
Apolipoprotein C3-Rich Low-Density Lipoprotein Induces Endothelial Cell Senescence via FBXO31 and Its Inhibition by Sesamol In Vitro and In Vivo


Abstract
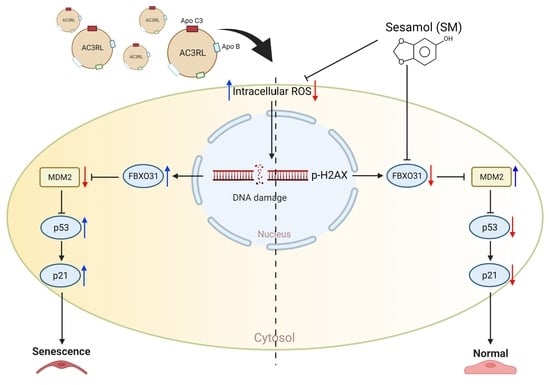
:
1. Introduction
2. Materials and Methods
2.1. LDL Isolation
2.2. Isolation and Measurement of AC3RL
2.3. Cell Culture and Treatment
2.4. SA-β-Gal Staining for HAECs
2.5. Western Blotting Analysis
2.6. Immunofluorescence Microscopy
2.7. Animals, Groupings, SA-β-Gal Staining, and Oil Red O Staining
2.8. Immunoprecipitation
2.9. mRNA Analysis Using Real-Time Quantitative PCR
2.10. Data Analysis and Statistical Procedures
3. Results
3.1. AC3RL Induces Endothelial Cell Senescence
3.2. ROS Is Involved in AC3RL-Induced EC Senescence
3.3. The Role of FBXO31 in AC3RL-Induced Cell Senescence
3.4. AC3RL-Induced Vascular Endothelial Senescence and the Inhibitory Effect of Sesamol
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lakatta, E.G.; Levy, D. Arterial and cardiac aging: Major shareholders in cardiovascular disease enterprises: Part I: Aging arteries: A “set up” for vascular disease. Circulation 2003, 107, 139–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minamino, T.; Komuro, I. Vascular cell senescence: Contribution to atherosclerosis. Circ. Res. 2007, 100, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Gerhard, M.; Roddy, M.A.; Creager, S.J.; Creager, M.A. Aging progressively impairs endothelium-dependent vasodilation in forearm resistance vessels of humans. Hypertension 1996, 27, 849–853. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.C.; Bennett, M. Aging and atherosclerosis: Mechanisms, functional consequences, and potential therapeutics for cellular senescence. Circ. Res. 2012, 111, 245–259. [Google Scholar] [CrossRef] [Green Version]
- Stojanovic, S.D.; Fiedler, J.; Bauersachs, J.; Thum, T.; Sedding, D.G. Senescence-induced inflammation: An important player and key therapeutic target in atherosclerosis. Eur. Heart J. 2020, 41, 2983–2996. [Google Scholar] [CrossRef] [Green Version]
- Minamino, T.; Miyauchi, H.; Yoshida, T.; Ishida, Y.; Yoshida, H.; Komuro, I. Endothelial cell senescence in human atherosclerosis: Role of telomere in endothelial dysfunction. Circulation 2002, 105, 1541–1544. [Google Scholar] [CrossRef] [Green Version]
- Shen, M.Y.; Hsu, J.F.; Chen, F.Y.; Lu, J.; Chang, C.M.; Madjid, M.; Dean, J.; Dixon, R.A.F.; Shayani, S.; Chou, T.C.; et al. Combined LDL and VLDL Electronegativity Correlates with Coronary Heart Disease Risk in Asymptomatic Individuals. J. Clin. Med. 2019, 8, 1193. [Google Scholar] [CrossRef] [Green Version]
- Hansson, G.K. Inflammation, atherosclerosis, and coronary artery disease. N. Engl. J. Med. 2005, 352, 1685–1695. [Google Scholar] [CrossRef] [Green Version]
- Levine, A.J. p53, the cellular gatekeeper for growth and division. Cell 1997, 88, 323–331. [Google Scholar] [CrossRef] [Green Version]
- Freedman, D.A.; Wu, L.; Levine, A.J. Functions of the MDM2 oncoprotein. Cell. Mol. Life Sci. CMLS 1999, 55, 96–107. [Google Scholar] [CrossRef]
- Kubbutat, M.H.; Jones, S.N.; Vousden, K.H. Regulation of p53 stability by Mdm2. Nature 1997, 387, 299–303. [Google Scholar] [CrossRef]
- Malonia, S.K.; Dutta, P.; Santra, M.K.; Green, M.R. F-box protein FBXO31 directs degradation of MDM2 to facilitate p53-mediated growth arrest following genotoxic stress. Proc. Natl. Acad. Sci. USA 2015, 112, 8632–8637. [Google Scholar] [CrossRef] [Green Version]
- Joshi, R.; Kumar, M.S.; Satyamoorthy, K.; Unnikrisnan, M.K.; Mukherjee, T. Free Radical Reactions and Antioxidant Activities of Sesamol: Pulse Radiolytic and Biochemical Studies. J. Agric. Food Chem. 2005, 53, 2696–2703. [Google Scholar] [CrossRef] [PubMed]
- Sacks, F.M.; Alaupovic, P.; Moye, L.A.; Cole, T.G.; Sussex, B.; Stampfer, M.J.; Pfeffer, M.A.; Braunwald, E. VLDL, apolipoproteins B, CIII, and E, and risk of recurrent coronary events in the Cholesterol and Recurrent Events (CARE) trial. Circulation 2000, 102, 1886–1892. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.S.; McConathy, W.J.; Kloer, H.U.; Alaupovic, P. Modulation of lipoprotein lipase activity by apolipoproteins. Effect of apolipoprotein C-III. J. Clin. Investig. 1985, 75, 384–390. [Google Scholar] [CrossRef]
- Clavey, V.; Lestavel-Delattre, S.; Copin, C.; Bard, J.M.; Fruchart, J.C. Modulation of lipoprotein B binding to the LDL receptor by exogenous lipids and apolipoproteins CI, CII, CIII, and E. Arter. Thromb. Vasc. Biol. 1995, 15, 963–971. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, A.; Aikawa, M.; Alcaide, P.; Luscinskas, F.W.; Libby, P.; Sacks, F.M. Apolipoprotein CIII induces expression of vascular cell adhesion molecule-1 in vascular endothelial cells and increases adhesion of monocytic cells. Circulation 2006, 114, 681–687. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, A.; Aikawa, M.; Libby, P.; Alcaide, P.; Luscinskas, F.W.; Sacks, F.M. Apolipoprotein CIII in apolipoprotein B lipoproteins enhances the adhesion of human monocytic cells to endothelial cells. Circulation 2006, 113, 691–700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, M.Y.; Chen, F.Y.; Hsu, J.F.; Fu, R.H.; Chang, C.M.; Chang, C.T.; Liu, C.H.; Wu, J.R.; Lee, A.S.; Chan, H.C.; et al. Plasma L5 levels are elevated in ischemic stroke patients and enhance platelet aggregation. Blood 2016, 127, 1336–1345. [Google Scholar] [CrossRef] [Green Version]
- Khoo, C.; Campos, H.; Judge, H.; Sacks, F.M. Effects of estrogenic oral contraceptives on the lipoprotein B particle system defined by apolipoproteins E and C-III content. J. Lipid Res. 1999, 40, 202–212. [Google Scholar] [CrossRef]
- Jo-Watanabe, A.; Ohse, T.; Nishimatsu, H.; Takahashi, M.; Ikeda, Y.; Wada, T.; Shirakawa, J.I.; Nagai, R.; Miyata, T.; Nagano, T.; et al. Glyoxalase I reduces glycative and oxidative stress and prevents age-related endothelial dysfunction through modulation of endothelial nitric oxide synthase phosphorylation. Aging Cell 2014, 13, 519–528. [Google Scholar] [CrossRef] [PubMed]
- Burnley, P.; Rahman, M.; Wang, H.; Zhang, Z.; Sun, X.; Zhuge, Q.; Su, D.M. Role of the p63-FoxN1 regulatory axis in thymic epithelial cell homeostasis during aging. Cell Death Dis. 2013, 4, e932. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.Y.; Chen, F.Y.; Lee, A.S.; Ting, K.H.; Chang, C.M.; Hsu, J.F.; Lee, W.S.; Sheu, J.R.; Chen, C.H.; Shen, M.Y. Sesamol reduces the atherogenicity of electronegative L5 LDL in vivo and in vitro. J. Nat. Prod. 2015, 78, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Klokov, D.; MacPhail, S.M.; Banath, J.P.; Byrne, J.P.; Olive, P.L. Phosphorylated histone H2AX in relation to cell survival in tumor cells and xenografts exposed to single and fractionated doses of X-rays. Radiother. Oncol. J. Eur. Soc. Ther. Radiol. Oncol. 2006, 80, 223–229. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.-S.; Tsai, P.-H.; Tseng, K.-F.; Chen, F.-Y.; Yang, W.-C.; Shen, M.-Y. Sesamol Ameliorates Renal Injury-Mediated Atherosclerosis via Inhibition of Oxidative Stress/IKKα/p53. Antioxidants 2021, 10, 1519. [Google Scholar] [CrossRef]
- Gu, Z.; Jiang, J.; Tan, W.; Xia, Y.; Cao, H.; Meng, Y.; Da, Z.; Liu, H.; Cheng, C. p53/p21 Pathway involved in mediating cellular senescence of bone marrow-derived mesenchymal stem cells from systemic lupus erythematosus patients. Clin. Dev. Immunol. 2013, 2013, 134243. [Google Scholar] [CrossRef]
- Mijit, M.; Caracciolo, V.; Melillo, A.; Amicarelli, F.; Giordano, A. Role of p53 in the Regulation of Cellular Senescence. Biomolecules 2020, 10, 420. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.C.; Lee, A.S.; Lu, L.S.; Ke, L.Y.; Chen, W.Y.; Dong, J.W.; Lu, J.; Chen, Z.; Chu, C.S.; Chan, H.C.; et al. Human electronegative LDL induces mitochondrial dysfunction and premature senescence of vascular cells in vivo. Aging Cell 2018, 17, e12792. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Liu, P.; Inuzuka, H.; Wei, W. Roles of F-box proteins in cancer. Nat. Rev. Cancer 2014, 14, 233–247. [Google Scholar] [CrossRef] [Green Version]
- Santra, M.K.; Wajapeyee, N.; Green, M.R. F-box protein FBXO31 mediates cyclin D1 degradation to induce G1 arrest after DNA damage. Nature 2009, 459, 722–725. [Google Scholar] [CrossRef]
- Wu, D.; Prives, C. Relevance of the p53-MDM2 axis to aging. Cell Death Differ. 2018, 25, 169–179. [Google Scholar] [CrossRef] [PubMed]
- Inuzuka, H.; Fukushima, H.; Shaik, S.; Wei, W. Novel insights into the molecular mechanisms governing Mdm2 ubiquitination and destruction. Oncotarget 2010, 1, 685–690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendivil, C.O.; Rimm, E.B.; Furtado, J.; Chiuve, S.E.; Sacks, F.M. Low-density lipoproteins containing apolipoprotein C-III and the risk of coronary heart disease. Circulation 2011, 124, 2065–2072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masucci-Magoulas, L.; Goldberg, I.J.; Bisgaier, C.L.; Serajuddin, H.; Francone, O.L.; Breslow, J.L.; Tall, A.R. A mouse model with features of familial combined hyperlipidemia. Science 1997, 275, 391–394. [Google Scholar] [CrossRef]
- Maeda, N.; Li, H.; Lee, D.; Oliver, P.; Quarfordt, S.H.; Osada, J. Targeted disruption of the apolipoprotein C-III gene in mice results in hypotriglyceridemia and protection from postprandial hypertriglyceridemia. J. Biol. Chem. 1994, 269, 23610–23616. [Google Scholar] [CrossRef]
- Boren, J.; Packard, C.J.; Taskinen, M.R. The Roles of ApoC-III on the Metabolism of Triglyceride-Rich Lipoproteins in Humans. Front. Endocrinol. 2020, 11, 474. [Google Scholar] [CrossRef]
- Wang, L.; Xu, F.; Song, Z.; Han, D.; Zhang, J.; Chen, L.; Na, L. A high fat diet with a high C18:0/C16:0 ratio induced worse metabolic and transcriptomic profiles in C57BL/6 mice. Lipids Health Dis. 2020, 19, 172. [Google Scholar] [CrossRef]
- Hou, R.C.; Chen, H.L.; Tzen, J.T.; Jeng, K.C. Effect of sesame antioxidants on LPS-induced NO production by BV2 microglial cells. Neuroreport 2003, 14, 1815–1819. [Google Scholar] [CrossRef]
- Hemalatha, G.; Pugalendi, K.V.; Saravanan, R. Modulatory effect of sesamol on DOCA-salt-induced oxidative stress in uninephrectomized hypertensive rats. Mol. Cell. Biochem. 2013, 379, 255–265. [Google Scholar] [CrossRef] [Green Version]
- Choppara, S.; Malonia, S.K.; Sankaran, G.; Green, M.R.; Santra, M.K. Degradation of FBXO31 by APC/C is regulated by AKT- and ATM-mediated phosphorylation. Proc. Natl. Acad. Sci. USA 2018, 115, 998–1003. [Google Scholar] [CrossRef] [Green Version]
- Mayo, L.D.; Donner, D.B. A phosphatidylinositol 3-kinase/Akt pathway promotes translocation of Mdm2 from the cytoplasm to the nucleus. Proc. Natl. Acad. Sci. USA 2001, 98, 11598–11603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonomini, F.; Rodella, L.F.; Rezzani, R. Metabolic syndrome, aging and involvement of oxidative stress. Aging Dis. 2015, 6, 109–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talebi, S.; Bagherniya, M.; Atkin, S.L.; Askari, G.; Orafai, H.M.; Sahebkar, A. The beneficial effects of nutraceuticals and natural products on small dense LDL levels, LDL particle number and LDL particle size: A clinical review. Lipids Health Dis. 2020, 19, 66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, G.-J.; Chang, C.-T.; Yang, C.-Y.; Chen, C.-H. Negatively charged L5 as a naturally occurring atherogenic low-density lipoprotein. BioMedicine 2012, 2, 147–154. [Google Scholar] [CrossRef]
- Shaito, A.; Thuan, D.T.B.; Phu, H.T.; Nguyen, T.H.D.; Hasan, H.; Halabi, S.; Abdelhady, S.; Nasrallah, G.K.; Eid, A.H.; Pintus, G. Herbal Medicine for Cardiovascular Diseases: Efficacy, Mechanisms, and Safety. Front. Pharm. 2020, 11, 422. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Primer (5′→3′) Forward | Primer (5′→3′) Reverse |
|---|---|---|
| GAPDH | TTGTTGCCATCAATGACCCCTT | CGTTCTCAGCCTTGACTGTGCCTT |
| FBXO31 | GTGGAGATCTTCGCCTCGCT | TCACAGACGCCATACTCCTCG |
| MDM2 | CACAGGTCCCTTTCCTTTGA | TGAATCCTGATCCAGCCAAT |
| p53 | CCCCCAAAGAGTGCTAAACGA | CAGTTCCAAGGCCTCATTCAA |
| p16(INK4A) | AGAGGTTCGGGCTTTGCT | CTACTTGGGTGTTGCCCATC |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsai, P.-H.; Chen, L.-Z.; Tseng, K.-F.; Chen, F.-Y.; Shen, M.-Y. Apolipoprotein C3-Rich Low-Density Lipoprotein Induces Endothelial Cell Senescence via FBXO31 and Its Inhibition by Sesamol In Vitro and In Vivo. Biomedicines 2022, 10, 854. https://doi.org/10.3390/biomedicines10040854
Tsai P-H, Chen L-Z, Tseng K-F, Chen F-Y, Shen M-Y. Apolipoprotein C3-Rich Low-Density Lipoprotein Induces Endothelial Cell Senescence via FBXO31 and Its Inhibition by Sesamol In Vitro and In Vivo. Biomedicines. 2022; 10(4):854. https://doi.org/10.3390/biomedicines10040854
Chicago/Turabian StyleTsai, Ping-Hsuan, Li-Zhen Chen, Kuo-Feng Tseng, Fang-Yu Chen, and Ming-Yi Shen. 2022. "Apolipoprotein C3-Rich Low-Density Lipoprotein Induces Endothelial Cell Senescence via FBXO31 and Its Inhibition by Sesamol In Vitro and In Vivo" Biomedicines 10, no. 4: 854. https://doi.org/10.3390/biomedicines10040854
APA StyleTsai, P. -H., Chen, L. -Z., Tseng, K. -F., Chen, F. -Y., & Shen, M. -Y. (2022). Apolipoprotein C3-Rich Low-Density Lipoprotein Induces Endothelial Cell Senescence via FBXO31 and Its Inhibition by Sesamol In Vitro and In Vivo. Biomedicines, 10(4), 854. https://doi.org/10.3390/biomedicines10040854