
Oncology Drug Repurposing for Sepsis Treatment


Abstract
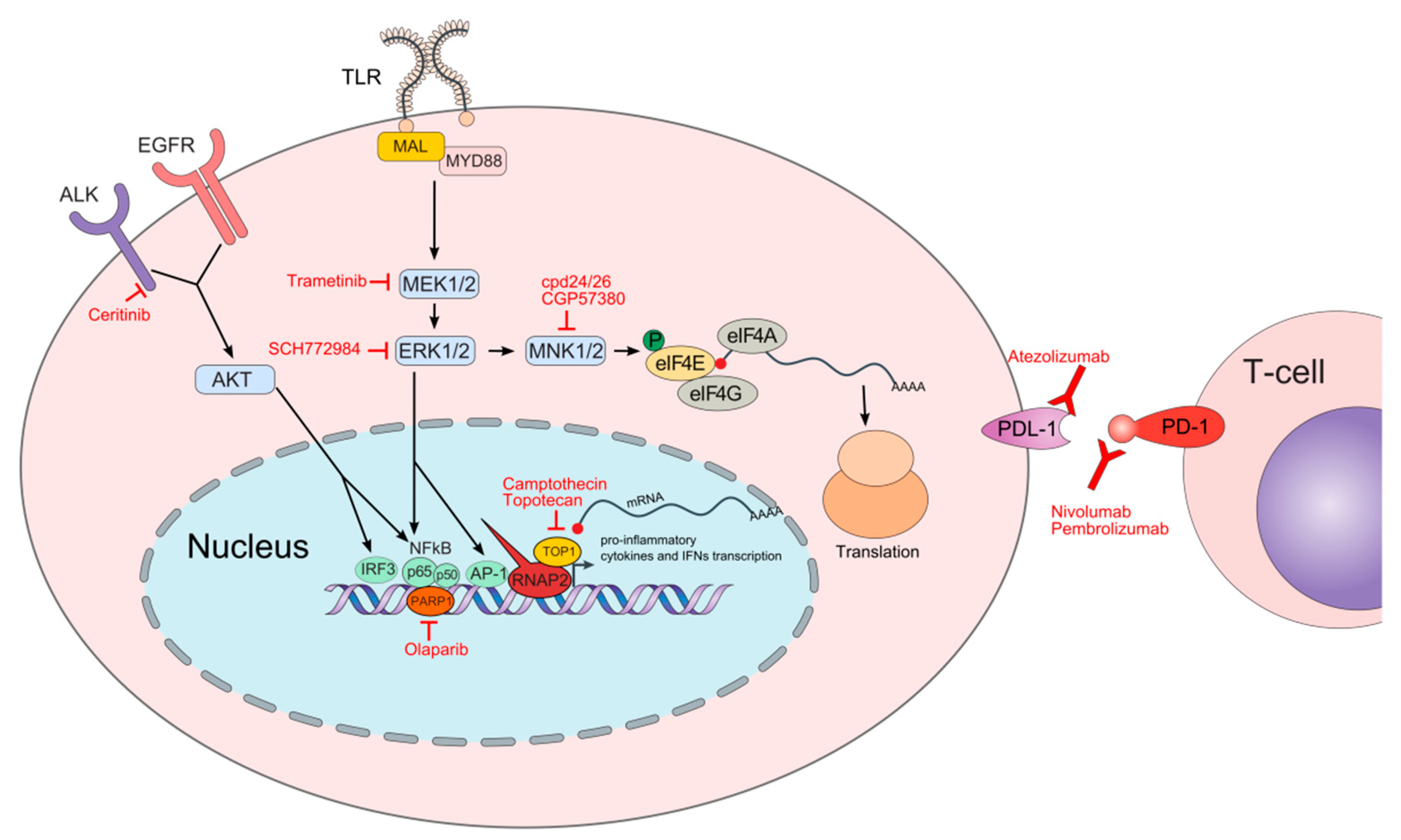
:1. Introduction
2. Sepsis Overview and Failure in the Drug Development Efforts
3. Drugs Development and Repurposing
4. Examples of Oncology Drug Repurposing for Sepsis Treatment
4.1. Topoisomerase 1 Inhibitors
4.2. Poly (ADP-ribose) Polymerase (PARP) Inhibitors
4.3. MAPK Pathway Inhibitors
4.3.1. MEK-ERK Inhibitors
4.3.2. MNK Inhibitors
4.4. Anaplastic Lymphoma Kinase (ALK) Inhibitors
4.5. Immune Checkpoint Inhibitors
5. Perspective and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Singer, M.; Deutschman, C.S.; Seymour, C.W.; Shankar-Hari, M.; Annane, D.; Bauer, M.; Bellomo, R.; Bernard, G.R.; Chiche, J.-D.; Coopersmith, C.M.; et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA 2016, 315, 801–810. [Google Scholar] [CrossRef] [PubMed]
- Shankar-Hari, M.; Phillips, G.S.; Levy, M.L.; Seymour, C.W.; Liu, V.X.; Deutschman, C.S.; Angus, D.C.; Rubenfeld, G.D.; Singer, M.; for the Sepsis Definitions Task Force. Developing a New Definition and Assessing New Clinical Criteria for Septic Shock: For the Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA 2016, 315, 775–787. [Google Scholar] [CrossRef] [PubMed]
- Rudd, K.E.; Johnson, S.C.; Agesa, K.M.; Shackelford, K.A.; Tsoi, D.; Kievlan, D.R.; Colombara, D.V.; Ikuta, K.S.; Kissoon, N.; Finfer, S.; et al. Global, regional, and national sepsis incidence and mortality, 1990–2017: Analysis for the Global Burden of Disease Study. Lancet 2020, 395, 200–211. [Google Scholar] [CrossRef] [Green Version]
- Vincent, J.-L.; Jones, G.; David, S.; Olariu, E.; Cadwell, K.K. Frequency and mortality of septic shock in Europe and North America: A systematic review and meta-analysis. Crit. Care 2019, 23, 196. [Google Scholar] [CrossRef] [Green Version]
- Reinhart, K.; Daniels, R.; Kissoon, N.; Machado, F.R.; Schachter, R.D.; Finfer, S. Recognizing Sepsis as a Global Health Priority—A WHO Resolution. N. Engl. J. Med. 2017, 377, 414–417. [Google Scholar] [CrossRef]
- Evans, L.; Rhodes, A.; Alhazzani, W.; Antonelli, M.; Coopersmith, C.M.; French, C.; Machado, F.R.; Mcintyre, L.; Ostermann, M.; Prescott, H.C.; et al. Surviving sepsis campaign: International guidelines for management of sepsis and septic shock 2021. Intensive Care Med. 2021, 47, 1181–1247. [Google Scholar] [CrossRef]
- Heming, N.; Lamothe, L.; Ambrosi, X.; Annane, D. Emerging drugs for the treatment of sepsis. Expert Opin. Emerg. Drugs 2016, 21, 27–37. [Google Scholar] [CrossRef]
- Medzhitov, R.; Janeway, C.A., Jr. Innate immunity: Impact on the adaptive immune response. Curr. Opin. Immunol. 1997, 9, 4–9. [Google Scholar] [CrossRef]
- Arthur, J.S.C.; Ley, S.C. Mitogen-activated protein kinases in innate immunity. Nat. Rev. Immunol. 2013, 13, 679–692. [Google Scholar] [CrossRef]
- Chousterman, B.G.; Swirski, F.; Weber, G.F. Cytokine storm and sepsis disease pathogenesis. Semin. Immunopathol. 2017, 39, 517–528. [Google Scholar] [CrossRef]
- Opal, S.M.; Van Der Poll, T. Endothelial barrier dysfunction in septic shock. J. Intern. Med. 2015, 277, 277–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hotchkiss, R.R.; Moldawer, L.L.L.; Opal, S.M.; Reinhart, K.; Turnbull, I.I.; Vincent, J.-L. Sepsis and septic shock. Nat. Rev. Dis. Primers 2016, 2, 16045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fink, M.P.; Warren, H.S. Strategies to improve drug development for sepsis. Nat. Rev. Drug Discov. 2014, 13, 741–758. [Google Scholar] [CrossRef] [PubMed]
- Tindal, E.W.; Armstead, B.E.; Monaghan, S.F.; Heffernan, D.S.; Ayala, A. Emerging therapeutic targets for sepsis. Expert Opin. Ther. Targets 2021, 25, 175–189. [Google Scholar] [CrossRef] [PubMed]
- Cavaillon, J.; Singer, M.; Skirecki, T. Sepsis therapies: Learning from 30 years of failure of translational research to propose new leads. EMBO Mol. Med. 2020, 12, e10128. [Google Scholar] [CrossRef] [PubMed]
- De Maio, A. Do Not Blame the Rodent for the Failure of Developing Sepsis Therapies. Shock 2020, 54, 631–632. [Google Scholar] [CrossRef]
- Ashburn, T.T.; Thor, K.B. Drug repositioning: Identifying and developing new uses for existing drugs. Nat. Rev. Drug Discov. 2004, 3, 673–683. [Google Scholar] [CrossRef]
- Gupta, S.C.; Sung, B.; Prasad, S.; Webb, L.J.; Aggarwal, B.B. Cancer drug discovery by repurposing: Teaching new tricks to old dogs. Trends Pharmacol. Sci. 2013, 34, 508–517. [Google Scholar] [CrossRef]
- Jin, G.; Wong, S.T. Toward better drug repositioning: Prioritizing and integrating existing methods into efficient pipelines. Drug Discov. Today 2014, 19, 637–644. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.Y.; Jones, S.J. Drug repositioning for personalized medicine. Genome Med. 2012, 4, 27. [Google Scholar] [CrossRef] [Green Version]
- Druker, B.J. Imatinib as a Paradigm of Targeted Therapies. Adv. Cancer Res. 2004, 91, 1–30. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.C. Cellular roles of DNA topoisomerases: A molecular perspective. Nat. Rev. Mol. Cell Biol. 2002, 3, 430–440. [Google Scholar] [CrossRef] [PubMed]
- Koster, D.A.; Croquette, V.; Dekker, C.; Shuman, S.; Dekker, N. Friction and torque govern the relaxation of DNA supercoils by eukaryotic topoisomerase IB. Nature 2005, 434, 671–674. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Jiang, T.; Li, Q.; Ling, X. Camptothecin (CPT) and its derivatives are known to target topoisomerase I (Top1) as their mechanism of action: Did we miss something in CPT analogue molecular targets for treating human disease such as cancer? Am. J. Cancer Res. 2017, 7, 2350–2394. [Google Scholar] [PubMed]
- Hsiang, Y.H.; Hertzberg, R.; Hecht, S.; Liu, L. Camptothecin induces protein-linked DNA breaks via mammalian DNA topoisomerase I. J. Biol. Chem. 1985, 260, 14873–14878. [Google Scholar] [CrossRef]
- Bao, X.; Wu, J.; Kim, S.; Lorusso, P.; Li, J. Pharmacometabolomics Reveals Irinotecan Mechanism of Action in Cancer Patients. J. Clin. Pharmacol. 2019, 59, 20–34. [Google Scholar] [CrossRef] [Green Version]
- Khaiwa, N.; Maarouf, N.R.; Darwish, M.H.; Alhamad, D.W.; Sebastian, A.; Hamad, M.; Omar, H.A.; Orive, G.; Al-Tel, T.H. Camptothecin’s Journey from Discovery to WHO Essential Medicine: Fifty Years of Promise. Eur. J. Med. Chem. 2021, 223, 113639. [Google Scholar] [CrossRef]
- Rialdi, A.; Campisi, L.; Zhao, N.; Lagda, A.C.; Pietzsch, C.; Ho, J.S.Y.; Martinez-Gil, L.; Fenouil, R.; Chen, X.; Edwards, M.; et al. Topoisomerase 1 inhibition suppresses inflammatory genes and protects from death by inflammation. Science 2016, 352, aad7993. [Google Scholar] [CrossRef] [Green Version]
- Kapoor, K.; Singla, E.; Sahu, B.; Naura, A.S. PARP inhibitor, olaparib ameliorates acute lung and kidney injury upon intratracheal administration of LPS in mice. Mol. Cell. Biochem. 2015, 400, 153–162. [Google Scholar] [CrossRef]
- Ahmad, A.; Vieira, J.D.C.; de Mello, A.H.; de Lima, T.M.; Ariga, S.K.; Barbeiro, D.F.; Barbeiro, H.V.; Szczesny, B.; Törö, G.; Druzhyna, N.; et al. The PARP inhibitor olaparib exerts beneficial effects in mice subjected to cecal ligature and puncture and in cells subjected to oxidative stress without impairing DNA integrity: A potential opportunity for repurposing a clinically used oncological drug for the experimental therapy of sepsis. Pharmacol. Res. 2019, 145, 104263. [Google Scholar] [CrossRef] [Green Version]
- Shi-Lin, D.; Yuan, X.; Zhan, S.; Luo-Jia, T.; Chao-Yang, T. Trametinib, a novel MEK kinase inhibitor, suppresses lipopolysaccharide-induced tumor necrosis factor (TNF)-α production and endotoxin shock. Biochem. Biophys. Res. Commun. 2015, 458, 667–673. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.A.; Mayeux, P.R.; Schnellmann, R.G. Delayed Mitogen-Activated Protein Kinase/Extracellular Signal–Regulated Kinase Inhibition by Trametinib Attenuates Systemic Inflammatory Responses and Multiple Organ Injury in Murine Sepsis. Crit. Care Med. 2016, 44, e711–e720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kopczynski, M.; Rumienczyk, I.; Kulecka, M.; Statkiewicz, M.; Pysniak, K.; Sandowska-Markiewicz, Z.; Wojcik-Trechcinska, U.; Goryca, K.; Pyziak, K.; Majewska, E.; et al. Selective Extracellular Signal-Regulated Kinase 1/2 (ERK1/2) Inhibition by the SCH772984 Compound Attenuates In Vitro and In Vivo Inflammatory Responses and Prolongs Survival in Murine Sepsis Models. Int. J. Mol. Sci. 2021, 22, 10204. [Google Scholar] [CrossRef] [PubMed]
- Dreas, A.; Kucwaj-Brysz, K.; Pyziak, K.; Kulesza, U.; Wincza, E.; Fabritius, C.-H.; Michalik, K.; Gabor-Worwa, E.; Gołas, A.; Milik, M.; et al. Discovery of indazole-pyridinone derivatives as a novel class of potent and selective MNK1/2 kinase inhibitors that protecting against endotoxin-induced septic shock. Eur. J. Med. Chem. 2021, 213, 113057. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Teng, L.; Yang, S.; Huang, S.; Li, L.; Zhou, L.; Liu, G.; Tang, H. MNK as a potential pharmacological target for suppressing LPS-induced acute lung injury in mice. Biochem. Pharmacol. 2021, 186, 114499. [Google Scholar] [CrossRef] [PubMed]
- Zeng, L.; Kang, R.; Zhu, S.; Wang, X.; Cao, L.; Wang, H.; Billiar, T.R.; Jiang, J.; Tang, D. ALK is a therapeutic target for lethal sepsis. Sci. Transl. Med. 2017, 9, eaan5689. [Google Scholar] [CrossRef] [Green Version]
- Ge, W.; Hu, Q.; Fang, X.; Liu, J.; Xu, J.; Hu, J.; Liu, X.; Ling, Q.; Wang, Y.; Li, H.; et al. LDK378 improves micro- and macro-circulation via alleviating STING-mediated inflammatory injury in a Sepsis rat model induced by Cecal ligation and puncture. J. Inflamm. 2019, 16, 3. [Google Scholar] [CrossRef] [Green Version]
- Brahmamdam, P.; Inoue, S.; Unsinger, J.; Chang, K.C.; McDunn, J.E.; Hotchkiss, R.S. Delayed administration of anti-PD-1 antibody reverses immune dysfunction and improves survival during sepsis. J. Leukoc. Biol. 2010, 88, 233–240. [Google Scholar] [CrossRef]
- Chang, K.C.; Burnham, C.-A.; Compton, S.M.; Rasche, D.P.; Mazuski, R.; SMcDonough, J.; Unsinger, J.; Korman, A.J.; Green, J.M.; Hotchkiss, R.S. Blockade ofthe negative co-stimulatory molecules PD-1 and CTLA-4 improves survival in primary and secondary fungal sepsis. Crit. Care 2013, 17, R85. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Zhou, Y.; Lou, J.; Li, J.; Bo, L.; Zhu, K.; Wan, X.; Deng, X.; Cai, Z. PD-L1 blockade improves survival in experimental sepsis by inhibiting lymphocyte apoptosis and reversing monocyte dysfunction. Crit. Care 2010, 14, R220. [Google Scholar] [CrossRef] [Green Version]
- Fujita, K.-I.; Kubota, Y.; Ishida, H.; Sasaki, Y. Irinotecan, a key chemotherapeutic drug for metastatic colorectal cancer. World J. Gastroenterol. 2015, 21, 12234–12248. [Google Scholar] [CrossRef] [PubMed]
- Hamano, H.; Mitsui, M.; Zamami, Y.; Takechi, K.; Nimura, T.; Okada, N.; Fukushima, K.; Imanishi, M.; Chuma, M.; Horinouchi, Y.; et al. Irinotecan-induced neutropenia is reduced by oral alkalization drugs: Analysis using retrospective chart reviews and the spontaneous reporting database. Support. Care Cancer 2018, 27, 849–856. [Google Scholar] [CrossRef] [PubMed]
- Vrdoljak, A.L.; Fuchs, N.; Mikolić, A.; Žunec, S.; Karačonji, I.B.; Jurič, A.; Prester, L.; Micek, V.; Neuberg, M.; Čanović, S.; et al. Irinotecan and Δ9-Tetrahydrocannabinol Interactions in Rat Liver: A Preliminary Evaluation Using Biochemical and Genotoxicity Markers. Molecules 2018, 23, 1332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curtin, N.J.; Szabo, C. Poly(ADP-ribose) polymerase inhibition: Past, present and future. Nat. Rev. Drug Discov. 2020, 19, 711–736. [Google Scholar] [CrossRef]
- Eisemann, T.; Pascal, J.M. Poly(ADP-ribose) polymerase enzymes and the maintenance of genome integrity. Cell. Mol. Life Sci. 2020, 77, 19–33. [Google Scholar] [CrossRef]
- Curtin, N.J.; Szabo, C. Therapeutic applications of PARP inhibitors: Anticancer therapy and beyond. Mol. Asp. Med. 2013, 34, 1217–1256. [Google Scholar] [CrossRef] [Green Version]
- Hurtado-Bagès, S.; Knobloch, G.; Ladurner, A.G.; Buschbeck, M. The taming of PARP1 and its impact on NAD+ metabolism. Mol. Metab. 2020, 38, 100950. [Google Scholar] [CrossRef]
- Durkacz, B.W.; Omidiji, O.; Gray, D.A.; Shall, S. (ADP-ribose)n participates in DNA excision repair. Nature 1980, 283, 593–596. [Google Scholar] [CrossRef]
- Ben-Hur, E.; Chen, C.C.; Elkind, M.M. Inhibitors of poly(adenosine diphosphoribose) synthetase, examination of metabolic perturbations, and enhancement of radiation response in Chinese hamster cells. Cancer Res. 1985, 45, 2123–2127. [Google Scholar]
- Bowman, K.J.; Newell, D.R.; Calvert, A.H.; Curtin, N.J. Differential effects of the poly (ADP-ribose) polymerase (PARP) inhibitor NU1025 on topoisomerase I and II inhibitor cytotoxicity in L1210 cells in vitro. Br. J. Cancer 2001, 84, 106–112. [Google Scholar] [CrossRef] [Green Version]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434, 913–917. [Google Scholar] [CrossRef] [PubMed]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.J.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef] [PubMed]
- Wasyluk, W.; Zwolak, A. PARP Inhibitors: An Innovative Approach to the Treatment of Inflammation and Metabolic Disorders in Sepsis. J. Inflamm. Res. 2021, 14, 1827–1844. [Google Scholar] [CrossRef] [PubMed]
- Bai, P.; Virág, L. Role of poly(ADP-ribose) polymerases in the regulation of inflammatory processes. FEBS Lett. 2012, 586, 3771–3777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmad, A.; Olah, G.; Herndon, D.N.; Szabo, C. The clinically used PARP inhibitor olaparib improves organ function, suppresses inflammatory responses and accelerates wound healing in a murine model of third-degree burn injury. Br. J. Pharmacol. 2018, 175, 232–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soriano, F.G.; Liaudet, L.; Szabó, É.; Virág, L.; Mabley, J.G.; Pacher, P.; Szabó, C. Resistance to Acute Septic Peritonitis in Poly(ADP-ribose) Polymerase-1-Deficient Mice. Shock 2002, 17, 286–292. [Google Scholar] [CrossRef]
- Kim, E.K.; Choi, E.-J. Pathological roles of MAPK signaling pathways in human diseases. Biochim. Biophys. Acta Mol. Basis Dis. 2010, 1802, 396–405. [Google Scholar] [CrossRef] [Green Version]
- Braicu, C.; Buse, M.; Busuioc, C.; Drula, R.; Gulei, D.; Raduly, L.; Rusu, A.; Irimie, A.; Atanasov, A.G.; Slaby, O.; et al. A Comprehensive Review on MAPK: A Promising Therapeutic Target in Cancer. Cancers 2019, 11, 1618. [Google Scholar] [CrossRef] [Green Version]
- Mikula, M.; Skrzypczak, M.; Goryca, K.; Paczkowska, K.; Ledwon, J.; Statkiewicz, M.; Kulecka, M.; Grzelak, M.; Dabrowska, M.; Kuklinska, U.; et al. Genome-wide co-localization of active EGFR and downstream ERK pathway kinases mirrors mitogen-inducible RNA polymerase 2 genomic occupancy. Nucleic Acids Res. 2016, 44, 10150–10164. [Google Scholar] [CrossRef] [Green Version]
- Cargnello, M.; Roux, P.P. Activation and Function of the MAPKs and Their Substrates, the MAPK-Activated Protein Kinases. Microbiol. Mol. Biol. Rev. 2011, 75, 50–83, Correction in Microbiol. Mol. Biol. Rev. 2012, 76, 496. [Google Scholar] [CrossRef] [Green Version]
- Du, Z.; Lovly, C.M. Mechanisms of receptor tyrosine kinase activation in cancer. Mol. Cancer 2018, 17, 58. [Google Scholar] [CrossRef] [PubMed]
- Chapman, P.B.; Hauschild, A.; Robert, C.; Haanen, J.B.; Ascierto, P.; Larkin, J.; Dummer, R.; Garbe, C.; Testori, A.; Maio, M.; et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N. Engl. J. Med. 2011, 364, 2507–2516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahapatra, D.K.; Asati, V.; Bharti, S.K. Discovery of small-molecule ATR inhibitors for potential cancer treatment: A patent review from 2014 to present. Expert Opin. Ther. Pat. 2017, 27, 887–906. [Google Scholar] [CrossRef]
- Reimann, T.; Büscher, D.; Hipskind, R.A.; Krautwald, S.; Lohmann-Matthes, M.L.; Baccarini, M. Lipopolysaccharide induces activation of the Raf-1/MAP kinase pathway. A putative role for Raf-1 in the induction of the IL-1 beta and the TNF-alpha genes. J. Immunol. 1994, 153, 5740–5749. [Google Scholar]
- Dumitru, C.D.; Ceci, J.D.; Tsatsanis, C.; Kontoyiannis, D.; Stamatakis, K.; Lin, J.-H.; Patriotis, C.; Jenkins, N.A.; Copeland, N.G.; Kollias, G.; et al. TNF-α Induction by LPS Is Regulated Posttranscriptionally via a Tpl2/ERK-Dependent Pathway. Cell 2000, 103, 1071–1083. [Google Scholar] [CrossRef] [Green Version]
- Wu, P.K.; Park, J.-I. MEK1/2 Inhibitors: Molecular Activity and Resistance Mechanisms. Semin. Oncol. 2015, 42, 849–862. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Han, G.; Wang, R.; Chen, G.; Xu, R.; Xiao, H.; Li, X.; Geng, S.; Li, Y.; Li, X.; et al. Regulation of IL-8 production by complement-activated product, C5a, in vitro and in vivo during sepsis. Clin. Immunol. 2010, 137, 157–165. [Google Scholar] [CrossRef]
- Brereton, C.F.; Sutton, C.E.; Lalor, S.; Lavelle, E.; Mills, K. Inhibition of ERK MAPK Suppresses IL-23- and IL-1-Driven IL-17 Production and Attenuates Autoimmune Disease. J. Immunol. 2009, 183, 1715–1723. [Google Scholar] [CrossRef]
- Schuh, K.; Pahl, A. Inhibition of the MAP kinase ERK protects from lipopolysaccharide-induced lung injury. Biochem. Pharmacol. 2009, 77, 1827–1834. [Google Scholar] [CrossRef] [Green Version]
- Han, J.; Liu, Y.; Yang, S.; Wu, X.; Li, H.; Wang, Q. MEK inhibitors for the treatment of non-small cell lung cancer. J. Hematol. Oncol. 2021, 14, 1. [Google Scholar] [CrossRef]
- Chen, S.; Xu, H.; Ye, P.; Wu, C.; Ding, X.; Zhang, H.; Zou, Y.; Zhao, J.; Le, S.; Wu, J.; et al. Trametinib alleviates lipopolysaccharide-induced acute lung injury by inhibiting the MEK-ERK-Egr-1 pathway. Int. Immunopharmacol. 2020, 80, 106152. [Google Scholar] [CrossRef] [PubMed]
- Roberts, P.J.; Der, C.J. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene 2007, 26, 3291–3310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ullah, R.; Yin, Q.; Snell, A.H.; Wan, L. RAF-MEK-ERK pathway in cancer evolution and treatment. Semin. Cancer Biol. 2021; in press. [Google Scholar] [CrossRef] [PubMed]
- Chin, H.M.; Lai, D.K.; Falchook, G.S. Extracellular Signal-Regulated Kinase (ERK) Inhibitors in Oncology Clinical Trials. J. Immunother. Precis. Oncol. 2020, 2, 10–16. [Google Scholar] [CrossRef]
- Wang, X.; Flynn, A.; Waskiewicz, A.J.; Webb, B.; Vries, R.G.; Baines, I.A.; Cooper, J.A.; Proud, C. The Phosphorylation of Eukaryotic Initiation Factor eIF4E in Response to Phorbol Esters, Cell Stresses, and Cytokines Is Mediated by Distinct MAP Kinase Pathways. J. Biol. Chem. 1998, 273, 9373–9377. [Google Scholar] [CrossRef]
- Carroll, M.; Borden, K.L. The Oncogene eIF4E: Using Biochemical Insights to Target Cancer. J. Interf. Cytokine Res. 2013, 33, 227–238. [Google Scholar] [CrossRef] [Green Version]
- Dreas, A.; Mikulski, M.; Milik, M.; Fabritius, C.-H.; Brzózka, K.; Rzymski, T.; Dreas, M.M.A. Mitogen-activated Protein Kinase (MAPK) Interacting Kinases 1 and 2 (MNK1 and MNK2) as Targets for Cancer Therapy: Recent Progress in the Development of MNK Inhibitors. Curr. Med. Chem. 2017, 24, 3025–3053. [Google Scholar] [CrossRef]
- Jin, X.; Yu, R.; Wang, X.; Proud, C.G.; Jiang, T. Progress in developing MNK inhibitors. Eur. J. Med. Chem. 2021, 219, 113420. [Google Scholar] [CrossRef]
- Yang, H.Y.; Chennamaneni, L.R.; Ho, M.W.T.; Ang, S.H.; Tan, E.S.W.; Jeyaraj, D.A.; Yeap, Y.S.; Liu, B.; Ong, E.H.; Joy, J.K.; et al. Optimization of Selective Mitogen-Activated Protein Kinase Interacting Kinases 1 and 2 Inhibitors for the Treatment of Blast Crisis Leukemia. J. Med. Chem. 2018, 61, 4348–4369. [Google Scholar] [CrossRef]
- Reich, S.H.; Sprengeler, P.A.; Chiang, G.G.; Appleman, J.R.; Chen, J.; Clarine, J.; Eam, B.; Ernst, J.T.; Han, Q.; Goel, V.K.; et al. Structure-based Design of Pyridone–Aminal eFT508 Targeting Dysregulated Translation by Selective Mitogen-activated Protein Kinase Interacting Kinases 1 and 2 (MNK1/2) Inhibition. J. Med. Chem. 2018, 61, 3516–3540. [Google Scholar] [CrossRef]
- Santag, S.; Siegel, F.; Wengner, A.M.; Lange, C.; Bömer, U.; Eis, K.; Pühler, F.; Lienau, P.; Bergemann, L.; Michels, M.; et al. BAY 1143269, a novel MNK1 inhibitor, targets oncogenic protein expression and shows potent anti-tumor activity. Cancer Lett. 2017, 390, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Buxadé, M.; Parra, J.L.; Rousseau, S.; Shpiro, N.; Marquez, R.; Morrice, N.; Bain, J.; Espel, E.; Proud, C.G. The Mnks Are Novel Components in the Control of TNFα Biosynthesis and Phosphorylate and Regulate hnRNP A1. Immunity 2005, 23, 177–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rowlett, R.M.; Chrestensen, C.A.; Nyce, M.; Harp, M.G.; Pelo, J.W.; Cominelli, F.; Ernst, P.B.; Pizarro, T.T.; Sturgill, T.W.; Worthington, M.T. MNK kinases regulate multiple TLR pathways and innate proinflammatory cytokines in macrophages. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 294, G452–G459. [Google Scholar] [CrossRef] [Green Version]
- Noubade, R.; Krementsov, D.N.; Del Rio, R.; Thornton, T.; Nagaleekar, V.K.; Saligrama, N.; Spitzack, A.; Spach, K.; Sabio, G.; Davis, R.J.; et al. Activation of p38 MAPK in CD4 T cells controls IL-17 production and autoimmune encephalomyelitis. Blood 2011, 118, 3290–3300. [Google Scholar] [CrossRef] [PubMed]
- Cherla, R.P.; Lee, S.-Y.; Mees, P.L.; Tesh, V.L. Shiga toxin 1-induced cytokine production is mediated by MAP kinase pathways and translation initiation factor eIF4E in the macrophage-like THP-1 cell line. J. Leukoc. Biol. 2006, 79, 397–407. [Google Scholar] [CrossRef] [PubMed]
- Hallberg, B.; Palmer, R.H. The role of the ALK receptor in cancer biology. Ann. Oncol. 2016, 27, iii4–iii15. [Google Scholar] [CrossRef]
- Katayama, R.; Lovly, C.; Shaw, A.T. Therapeutic Targeting of Anaplastic Lymphoma Kinase in Lung Cancer: A Paradigm for Precision Cancer Medicine. Clin. Cancer Res. 2015, 21, 2227–2235. [Google Scholar] [CrossRef] [Green Version]
- Kong, X.; Pan, P.; Sun, H.; Xia, H.; Wang, X.; Li, Y.; Hou, T. Drug Discovery Targeting Anaplastic Lymphoma Kinase (ALK). J. Med. Chem. 2019, 62, 10927–10954. [Google Scholar] [CrossRef]
- Gristina, V.; La Mantia, M.; Iacono, F.; Galvano, A.; Russo, A.; Bazan, V. The Emerging Therapeutic Landscape of ALK Inhibitors in Non-Small Cell Lung Cancer. Pharmaceuticals 2020, 13, 474. [Google Scholar] [CrossRef]
- Drew, L.; Cheng, J.; Engelman, J.; Ferguson, D.; Katayama, R.; McDermott, B.; Saeh, J.; Shaw, A.; Shen, M.; Widzowski, D.; et al. Abstract 919: AZD3463, a novel ALK/IGF1R inhibitor, overcomes multiple mechanisms of acquired resistance to crizotinib. Cancer Res. 2013, 73, 919. [Google Scholar] [CrossRef]
- Dayang, E.-Z.; Luxen, M.; Kuiper, T.; Yan, R.; Rangarajan, S.; van Meurs, M.; Moser, J.; Molema, G. Pharmacological inhibition of focal adhesion kinase 1 (FAK1) and anaplastic lymphoma kinase (ALK) identified via kinome profile analysis attenuates lipopolysaccharide-induced endothelial inflammatory activation. Biomed. Pharmacother. 2021, 133, 111073. [Google Scholar] [CrossRef] [PubMed]
- Kuenzi, B.; Rix, L.L.R.; Stewart, P.A.; Fang, B.; Kinose, F.; Bryant, A.; Boyle, T.A.; Koomen, J.M.; Haura, E.B.; Rix, U. Polypharmacology-based ceritinib repurposing using integrated functional proteomics. Nat. Chem. Biol. 2017, 13, 1222–1231. [Google Scholar] [CrossRef] [PubMed]
- Marsilje, T.H.; Pei, W.; Chen, B.; Lu, W.; Uno, T.; Jin, Y.; Jiang, T.; Kim, S.; Li, N.; Warmuth, M.; et al. Synthesis, Structure–Activity Relationships, and in Vivo Efficacy of the Novel Potent and Selective Anaplastic Lymphoma Kinase (ALK) Inhibitor 5-Chloro-N2-(2-isopropoxy-5-methyl-4-(piperidin-4-yl)phenyl)-N4-(2-(isopropylsulfonyl)phenyl)pyrimidine-2,4-diamine (LDK378) Currently in Phase 1 and Phase 2 Clinical Trials. J. Med. Chem. 2013, 56, 5675–5690. [Google Scholar] [CrossRef] [PubMed]
- Turnis, M.E.; Andrews, L.P.; Vignali, D.A.A. Inhibitory receptors as targets for cancer immunotherapy. Eur. J. Immunol. 2015, 45, 1892–1905. [Google Scholar] [CrossRef]
- Jacob, J.B.; Jacob, M.K.; Parajuli, P. Review of immune checkpoint inhibitors in immuno-oncology. In Advances in Pharmacology; Copple, B.L., Rockwell, C.E., Eds.; Academic Press: Cambridge, MA, USA, 2021; Chapter 3; pp. 111–139. [Google Scholar] [CrossRef]
- Huang, X.; Venet, F.; Wang, Y.L.; Lepape, A.; Yuan, Z.; Chen, Y.; Swan, R.; Kherouf, H.; Monneret, G.; Chung, C.-S.; et al. PD-1 expression by macrophages plays a pathologic role in altering microbial clearance and the innate inflammatory response to sepsis. Proc. Natl. Acad. Sci. USA 2009, 106, 6303–6308. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.; Chen, Y.; Chung, C.-S.; Yuan, Z.; Monaghan, S.F.; Wang, F.; Ayala, A. Identification of B7-H1 as a novel mediator of the innate immune/proinflammatory response as well as a possible myeloid cell prognostic biomarker in sepsis. J. Immunol. 2014, 192, 1091–1099. [Google Scholar] [CrossRef] [Green Version]
- Guignant, C.; Lepape, A.; Huang, X.; Kherouf, H.; Denis, L.; Poitevin, F.; Malcus, C.; Chéron, A.; Allaouchiche, B.; Gueyffier, F.; et al. Programmed death-1 levels correlate with increased mortality, nosocomial infection and immune dysfunctions in septic shock patients. Crit. Care 2011, 15, R99. [Google Scholar] [CrossRef] [Green Version]
- Patil, N.K.; Bohannon, J.K.; Sherwood, E.R. Immunotherapy: A promising approach to reverse sepsis-induced immunosuppression. Pharmacol. Res. 2016, 111, 688–702. [Google Scholar] [CrossRef]
- Rodrigues, P.R.; Picco, N.; Morgan, B.P.; Ghazal, P. Sepsis target validation for repurposing and combining complement and immune checkpoint inhibition therapeutics. Expert Opin. Drug Discov. 2021, 16, 537–551. [Google Scholar] [CrossRef]
- Patera, A.C.; Drewry, A.M.; Chang, K.; Beiter, E.R.; Osborne, D.; Hotchkiss, R.S. Frontline Science: Defects in immune function in patients with sepsis are associated with PD-1 or PD-L1 expression and can be restored by antibodies targeting PD-1 or PD-L1. J. Leukoc. Biol. 2016, 100, 1239–1254. [Google Scholar] [CrossRef]
- Chang, K.; Svabek, C.; Guillamet, M.C.V.; Sato, B.; Rasche, D.; Wilson, S.; Robbins, P.; Ulbrandt, N.; Suzich, J.; Green, J.; et al. Targeting the programmed cell death 1: Programmed cell death ligand 1 pathway reverses T cell exhaustion in patients with sepsis. Crit. Care 2014, 18, R3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mullard, A. Sepsis researchers set sights on immunotherapeutic strategies. Nat. Rev. Drug Discov. 2018, 17, 381–383. [Google Scholar] [CrossRef] [PubMed]
- Busch, L.M.; Sun, J.; Cui, X.; Eichacker, P.Q.; Torabi-Parizi, P. Checkpoint inhibitor therapy in preclinical sepsis models: A systematic review and meta-analysis. Intensive Care Med. Exp. 2020, 8, 7. [Google Scholar] [CrossRef] [PubMed]
- Vignon, P.; Laterre, P.-F.; Daix, T.; François, B. New Agents in Development for Sepsis: Any Reason for Hope? Drugs 2020, 80, 1751–1761. [Google Scholar] [CrossRef] [PubMed]
- Ho, J.S.Y.; Mok, B.W.-Y.; Campisi, L.; Jordan, T.; Yildiz, S.; Parameswaran, S.; Wayman, J.A.; Gaudreault, N.N.; Meekins, D.A.; Indran, S.V.; et al. TOP1 inhibition therapy protects against SARS-CoV-2-induced lethal inflammation. Cell 2021, 184, 2618–2632.e17. [Google Scholar] [CrossRef]
- Zhou, L.; Huntington, K.; Zhang, S.; Carlsen, L.; So, E.-Y.; Parker, C.; Sahin, I.; Safran, H.; Kamle, S.; Lee, C.-M.; et al. MEK inhibitors reduce cellular expression of ACE2, pERK, pRb while stimulating NK-mediated cytotoxicity and attenuating inflammatory cytokines relevant to SARS-CoV-2 infection. Oncotarget 2020, 11, 4201–4223. [Google Scholar] [CrossRef]
- Schreiber, A.; Viemann, D.; Schöning, J.; Schloer, S.; Zambrano, A.M.; Brunotte, L.; Faist, A.; Schöfbänker, M.; Hrincius, E.; Hoffmann, H.; et al. The MEK1/2-inhibitor ATR-002 efficiently blocks SARS-CoV-2 propagation and alleviates pro-inflammatory cytokine/chemokine responses. Cell. Mol. Life Sci. 2022, 79, 65. [Google Scholar] [CrossRef]
- Curtin, N.; Bányai, K.; Thaventhiran, J.; Le Quesne, J.; Helyes, Z.; Bai, P. Repositioning PARP inhibitors for SARS-CoV-2 infection(COVID-19): A new multi-pronged therapy for acute respiratory distress syndrome? Br. J. Pharmacol. 2020, 177, 3635–3645. [Google Scholar] [CrossRef]
- Fisher, C.J., Jr.; Agosti, J.M.; Opal, S.M.; Lowry, S.F.; Balk, R.A.; Sadoff, J.C.; Abraham, E.; Schein, R.M.; Benjamin, E.; for The Soluble TNF Receptor Sepsis Study Group. Treatment of septic shock with the tumor necrosis factor receptor:Fc fusion protein. N. Engl. J. Med. 1996, 334, 1697–1702. [Google Scholar] [CrossRef]
- López, A.; Lorente, J.A.; Steingrub, J.; Bakker, J.; McLuckie, A.; Willatts, S.; Brockway, M.; Anzueto, A.; Holzapfel, L.; Breen, D.; et al. Multiple-center, randomized, placebo-controlled, double-blind study of the nitric oxide synthase inhibitor 546C88: Effect on survival in patients with septic shock. Crit. Care Med. 2004, 32, 21–30. [Google Scholar] [CrossRef]
- Dellinger, R.P.; Parrillo, J.E. Mediator modulation therapy of severe sepsis and septic shock: Does it work? Crit. Care Med. 2004, 32, 282–286. [Google Scholar] [CrossRef] [PubMed]
- Rumienczyk, I.; Kulecka, M.; Ostrowski, J.; Mar, D.; Bomsztyk, K.; Standage, S.W.; Mikula, M. Multi-Organ Transcriptome Dynamics in a Mouse Model of Cecal Ligation and Puncture-Induced Polymicrobial Sepsis. J. Inflamm. Res. 2021, 14, 2377–2388. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Host Protein | Name of the Compound | Oncology Indication | Sepsis Model | Reference |
|---|---|---|---|---|
| TOPO1 | Topotecan Camptothecin | ovarian cancer, small cell lung cancer, cervical cancer | LPS S. aureus infection | [28] |
| PARP | Olaparib | ovarian cancer, breast cancer, prostate cancer, and pancreatic cancer | CLP LPS | [29,30] |
| MEK1/2 | Trametinib | melanoma | CLP LPS | [31,32] |
| ERK1/2 | SCH772984 | melanoma colon cancer | CLP LPS | [33] |
| MNK1/2 | Cpd 24/26 CGP57380 | breast cancer, colorectal cancer, diffuse large B cell lymphoma | LPS | [34,35] |
| ALK | Ceritinib | non-small-cell lung cancer | CLP LPS | [36,37] |
| PD-1 PD-L1 | Anti PD-1 Anti PD-L1 | cancers with high tumor mutational burden | CLP | [38,39,40] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rumienczyk, I.; Kulecka, M.; Statkiewicz, M.; Ostrowski, J.; Mikula, M. Oncology Drug Repurposing for Sepsis Treatment. Biomedicines 2022, 10, 921. https://doi.org/10.3390/biomedicines10040921
Rumienczyk I, Kulecka M, Statkiewicz M, Ostrowski J, Mikula M. Oncology Drug Repurposing for Sepsis Treatment. Biomedicines. 2022; 10(4):921. https://doi.org/10.3390/biomedicines10040921
Chicago/Turabian StyleRumienczyk, Izabela, Maria Kulecka, Małgorzata Statkiewicz, Jerzy Ostrowski, and Michal Mikula. 2022. "Oncology Drug Repurposing for Sepsis Treatment" Biomedicines 10, no. 4: 921. https://doi.org/10.3390/biomedicines10040921
APA StyleRumienczyk, I., Kulecka, M., Statkiewicz, M., Ostrowski, J., & Mikula, M. (2022). Oncology Drug Repurposing for Sepsis Treatment. Biomedicines, 10(4), 921. https://doi.org/10.3390/biomedicines10040921