017, 4, e1264351. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Su, K.; Sun, X.; Jiang, Y.; Wang, L.; Hu, C.; Zhang, C.; Lu, M.; Du, X.; Xing, B. Sec62 promotes stemness and chemoresistance of human colorectal cancer through activating Wnt/β-catenin pathway. J. Exp. Clin. Cancer Res. 2021, 40, 132. [Google Scholar] [CrossRef]
- Meng, Y.; Zhao, H.; Zhao, Z.; Yin, Z.; Chen, Z.; Du, J. Sec62 promotes pro-angiogenesis of hepatocellular carcinoma cells under hypoxia. Cell Biochem. Biophys. 2021, 79, 747–755. [Google Scholar] [CrossRef]
- Grumati, P.; Morozzi, G.; Holper, S.; Mari, M.; Harwardt, M.I.; Yan, R.; Muller, S.; Reggiori, F.; Heilemann, M.; Dikic, I. Full length RTN3 regulates turnover of tubular endoplasmic reticulum via selective autophagy. eLife 2017, 6, e25555. [Google Scholar] [CrossRef]
- Grumati, P.; Dikic, I.; Stolz, A. ER-phagy at a glance. J. Cell Sci. 2018, 131, jcs217364. [Google Scholar] [CrossRef] [Green Version]
- Fregno, I.; Molinari, M. Endoplasmic reticulum turnover ER-phagy and other flavors in selective and non-selective ER clearance. F1000Research 2018, 7, 454. [Google Scholar] [CrossRef]
- D’Eletto, M.; Risuglia, A.; Oliverio, S.; Mehdawy, B.; Nardacci, R.; Bordi, M.; Di Sano, F. Modulation of autophagy by RTN-1C Role in autophagosome biogenesis. Cell Death Dis. 2019, 10, 868. [Google Scholar] [CrossRef]
- Song, S.; Shi, Y.; Wu, W.; Wu, H.; Chang, L.; Peng, P.; Zhang, L.; Fan, J.; Gu, J.; Ruan, Y. Reticulon 3-mediated Chk2/p53 activation suppresses hepatocellular carcinogenesis and is blocked by hepatitis B virus. Gut 2021, 70, 2159–2171. [Google Scholar] [CrossRef]
- Smith, M.D.; Harley, M.E.; Kemp, A.J.; Wills, J.; Lee, M.; Arends, M.; von Kriegsheim, A.; Behrends, C.; Wilkinson, S. CCPG1 Is a Non-canonical Autophagy Cargo Receptor Essential for ER-Phagy and Pancreatic ER Proteostasis. Dev. Cell 2018, 44, 217–232.e211. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Wei, N.; Wang, L.; Wang, X.; Liu, Q.H. miR-498 promotes cell proliferation and inhibits cell apoptosis in retinoblastoma by directly targeting CCPG1. Childs Nerv. Syst. 2018, 34, 417–422. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.R.; Lingeman, E.; Ahmed, S.; Corn, J.E. Atlastins remodel the endoplasmic reticulum for selective autophagy. J. Cell Biol. 2018, 217, 3354–3367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Q.; Xiao, Y.; Chai, P.; Zheng, P.; Teng, J.; Chen, J. ATL3 Is a Tubular ER-Phagy Receptor for GABARAP-Mediated Selective Autophagy. Curr. Biol. 2019, 29, 846–855.e846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, X.; Wu, F.; Sun, S.; Yu, W.; Hu, J. Human atlastin GTPases mediate differentiated fusion of endoplasmic reticulum membranes. Protein Cell 2015, 6, 307–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Q.; Teng, J. ATL3, a cargo receptor for reticulophagy. Autophagy 2019, 15, 1465–1466. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, S. Emerging Principles of Selective ER Autophagy. J. Mol. Biol. 2020, 432, 185–205. [Google Scholar] [CrossRef] [PubMed]
- An, H.; Ordureau, A.; Paulo, J.A.; Shoemaker, C.J.; Denic, V.; Harper, J.W. TEX264 Is an Endoplasmic Reticulum-Resident ATG8-Interacting Protein Critical for ER Remodeling during Nutrient Stress. Mol. Cell 2019, 74, 891–908.e810. [Google Scholar] [CrossRef] [PubMed]
- Chino, H.; Hatta, T.; Natsume, T.; Mizushima, N. Intrinsically Disordered Protein TEX264 Mediates ER-phagy. Mol. Cell 2019, 74, 909–921.e906. [Google Scholar] [CrossRef]
- Nthiga, T.M.; Shrestha, B.K.; Lamark, T.; Johansen, T. CALCOCO1 is a soluble reticulophagy receptor. Autophagy 2020, 16, 1729–1731. [Google Scholar] [CrossRef] [PubMed]
- Nthiga, T.M.; Kumar Shrestha, B.; Sjøttem, E.; Bruun, J.A.; Bowitz Larsen, K.; Bhujabal, Z.; Lamark, T.; Johansen, T. CALCOCO1 acts with VAMP-associated proteins to mediate ER-phagy. EMBO J. 2020, 39, e103649. [Google Scholar] [CrossRef]
- He, L.; Qian, X.; Cui, Y. Advances in ER-Phagy and Its Diseases Relevance. Cells 2021, 10, 2328. [Google Scholar] [CrossRef]
- Yu, G.; Xiong, D.; Liu, Z.; Li, Y.; Chen, K.; Tang, H. Long noncoding RNA LINC00052 inhibits colorectal cancer metastasis by sponging microRNA-574-5p to modulate CALCOCO1 expression. J. Cell. Biochem. 2019, 120, 17258–17272. [Google Scholar] [CrossRef]
- Stephani, M.; Picchianti, L.; Gajic, A.; Beveridge, R.; Skarwan, E.; Sanchez de Medina Hernandez, V.; Mohseni, A.; Clavel, M.; Zeng, Y.; Naumann, C.; et al. A cross-kingdom conserved ER-phagy receptor maintains endoplasmic reticulum homeostasis during stress. eLife 2020, 9, e58396. [Google Scholar] [CrossRef]
- Stephani, M.; Picchianti, L.; Dagdas, Y. C53 is a cross-kingdom conserved reticulophagy receptor that bridges the gap betweenselective autophagy and ribosome stalling at the endoplasmic reticulum. Autophagy 2021, 17, 586–587. [Google Scholar] [CrossRef]
- Mak, G.W.; Chan, M.M.; Leong, V.Y.; Lee, J.M.; Yau, T.O.; Ng, I.O.; Ching, Y.P. Overexpression of a novel activator of PAK4, the CDK5 kinase-associated protein CDK5RAP3, promotes hepatocellular carcinoma metastasis. Cancer Res. 2011, 71, 2949–2958. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.J.; Ye, L.; Huang, W.F.; Guo, L.J.; Xu, Z.G.; Wu, H.L.; Yang, C.; Liu, H.F. p62 links the autophagy pathway and the ubiqutin-proteasome system upon ubiquitinated protein degradation. Cell. Mol. Biol. Lett. 2016, 21, 29. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Mun, S.R.; Linares, J.F.; Ahn, J.; Towers, C.G.; Ji, C.H.; Fitzwalter, B.E.; Holden, M.R.; Mi, W.; Shi, X.; et al. ZZ-dependent regulation of p62/SQSTM1 in autophagy. Nat. Commun. 2018, 9, 4373. [Google Scholar] [CrossRef] [Green Version]
- Ji, C.H.; Kim, H.Y.; Heo, A.J.; Lee, S.H.; Lee, M.J.; Kim, S.B.; Srinivasrao, G.; Mun, S.R.; Cha-Molstad, H.; Ciechanover, A.; et al. The N-Degron Pathway Mediates ER-phagy. Mol. Cell 2019, 75, 1058–1072.e1059. [Google Scholar] [CrossRef]
- Islam, M.A.; Sooro, M.A.; Zhang, P. Autophagic Regulation of p62 is Critical for Cancer Therapy. Int. J. Mol. Sci. 2018, 19, 1405. [Google Scholar] [CrossRef] [Green Version]
- Sano, R.; Reed, J.C. ER stress-induced cell death mechanisms. Biochim. Biophys. Acta 2013, 1833, 3460–3470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siwecka, N.; Rozpędek, W.; Pytel, D.; Wawrzynkiewicz, A.; Dziki, A.; Dziki, Ł.; Diehl, J.A.; Majsterek, I. Dual role of Endoplasmic Reticulum Stress-Mediated Unfolded Protein Response Signaling Pathway in Carcinogenesis. Int. J. Mol. Sci. 2019, 20, 4354. [Google Scholar] [CrossRef] [Green Version]
- Kimmelman, A.C.; White, E. Autophagy and Tumor Metabolism. Cell Metab. 2017, 25, 1037–1043. [Google Scholar] [CrossRef] [PubMed]
- Papaioannou, A.; Chevet, E. Driving cancer tumorigenesis and metastasis through UPR signaling. In Coordinating Organismal Physiology Through the Unfolded Protein Response; Wiseman, R.L., Haynes, C.M., Eds.; Springer International Publishing: Cham, Switzerland, 2018; pp. 159–192. [Google Scholar]
- Mujcic, H.; Nagelkerke, A.; Rouschop, K.M.; Chung, S.; Chaudary, N.; Span, P.N.; Clarke, B.; Milosevic, M.; Sykes, J.; Hill, R.P.; et al. Hypoxic activation of the PERK/eIF2α arm of the unfolded protein response promotes metastasis through induction of LAMP3. Clin. Cancer Res. 2013, 19, 6126–6137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jamison, S.; Lin, Y.; Lin, W. Pancreatic endoplasmic reticulum kinase activation promotes medulloblastoma cell migration and invasion through induction of vascular endothelial growth factor A. PLoS ONE 2015, 10, e0120252. [Google Scholar] [CrossRef] [PubMed]
- Linxweiler, M.; Linxweiler, J.; Barth, M.; Benedix, J.; Jung, V.; Kim, Y.J.; Bohle, R.M.; Zimmermann, R.; Greiner, M. Sec62 bridges the gap from 3q amplification to molecular cell biology in non-small cell lung cancer. Am. J. Pathol. 2012, 180, 473–483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dauer, P.; Sharma, N.S.; Gupta, V.K.; Durden, B.; Hadad, R.; Banerjee, S.; Dudeja, V.; Saluja, A.; Banerjee, S. ER stress sensor, glucose regulatory protein 78 (GRP78) regulates redox status in pancreatic cancer thereby maintaining “stemness”. Cell Death Dis. 2019, 10, 132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madden, E.; Logue, S.E.; Healy, S.J.; Manie, S.; Samali, A. The role of the unfolded protein response in cancer progression from oncogenesis to chemoresistance. Biol. Cell 2019, 111, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Logue, S.E.; McGrath, E.P.; Cleary, P.; Greene, S.; Mnich, K. Inhibition of IRE1 RNase activity modulates the tumor cell secretome and enhances response to chemotherapy. Nat. Commun. 2018, 9, 3267. [Google Scholar] [CrossRef] [PubMed]
- Urra, H.; Dufey, E.; Avril, T.; Chevet, E.; Hetz, C. Endoplasmic Reticulum Stress and the Hallmarks of Cancer. Trends Cancer 2016, 2, 252–262. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Alam, G.N.; Ning, Y.; Visioli, F.; Dong, Z.; Nör, J.E.; Polverini, P.J. The unfolded protein response induces the angiogenic switch in human tumor cells through the PERK/ATF4 pathway. Cancer Res. 2012, 72, 5396–5406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walczak, A.; Gradzik, K.; Kabzinski, J. The Role of the ER-Induced UPR Pathway and the Efficacy of Its Inhibitors and Inducers in the Inhibition of Tumor Progression. Oxidative Med. Cell. Longev. 2019, 2019, 5729710. [Google Scholar] [CrossRef] [Green Version]
- Ghavimi, S.; Apfel, T.; Azimi, H.; Persaud, A.; Pyrsopoulos, N.T. Management and Treatment of Hepatocellular Carcinoma with Immunotherapy A Review of Current and Future Options. J. Clin. Transl. Hepatol. 2020, 8, 168–176. [Google Scholar] [CrossRef] [PubMed]
- Cubillos-Ruiz, J.R.; Bettigole, S.E.; Glimcher, L.H. Tumorigenic and Immunosuppressive Effects of Endoplasmic Reticulum Stress in Cancer. Cell 2017, 168, 692–706. [Google Scholar] [CrossRef] [Green Version]
- Hsu, S.K.; Chiu, C.C. Unfolded Protein Response (UPR) in Survival, Dormancy, Immunosuppression, Metastasis, and Treatments of Cancer Cells. Int. J. Mol. Sci. 2019, 20, 2518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.; Kaufman, R.J. The impact of the endoplasmic reticulum protein-folding environment on cancer development. Nat. Rev. Cancer 2014, 14, 581–597. [Google Scholar] [CrossRef]
- Cybulsky, A.V. Endoplasmic reticulum stress, the unfolded protein response and autophagy in kidney diseases. Nat. Rev. Nephrol. 2017, 13, 681–696. [Google Scholar] [CrossRef]
- Kroemer, G.; Marino, G.; Levine, B. Autophagy and the integrated stress response. Mol. Cell 2010, 40, 280–293. [Google Scholar] [CrossRef] [Green Version]
- Holczer, M.; Marton, M.; Kurucz, A.; Banhegyi, G.; Kapuy, O. A Comprehensive Systems Biological Study of Autophagy-Apoptosis Crosstalk during Endoplasmic Reticulum Stress. Biomed. Res. Int. 2015, 2015, 319589. [Google Scholar] [CrossRef] [Green Version]
- Galluzzi, L.; Aaronson, S.A.; Abrams, J.; Alnemri, E.S.; Andrews, D.W.; Baehrecke, E.H.; Bazan, N.G.; Blagosklonny, M.V.; Blomgren, K.; Borner, C.; et al. Guidelines for the use and interpretation of assays for monitoring cell death in higher eukaryotes. Cell Death Differ. 2009, 16, 1093–1107. [Google Scholar] [CrossRef] [Green Version]
- Kasem, K.; Gopalan, V.; Salajegheh, A.; Lu, C.-T.; Smith, R.A.; Lam, A.K.Y. The roles of JK-1 (FAM134B) expressions in colorectal cancer. Exp. Cell Res. 2014, 326, 166–173. [Google Scholar] [CrossRef]
- Islam, F.; Gopalan, V.; Lam, A.K.-Y. RETREG1 (FAM134B) A new player in human diseases 15 years after the discovery in cancer. J. Cell. Physiol. 2018, 233, 4479–4489. [Google Scholar] [CrossRef] [Green Version]
- Kasem, K.; Sullivan, E.; Gopalan, V.; Salajegheh, A.; Smith, R.A.; Lam, A.K.Y. JK1 (FAM134B) represses cell migration in colon cancer A functional study of a novel gene. Exp. Mol. Pathol. 2014, 97, 99–104. [Google Scholar] [CrossRef]
- Saygin, C.; Matei, D.; Majeti, R.; Reizes, O.; Lathia, J.D. Targeting Cancer Stemness in the Clinic from Hype to Hope. Cell Stem Cell 2019, 24, 25–40. [Google Scholar] [CrossRef] [Green Version]
- Prasad, S.; Ramachandran, S.; Gupta, N.; Kaushik, I.; Srivastava, S.K. Cancer cells stemness A doorstep to targeted therapy. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2020, 1866, 165424. [Google Scholar] [CrossRef]
- Spaan, C.N.; Smit, W.L.; van Lidth de Jeude, J.F.; Meijer, B.J.; Muncan, V.; van den Brink, G.R.; Heijmans, J. Expression of UPR effector proteins ATF6 and XBP1 reduce colorectal cancer cell proliferation and stemness by activating PERK signaling. Cell Death Dis. 2019, 10, 490. [Google Scholar] [CrossRef]
- Klymenko, O.; Huehn, M.; Wilhelm, J.; Wasnick, R.; Shalashova, I.; Ruppert, C.; Henneke, I.; Hezel, S.; Guenther, K.; Mahavadi, P.; et al. Regulation and role of the ER stress transcription factor CHOP in alveolar epithelial type-II cells. J. Mol. Med. 2019, 97, 973–990. [Google Scholar] [CrossRef] [Green Version]
- Rozpedek, W.; Pytel, D.; Mucha, B.; Leszczynska, H.; Diehl, J.A.; Majsterek, I. The Role of the PERK/eIF2alpha/ATF4/CHOP Signaling Pathway in Tumor Progression During Endoplasmic Reticulum Stress. Curr. Mol. Med. 2016, 16, 533–544. [Google Scholar] [CrossRef]
- Tuzlak, S.; Kaufmann, T.; Villunger, A. Interrogating the relevance of mitochondrial apoptosis for vertebrate development and postnatal tissue homeostasis. Genes Dev. 2016, 30, 2133–2151. [Google Scholar] [CrossRef]
- Yuan, X.; Gajan, A.; Chu, Q.; Xiong, H.; Wu, K.; Wu, G.S. Developing TRAIL/TRAIL death receptor-based cancer therapies. Cancer Metastasis Rev. 2018, 37, 733–748. [Google Scholar] [CrossRef]
- Hu, H.; Tian, M.; Ding, C.; Yu, S. The C/EBP Homologous Protein (CHOP) Transcription Factor Functions in Endoplasmic Reticulum Stress-Induced Apoptosis and Microbial Infection. Front. Immunol. 2018, 9, 3083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Debernardi, J.; Hollville, E.; Lipinski, M.; Wiels, J.; Robert, A. Differential role of FL-BID and t-BID during verotoxin-1-induced apoptosis in Burkitt’s lymphoma cells. Oncogene 2018, 37, 2410–2421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Upton, J.P.; Austgen, K.; Nishino, M.; Coakley, K.M.; Hagen, A.; Han, D.; Papa, F.R.; Oakes, S.A. Caspase-2 cleavage of BID is a critical apoptotic signal downstream of endoplasmic reticulum stress. Mol. Cell. Biol. 2008, 28, 3943–3951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oakes, S.A.; Papa, F.R. The role of endoplasmic reticulum stress in human pathology. Annu. Rev. Pathol. 2015, 10, 173–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Brandizzi, F. IRE1 ER stress sensor and cell fate executor. Trends Cell Biol. 2013, 23, 547–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moilanen, A.; Korhonen, K.; Saaranen, M.J.; Ruddock, L.W. Molecular analysis of human Ero1 reveals novel regulatory mechanisms for oxidative protein folding. Life Sci. Alliance 2018, 1, e201800090. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Xie, Y.; Yang, D.; Ren, D. Oxidative stress-induced apoptosis in granulosa cells involves JNK, p53 and Puma. Oncotarget 2017, 8, 25310–25322. [Google Scholar] [CrossRef] [Green Version]
- Timmins, J.M.; Ozcan, L.; Seimon, T.A.; Li, G.; Malagelada, C.; Backs, J.; Backs, T.; Bassel-Duby, R.; Olson, E.N.; Anderson, M.E.; et al. Calcium/calmodulin-dependent protein kinase II links ER stress with Fas and mitochondrial apoptosis pathways. J. Clin. Investig. 2009, 119, 2925–2941. [Google Scholar] [CrossRef] [Green Version]
- Rojas, M.; Vasconcelos, G.; Dever, T.E. An eIF2alpha-binding motif in protein phosphatase 1 subunit GADD34 and its viral orthologs is required to promote dephosphorylation of eIF2alpha. Proc. Natl. Acad. Sci. USA 2015, 112, E3466–E3475. [Google Scholar] [CrossRef] [Green Version]
- Han, J.; Back, S.H.; Hur, J.; Lin, Y.H.; Gildersleeve, R.; Shan, J.; Yuan, C.L.; Krokowski, D.; Wang, S.; Hatzoglou, M.; et al. ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat. Cell Biol. 2013, 15, 481–490. [Google Scholar] [CrossRef]
- Sisinni, L.; Pietrafesa, M.; Lepore, S.; Maddalena, F.; Condelli, V.; Esposito, F.; Landriscina, M. Endoplasmic Reticulum Stress and Unfolded Protein Response in Breast Cancer: The Balance between Apoptosis and Autophagy and Its Role in Drug Resistance. Int. J. Mol. Sci. 2019, 20, 857. [Google Scholar] [CrossRef] [Green Version]
- Puthalakath, H.; O’Reilly, L.A.; Gunn, P.; Lee, L.; Kelly, P.N.; Huntington, N.D.; Hughes, P.D.; Michalak, E.M.; McKimm-Breschkin, J.; Motoyama, N.; et al. ER stress triggers apoptosis by activating BH3-only protein Bim. Cell 2007, 129, 1337–1349. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.Y.; Li, Y.; Jiang, W.Q.; Zhou, L.F. MAPK/JNK signalling A potential autophagy regulation pathway. Biosci. Rep. 2015, 35, e00199. [Google Scholar] [CrossRef]
- Ojha, R.; Amaravadi, R.K. Targeting the unfolded protein response in cancer. Pharmacol. Res. 2017, 120, 258–266. [Google Scholar] [CrossRef]
- Zhang, X.; Hu, P.; Ding, S.Y.; Sun, T.; Liu, L.; Han, S.; DeLeo, A.B.; Sadagopan, A.; Guo, W.; Wang, X. Induction of autophagy-dependent apoptosis in cancer cells through activation of ER stress: An uncovered anti-cancer mechanism by anti-alcoholism drug disulfiram. Am. J. Cancer Res. 2019, 9, 1266–1281. [Google Scholar]
- Mossmann, D.; Park, S.; Hall, M.N. mTOR signalling and cellular metabolism are mutual determinants in cancer. Nat. Rev. Cancer 2018, 18, 744–757. [Google Scholar] [CrossRef]
- Schönthal, A.H. Pharmacological targeting of endoplasmic reticulum stress signaling in cancer. Biochem. Pharmacol. 2013, 85, 653–666. [Google Scholar] [CrossRef]
- Kim, C.; Kim, B. Anti-Cancer Natural Products and Their Bioactive Compounds Inducing ER Stress-Mediated Apoptosis A Review. Nutrients 2018, 10, 1021. [Google Scholar] [CrossRef] [Green Version]
- Levy, J.M.M.; Towers, C.G.; Thorburn, A. Targeting autophagy in cancer. Nat. Rev. Cancer 2017, 17, 528–542. [Google Scholar] [CrossRef]
- Wang, M.; Law, M.E.; Castellano, R.K.; Law, B.K. The unfolded protein response as a target for anticancer therapeutics. Crit. Rev. Oncol. Hematol. 2018, 127, 66–79. [Google Scholar] [CrossRef]
- Yun, C.W.; Lee, S.H. The Roles of Autophagy in Cancer. Int. J. Mol. Sci. 2018, 19, 3466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Onorati, A.V.; Dyczynski, M.; Ojha, R.; Amaravadi, R.K. Targeting autophagy in cancer. Nat. Rev. Cancer 2018, 124, 3307–3318. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, H.; Wang, K.; Wang, M.; Zhao, W.; Zhang, C.; Cai, M.; Qiu, Y.; Zhang, T.; Shao, R.; Zhao, W. ER-phagy in the Occurrence and Development of Cancer. Biomedicines 2022, 10, 707. https://doi.org/10.3390/biomedicines10030707
Zhou H, Wang K, Wang M, Zhao W, Zhang C, Cai M, Qiu Y, Zhang T, Shao R, Zhao W. ER-phagy in the Occurrence and Development of Cancer. Biomedicines. 2022; 10(3):707. https://doi.org/10.3390/biomedicines10030707
Chicago/Turabian StyleZhou, Huimin, Kexin Wang, Mengyan Wang, Wenxia Zhao, Conghui Zhang, Meilian Cai, Yuhan Qiu, Tianshu Zhang, Rongguang Shao, and Wuli Zhao. 2022. "ER-phagy in the Occurrence and Development of Cancer" Biomedicines 10, no. 3: 707. https://doi.org/10.3390/biomedicines10030707
APA StyleZhou, H., Wang, K., Wang, M., Zhao, W., Zhang, C., Cai, M., Qiu, Y., Zhang, T., Shao, R., & Zhao, W. (2022). ER-phagy in the Occurrence and Development of Cancer. Biomedicines, 10(3), 707. https://doi.org/10.3390/biomedicines10030707