
Hepatitis B Virus Variants with Multiple Insertions and/or Deletions in the X Open Reading Frame 3′ End: Common Members of Viral Quasispecies in Chronic Hepatitis B Patients

 , , ,
, , ,  , , , , , ,
, , , , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients and Samples
2.2. Serological and Virological Determinations
2.3. Amplification of HBV Genome Region Analyzed and Next-Generation Sequencing
2.4. Next-Generation Sequencing Data Treatment
2.5. Cloning to Confirm NGS Results
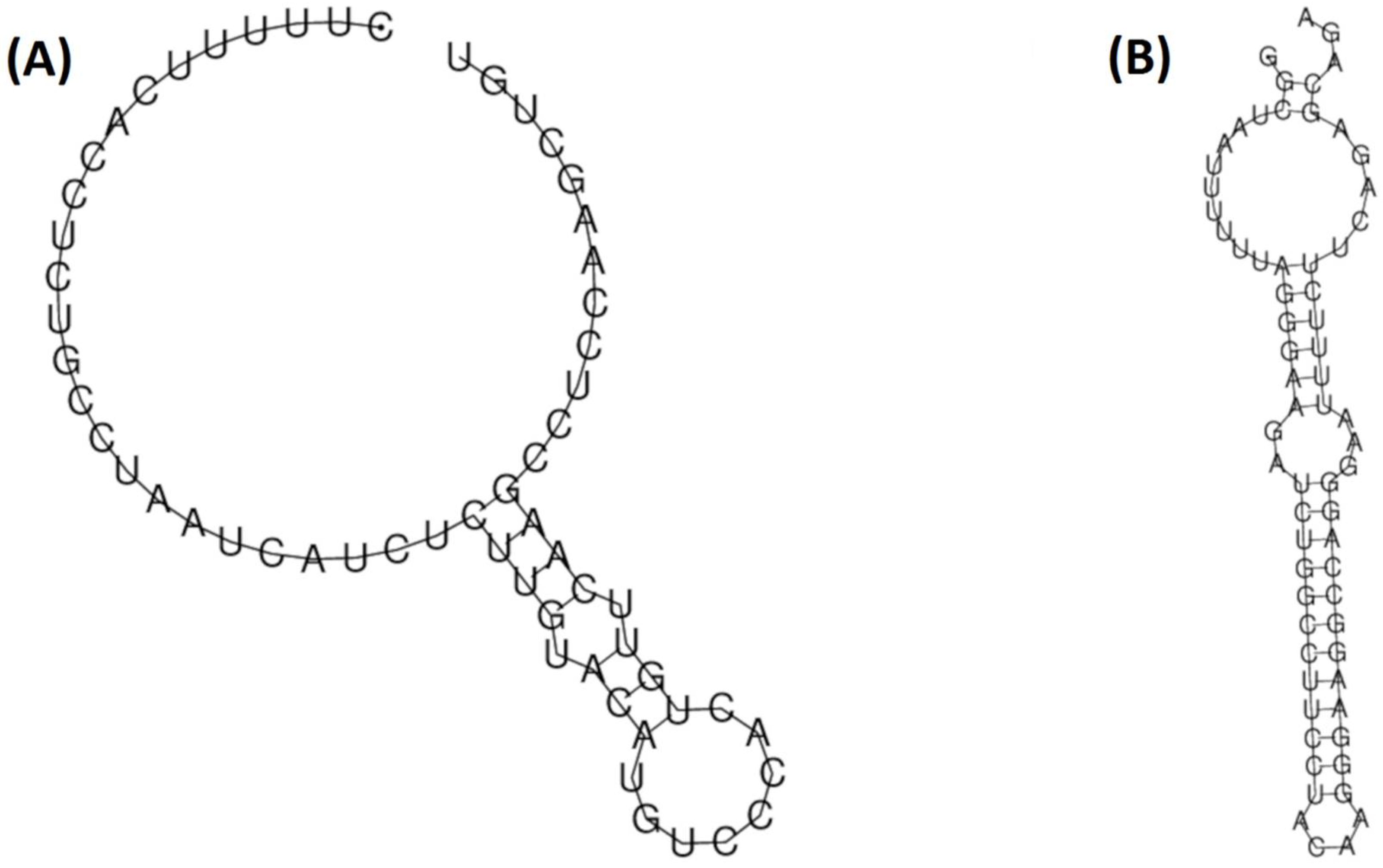
2.6. RNA Structural Modelling
2.7. Statistical Analysis
3. Results
3.1. Patients and Samples
3.2. Overview of Next-Generation Data Analyses
3.3. Alternative HBX Stop Codons
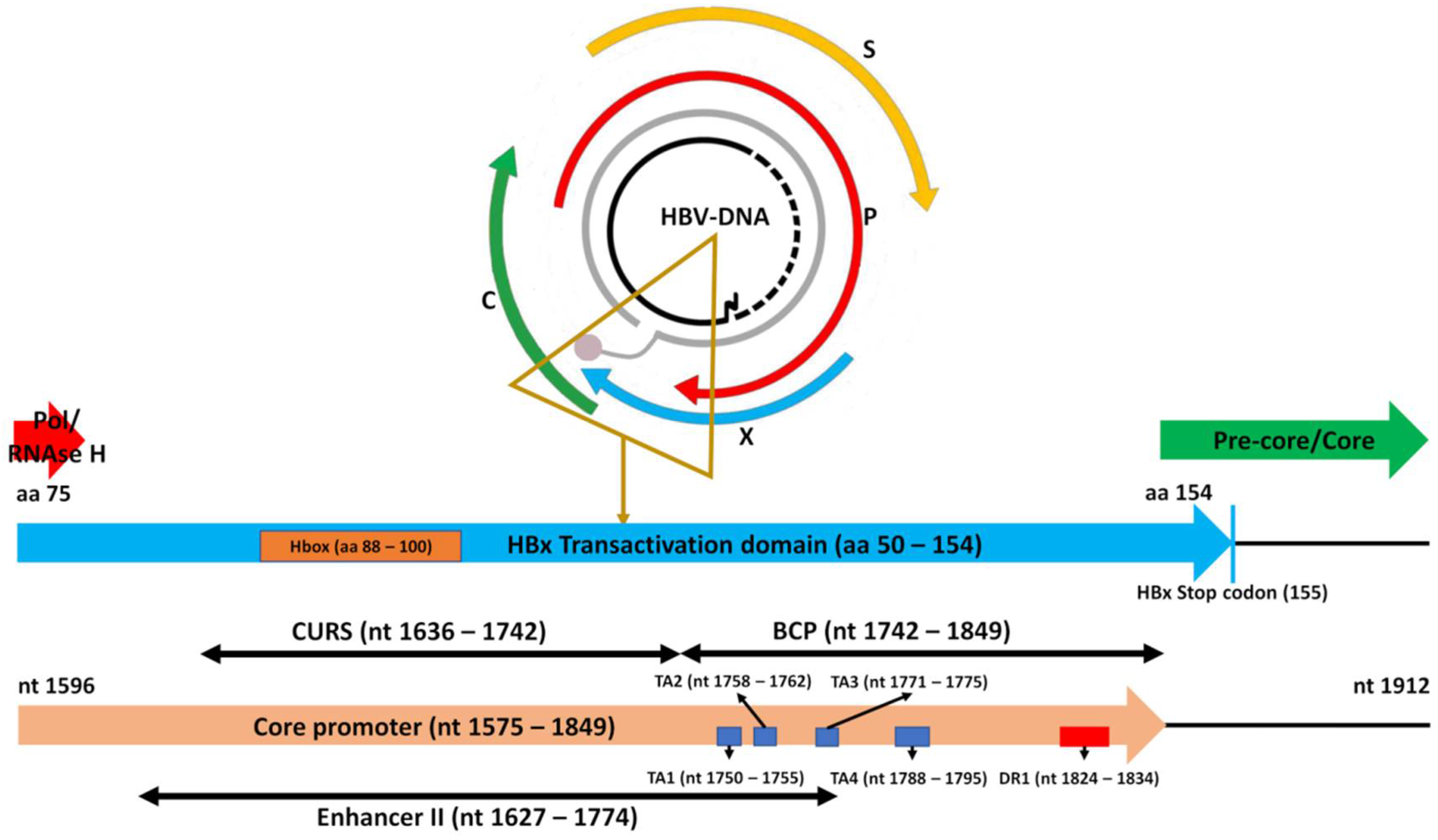
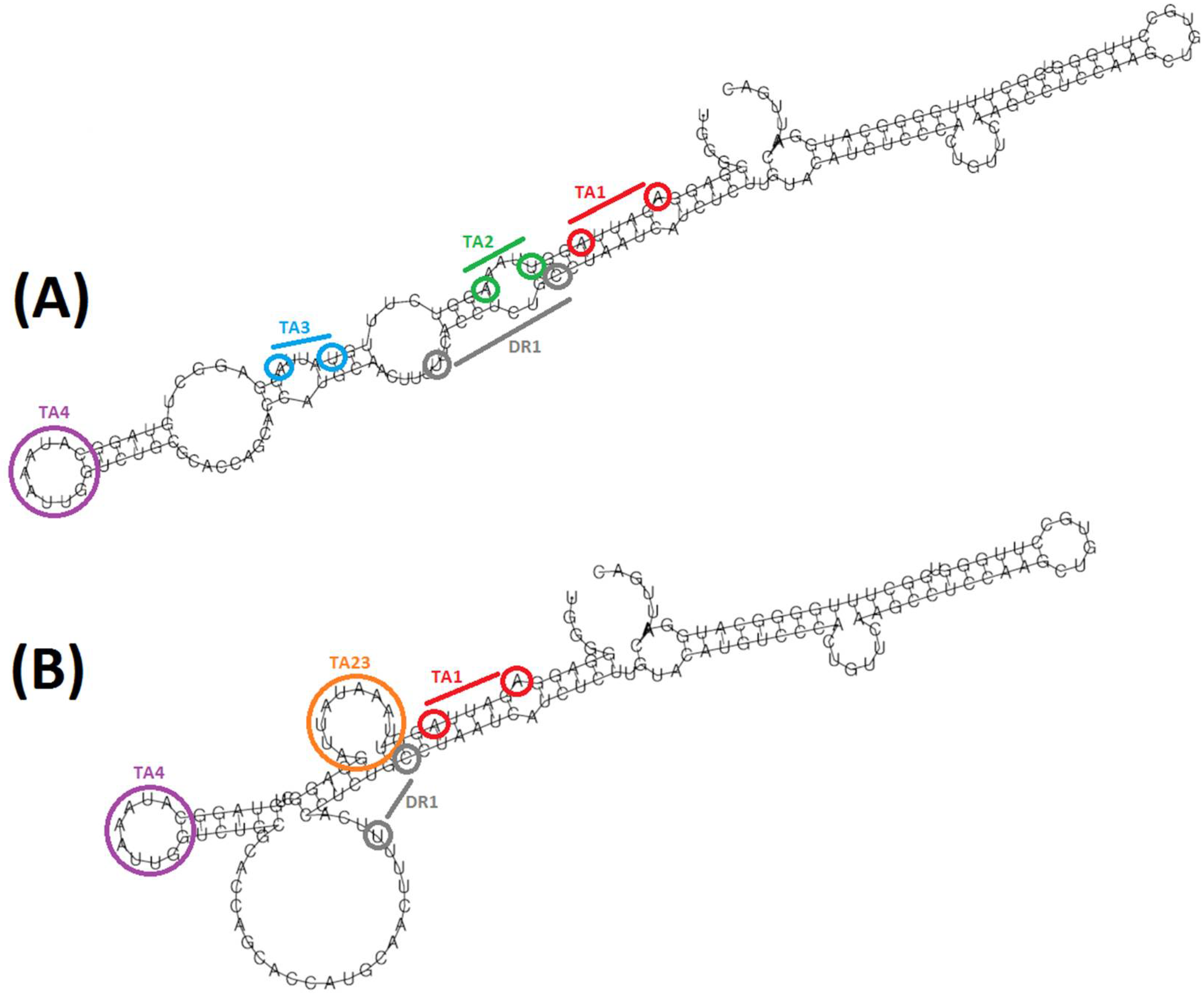
3.4. Indels Affecting the TATA-Like Box Region
3.5. Confirmation of Indel Variants by Cloning
3.6. Clinical Outcome of Patients Studied
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- World Health Organization. Hepatitis B. Available online: https://www.who.int/news-room/fact-sheets/detail/hepatitis-b (accessed on 3 March 2022).
- Martinez, M.G.; Testoni, B.; Zoulim, F. Biological Basis for Functional Cure of Chronic Hepatitis B. J. Viral Hepat. 2019, 26, 786–794. [Google Scholar] [CrossRef]
- Nassal, M. HBV CccDNA: Viral Persistence Reservoir and Key Obstacle for a Cure of Chronic Hepatitis B. Gut 2015, 64, 1972–1984. [Google Scholar] [CrossRef] [Green Version]
- Tu, T.; Budzinska, M.A.; Shackel, N.A.; Urban, S. HBV DNA Integration: Molecular Mechanisms and Clinical Implications. Viruses 2017, 9, 75. [Google Scholar] [CrossRef]
- Panjaworayan, N.; Roessner, S.K.; Firth, A.E.; Brown, C.M. HBVRegDB: Annotation, Comparison, Detection and Visualization of Regulatory Elements in Hepatitis B Virus Sequences. Virol. J. 2007, 4, 136. [Google Scholar] [CrossRef] [Green Version]
- Quarleri, J. Core Promoter: A Critical Region Where the Hepatitis B Virus Makes Decisions. World J. Gastroenterol. 2014, 20, 425–435. [Google Scholar] [CrossRef]
- Kramvis, A.; Kew, M.C. The Core Promoter of Hepatitis B Virus. J. Viral Hepat. 1999, 6, 415–427. [Google Scholar] [CrossRef]
- Levrero, M.; Zucman-Rossi, J. Mechanisms of HBV-Induced Hepatocellular Carcinoma. J. Hepatol. 2016, 64, S84–S101. [Google Scholar] [CrossRef]
- Xie, N.; Chen, X.; Zhang, T.; Liu, B.; Huang, C. Using Proteomics to Identify the HBx Interactome in Hepatitis B Virus: How Can This Inform the Clinic? Expert Rev. Proteom. 2014, 11, 59–74. [Google Scholar] [CrossRef]
- Kumar, V.; Jayasuryan, N.; Kumar, R. A Truncated Mutant (Residues 58–140) of the Hepatitis B Virus X Protein Retains Transactivation Function. Proc. Natl. Acad. Sci. USA 1996, 93, 5647–5652. [Google Scholar] [CrossRef] [Green Version]
- Zhang, A.Y.; Lai, C.L.; Poon, R.T.P.; Huang, F.Y.; Seto, W.K.; Fung, J.; Wong, D.K.H.; Yuen, M.F. Hepatitis B Virus Full-Length Genomic Mutations and Quasispecies in Hepatocellular Carcinoma. J. Gastroenterol. Hepatol. 2016, 31, 1638–1645. [Google Scholar] [CrossRef]
- Ning-Fang, M.; Lau, S.H.; Hu, L.; Xie, D.; Wu, J.; Yang, J.; Wang, Y.; Wu, M.C.; Fung, J.; Bai, X.; et al. COOH-Terminal Truncated HBV X Protein Plays Key Role in Hepatocarcinogenesis. Clin. Cancer Res. 2008, 14, 5061–5068. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Frias, F.; Buti, M.; Tabernero, D.; Homs, M. Quasispecies Structure, Cornerstone of Hepatitis B Virus Infection: Mass Sequencing Approach. World J. Gastroenterol. 2013, 19, 6995–7023. [Google Scholar] [CrossRef]
- Günther, S.; Piwon, N.; Iwanska, A.; Schilling, R.; Meisel, H.; Will, H. Type, Prevalence, and Significance of Core Promoter/Enhancer II Mutations in Hepatitis B Viruses from Immunosuppressed Patients with Severe Liver Disease. J. Virol. 1996, 70, 8318–8331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Preikschat, P.; Günther, S.; Reinhold, S.; Will, H.; Budde, K.; Neumayer, H.H.; Krüger, D.H.; Meisel, H. Complex HBV Populations with Mutations in Core Promoter, C Gene, and Pre-S Region Are Associated with Development of Cirrhosis in Long-Term Renal Transplant Recipients. Hepatology 2002, 35, 466–477. [Google Scholar] [CrossRef]
- Peng, Y.; Liu, B.; Hou, J.; Sun, J.; Hao, R.; Xiang, K.; Yan, L.; Zhang, J.; Zhuang, H.; Li, T. Naturally Occurring Deletions/Insertions in HBV Core Promoter Tend to Decrease in Hepatitis B e Antigen-Positive Chronic Hepatitis B Patients during Antiviral Therapy. Antivir. Ther. 2015, 20, 623–632. [Google Scholar] [CrossRef] [Green Version]
- Hao, R.; Xiang, K.; Peng, Y.; Hou, J.; Sun, J.; Li, Y.; Su, M.; Yan, L.; Zhuang, H.; Li, T. Naturally Occurring Deletion/Insertion Mutations within HBV Whole Genome Sequences in HBeAg-Positive Chronic Hepatitis B Patients Are Correlated with Baseline Serum HBsAg and HBeAg Levels and Might Predict a Shorter Interval to HBeAg Loss and Seroconversi. Infect. Genet. Evol. 2015, 33, 261–268. [Google Scholar] [CrossRef]
- Caballero, A.; Gregori, J.; Homs, M.; Tabernero, D.; Gonzalez, C.; Quer, J.; Blasi, M.; Casillas, R.; Nieto, L.; Riveiro-Barciela, M.; et al. Complex Genotype Mixtures Analyzed by Deep Sequencing in Two Different Regions of Hepatitis B Virus. PLoS ONE 2015, 10, e0144816. [Google Scholar] [CrossRef]
- Ramírez, C.; Gregori, J.; Buti, M.; Tabernero, D.; Camós, S.; Casillas, R.; Quer, J.; Esteban, R.; Homs, M.; Rodríguez-Frías, F.; et al. A Comparative Study of Ultra-Deep Pyrosequencing and Cloning to Quantitatively Analyze the Viral Quasispecies Using Hepatitis B Virus Infection as a Model. Antivir. Res. 2013, 98, 273–283. [Google Scholar] [CrossRef]
- Gregori, J.; Esteban, J.I.; Cubero, M.; Garcia-Cehic, D.; Perales, C.; Casillas, R.; Alvarez-Tejado, M.; Rodríguez-Frías, F.; Guardia, J.; Domingo, E.; et al. Ultra-Deep Pyrosequencing (UDPS) Data Treatment to Study Amplicon HCV Minor Variants. PLoS ONE 2013, 8, e83361. [Google Scholar] [CrossRef]
- Edgar, R.C.; Drive, R.M.; Valley, M. MUSCLE: Multiple Sequence Alignment with High Accuracy and High Throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [Green Version]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing. Vienna, Austria, 2013. Available online: https://www.yumpu.com/en/document/view/6853895/r-a-language-and-environment-for-statistical-computing (accessed on 18 May 2022).
- Pages, H.; Aboyoun, P.; Gentleman, R.; DebRoy, S. Biostrings: String Objects Representing Biological Sequences, and Matching Algorithms; R Package Version. 2.31.14; Fred Hutchinson Cancer Research Center: Seattle, WA, USA, 2011. [Google Scholar]
- Paradis, E.; Claude, J.; Strimmer, K. APE: Analyses of Phylogenetics and Evolution in R Language. Bioinformatics 2004, 20, 289–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gruber, A.R.; Lorenz, R.; Bernhart, S.H.; Neuböck, R.; Hofacker, I.L. The Vienna RNA Websuite. Nucleic Acids Res. 2008, 36, 70–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- R Core Team. A Language and Environment for Statistical Computing; R Foundation for Statistical Computing. Vienna, Austria, 2021. Available online: https://cran.asia/web/packages/dplR/vignettes/chron-dplR.pdf (accessed on 18 May 2022).
- Li, T.; Robert, E.I.; van Breugel, P.C.; Strubin, M.; Zheng, N. A Promiscuous Alpha-Helical Motif Anchors Viral Hijackers and Substrate Receptors to the CUL4-DDB1 Ubiquitin Ligase Machinery. Nat. Struct. Mol. Biol. 2010, 17, 105–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Firth, A.E.; Brierley, I. Non-Canonical Translation in RNA Viruses. J. Gen. Virol. 2012, 93, 1385–1409. [Google Scholar] [CrossRef] [PubMed]
- Mouzakis, K.D.; Lang, A.L.; Vander Meulen, K.A.; Easterday, P.D.; Butcher, S.E. HIV-1 Frameshift Efficiency Is Primarily Determined by the Stability of Base Pairs Positioned at the MRNA Entrance Channel of the Ribosome. Nucleic Acids Res. 2013, 41, 1901–1913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belew, A.T.; Meskauskas, A.; Musalgaonkar, S.; Adnavi, V.M.; Sulima, S.O.; Kasprzak, W.K.; Shapiro, B.A.; Dinman, J. Ribosomal Frameshifting in the CCR5 MRNA Is Regulated by MiRNAs and the NMD Pathway. Nature 2014, 512, 265–269. [Google Scholar] [CrossRef] [PubMed]
- Murphy, C.M.; Xu, Y.; Li, F.; Nio, K.; Reszka-Blanco, N.; Li, X.; Wu, Y.; Yu, Y.; Xiong, Y.; Su, L. Hepatitis B Virus X Protein Promotes Degradation of SMC5/6 to Enhance HBV Replication. Cell Rep. 2016, 16, 2846–2854. [Google Scholar] [CrossRef] [Green Version]
- Choi, B.H.; Park, G.T.; Rho, H.M. Interaction of Hepatitis B Viral X Protein and CCAAT/ Enhancer-Binding Protein Alpha Synergistically Activates the Hepatitis B Viral Enhancer II/Pregenomic Promoter. J. Biol. Chem. 1999, 274, 2858–2865. [Google Scholar] [CrossRef] [Green Version]
- Lee, T.Y.; Chang, W.C.; Hsu, J.B.K.; Chang, T.H.; Shien, D.M. GPMiner: An Integrated System for Mining Combinatorial Cis-Regulatory Elements in Mammalian Gene Group. Ser. Adv. Bioinform. Comput. Biol. 2012, 13, S3. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Zhang, D.; Li, Y.; Jiang, D.; Luo, S.; Du, N.; Chen, W.; Deng, L.; Zeng, C. Whole Genome Characterization of Hepatitis B Virus Quasispecies with Massively Parallel Pyrosequencing. Clin. Microbiol. Infect. 2015, 21, 280–287. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Garcia, S.; Cortese, M.F.; Rodríguez-Algarra, F.; Tabernero, D.; Rando-Segura, A.; Quer, J.; Buti, M.; Rodríguez-Frías, F. Next-Generation Sequencing for the Diagnosis of Hepatitis B: Current Status and Future Prospects. Expert Rev. Mol. Diagn. 2021, 21, 381–396. [Google Scholar] [CrossRef] [PubMed]
- Lucifora, J.; Arzberger, S.; Durantel, D.; Belloni, L.; Strubin, M.; Levrero, M.; Zoulim, F.; Hantz, O.; Protzer, U. Hepatitis B Virus X Protein Is Essential to Initiate and Maintain Virus Replication after Infection. J. Hepatol. 2011, 55, 996–1003. [Google Scholar] [CrossRef] [PubMed]
- Pál, J.; Nyárády, Z.; Marczinovits, I.; Pár, A.; Ali, Y.S.; Berencsi, G.; Kvell, K.; Németh, P. Comprehensive Regression Analysis of Hepatitis B Virus X Antigen Level and Anti-HBx Antibody Titer in the Sera of Patients with HBV Infection. Pathol. Oncol. Res. 2006, 12, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Feitelson, M.A. Products of the “X” Gene in Hepatitis B and Related Viruses. Hepatology 1986, 6, 191–198. [Google Scholar] [CrossRef]
- Kim, S.K.; Jang, S.K.; Rho, H.M. Effect of Frameshift Mutation in the Pre-C Region of Hepatitis B Virus on the X and C Genes. J. Gen. Virol. 1994, 75, 917–923. [Google Scholar] [CrossRef]
- Deng, H.; Dong, J.; Cheng, J.; Huangfu, K.J.; Shi, S.S.; Hong, Y.; Ren, X.M.; Li, L. Quasispecies Groups in the Core Promoter Region of Hepatitis B Virus. Hepatobiliary Pancreat. Dis. Int. 2002, 1, 392–396. [Google Scholar]
- Zhang, D.; Dong, P.; Zhang, K.; Deng, L.; Bach, C.; Chen, W.; Li, F.; Protzer, U.; Ding, H.; Zeng, C. Whole Genome HBV Deletion Profiles and the Accumulation of PreS Deletion Mutant during Antiviral Treatment. BMC Microbiol. 2012, 12, 307. [Google Scholar] [CrossRef] [Green Version]
- Ponjavic, J.; Lenhard, B.; Kai, C.; Kawai, J.; Carninci, P.; Hayashizaki, Y.; Sandelin, A. Transcriptional and Structural Impact of TATA-Initiation Site Spacing in Mammalian Core Promoters. Genome Biol. 2006, 7, R78. [Google Scholar] [CrossRef] [Green Version]
- Angers, S.; Li, T.; Yi, X.; MacCoss, M.J.; Moon, R.T.; Zheng, N. Molecular Architecture and Assembly of the DDB1-CUL4A Ubiquitin Ligase Machinery. Nature 2006, 443, 590–593. [Google Scholar] [CrossRef]
- Li, W.; Li, M.; Liao, D.; Lu, X.; Gu, X.; Zhang, Q. Carboxyl-Terminal Truncated HBx Contributes to Invasion and Metastasis via Deregulating Metastasis Suppressors in Hepatocellular Carcinoma. Oncotarget 2016, 7, 55110–55127. [Google Scholar] [CrossRef] [Green Version]
- Tu, H.; Bonura, C.; Giannini, C.; Mouly, H.; Soussan, P.; Kew, M.; Paterlini-Bréchot, P.; Bréchot, C.; Kremsdorf, D. Biological Impact of Natural COOH-Terminal Deletions of Hepatitis B Virus X Protein in Hepatocellular Carcinoma Tissues. Cancer Res. 2001, 61, 7803–7810. [Google Scholar] [PubMed]
- Tautz, N.; Meyers, G.; Stark, R.; Dubovi, E.J.; Thiel, H.J. Cytopathogenicity of a Pestivirus Correlates with a 27-Nucleotide Insertion. J. Virol. 1996, 70, 7851–7858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, J.X.; Peng, D.X.; Liu, Y.L.; Wu, Y.T.; Liu, X.F. Virulence of H5N1 Avian Influenza Virus Enhanced by a 15-Nucleotide Deletion in the Viral Nonstructural Gene. Virus Genes 2008, 36, 471–478. [Google Scholar] [CrossRef] [PubMed]
- Andres, C.; Garcia-Cehic, D.; Gregori, J.; Piñana, M.; Rodriguez-Frias, F.; Guerrero-Murillo, M.; Esperalba, J.; Rando, A.; Goterris, L.; Codina, M.G.; et al. Naturally Occurring SARS-CoV-2 Gene Deletions Close to the Spike S1/S2 Cleavage Site in the Viral Quasispecies of COVID19 Patients. Emerg. Microbes Infect. 2020, 9, 1900–1911. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Patients (N = 50) | |
|---|---|
| Age (median years, IQR) | 43 (35–55) |
| Gender (N Male, %) | 42 (84) |
| HBV-DNA (median logIU/mL, IQR) | 5.9 (5.3–7.9) |
| HBV genotype A (N, %) D (N, %) F (N, %) | |
| 39 (78) | |
| 8 (16) | |
| 3 (6) | |
| HBeAg-negative (N, %) | 33 (66) |
| ALT (median IU/L, IQR) | 80 (57–115) |
| Liver fibrosis 1 ≤F3 (%) >F3 (%) | |
| 45 (90) | |
| 5 (10) | |
| Treatment history Previous NA (N, %) Previous IFN (N, %) | |
| 17 (34) | |
| 2 (4) |
| ID | Deletions | Insertions | N Patients (%) | Median % Reads/Patient (IQR) | Median % HPL/Patient (IQR) |
|---|---|---|---|---|---|
| 11 | 1646 | - | 6 (12) | 0.5 (0.4–0.6) | 6.5 (2.8–10.4) |
| 30 | 1692 | 1697TT | 6 (12) | 0.5 (0.4–0.6) | 7.5 (6.1–14.6) |
| 37 | - | 1739G | 9 (18) | 0.3 (0.3–0.5) | 8.3 (7.1–14.3) |
| 38 | - | 1746G/T | 7 (14) | 0.4 (0.3–0.4) | 10 (6.7–12.7) |
| 40 | 1749 | - | 7 (14) | 0.4 (0.3–0.5) | 6.7 (5.2–11.3) |
| 51 | 1763–1770 | - | 10 (20) | 1.7 (0.9–2.1) | 5.5 (3.0–8.2) |
| 59 | - | 1781C | 5 (10) | 0.6 (0.6–0.7) | 8.3 (4.8–8.3) |
| 74 | - | 1820C | 7 (14) | 0.4 (0.4–0.9) | 4.3 (1.9–4.8) |
| 84 | - | 1825T | 19 (38) | 1.5 (0.7–2.1) | 4.8 (2.8–6.9) |
| 85 | 1825 | - | 10 (20) | 0.4 (0.4–0.8) | 4.1 (2.8–5.1) |
| 88 | - | 1826C/T | 9 (18) | 0.6 (0.4–0.9) | 4.8 (2.6–8.3) |
| 103 | - | 1838A | 5 (10) | 1.2 (0.9–2.7) | 2.3 (2.2–5.9) |
| HBX Stop Codon | IDs | N Patients (%) | Median % Reads/Patient (IQR) | Median % HPL/Patient (IQR) |
|---|---|---|---|---|
| 95 | 2, 3, 7, 8 | 5 (10) | 0.4 (0.3–0.5) | 5.9 (5.3–8.3) |
| 109 | 28, 29, 30, NA | 9 (18) | 0.5 (0.4–0.6) | 6.7 (4.3–16.7) |
| 125 | 18, NA | 8 (16) | 1.9 (0.6–3.8) | 3.8 (2.2–5.8) |
| 128 | 37, 38, 41 | 15 (30) | 0.4 (0.3–0.6) | 8.3 (5.6–18.3) |
| 129 | 6, 11, 23, 25, 34, 35, 36, 39, 40 | 21 (42) | 0.5 (0.4–0.6) | 7.1 (4.8–11.1) |
| 132 | 45, 46, 49, 57 | 5 (10) | 0.5 (0.5–0.8) | 2.6 (2.6–4.3) |
| 135 | 14, 15, 22, 47, 48, 51, 53, 55, 80, 81, 87, 90, 100 | 14 (28) | 2.0 (0.5–4.9) | 6.1 (3.0–11.8) |
| 138 | 59 | 5 (10) | 0.6 (0.6–0.7) | 8.3 (4.8–8.3) |
| 149 | 63, 72 | 5 (10) | 0.7 (0.4–1.0) | 4.5 (4.2–4.8) |
| 156 | 12, 13, 27, 58, 76, 82, 89 | 7 (14) | 2.6 (0.7–7.3) | 7.1 (5.5–7.7) |
| 179 | 85 | 10 (20) | 0.4 (0.4–0.8) | 4.1 (2.8–5.1) |
| 180 * | 75, 86, 98 | 5 (10) | 0.9 (0.6–1.2) | 5.1 (3.6–5.9) |
| 207 * | 101, NA | 6 (12) | 0.5 (0.4–0.6) | 3 (2.4–4.4) |
| 360 * | 70, 92 | 5 (10) | 0.6 (0.6–1.1) | 2.6 (2.3–4.3) |
| 362 * | 74, 78, 84, 88 | 24 (48) | 1.6 (0.8–2.1) | 5.8 (3.1–10.7) |
| TA Alteration | Cause of Alteration and Indels Involved | N Patients (%) | Median % Reads/Patient (IQR) | Median % HPL/Patient (IQR) |
|---|---|---|---|---|
| Insertion in TA1 (nt 1750–1755) | Ins: ID: 41 (Ins 1751G) | 3 (6) | 0.3–0.4 * | 3.4–5.9 * |
| Partial or total TA1 or TA2 (nt 1758–1762) elimination | 7 to 10 nt Del between nt 1754 and 1767: Dels: ID: 42, 45, 47, 48, 49, 50 Ins + Del: ID: 16 | 5 (10) | 0.5 (0.4–2.0) | 3.6 (2.2–4.5) |
| Partial or total TA2 + TA3 (nt 1758–1775) elimination | Dels between nt 1756 and 1787: Dels: ID: 43, 44, 46 Ins + Dels: ID: 9, 12, 13, 14, 15, 17, 18, 19, 20, 22, 26, 27 | 3 (6) | 0.9–71.2 * | 2.3–61.8 * |
| TA2 + TA3 Fusion | 8 nt Del between nt 1763 and 1770: Del: ID: 51 Ins + Dels: ID: 24, 80, 81, 87, 90, 100 | 10 (20) | 1.8 (0.9–4.3) | 5.5 (3.0–17.5) |
| Partial or total TA3 (nt 1771–1775) elimination | 8 to 10 nt Del between nt 1763 and 1776 Dels: ID: 52, 53, 55, 56, 57 | 7 (14) | 0.4 (0.4–2.1) | 2.6 (2.4–5.6) |
| No TA affected | Ins: ID: 58 (Ins 1768GTT/ATT), 59 (Ins 1781C), 60 (Ins 1785C) Del: ID: 54 (Del 1766) | 10 (20) | 0.6 (0.4–0.9) | 6.5 (4.3–8.3) |
| Patient (N Clones Analyzed) | Deletions | Insertions | N Clones (%) | ID | N Reads (%) |
|---|---|---|---|---|---|
| 2 (24) | 1627 + 1758 − 1777 | 1647 TCTTACATAAGAGGACTCTTGGAC | 12 (50) | 12 | 9554 (51.9) |
| - | 1820 C | 1 (4.2) | 74 | 75 (0.4) | |
| 1627 + 1726 + 1758 − 1777 | 1647 TCTTACATAAGAGGACTCTTGGAC | 2 (8) | - | - | |
| 1627 + 1758 − 1777 | 1647 TCTTACATAAGAGGACTCTTGGAC + 1822 ATTCAA + 1825 T | 1 (4.2) | - | - | |
| 1627 | 1600T + 1647 TCTTACATAAGAGGACTCTTGGAC | 1 (4.2) | - | - | |
| - | - | 7 (29.2) | - | 4692 (25.5) | |
| 17 (29) | - | 1825 T | 2 (6.9) | 84 | 3140 (14.2) |
| - | 1909 TG | 1 (3.4) | * | * | |
| - | - | 26 (89.7) | - | 18736 (84.8) | |
| 20 (18) | 1825 | - | 1 (5.6) | 85 | 3011 (15.5) |
| - | 1826 TTC | 6 (33.3) | 89 | 2171 (11.2) | |
| - | - | 11 (61) | - | 14064 (72.5) | |
| 39 (19) | - | 1605 T | 1 (5.3) | - | - |
| - | 1825 T | 7 (36.8) | 84 | 3242 (22.9) | |
| - | 1895 T | 1 (5.3) | * | * | |
| - | - | 10 (52.6) | - | 9701 (68.5) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
García-García, S.; Caballero-Garralda, A.; Tabernero, D.; Cortese, M.F.; Gregori, J.; Rodriguez-Algarra, F.; Quer, J.; Riveiro-Barciela, M.; Homs, M.; Rando-Segura, A.; et al. Hepatitis B Virus Variants with Multiple Insertions and/or Deletions in the X Open Reading Frame 3′ End: Common Members of Viral Quasispecies in Chronic Hepatitis B Patients. Biomedicines 2022, 10, 1194. https://doi.org/10.3390/biomedicines10051194
García-García S, Caballero-Garralda A, Tabernero D, Cortese MF, Gregori J, Rodriguez-Algarra F, Quer J, Riveiro-Barciela M, Homs M, Rando-Segura A, et al. Hepatitis B Virus Variants with Multiple Insertions and/or Deletions in the X Open Reading Frame 3′ End: Common Members of Viral Quasispecies in Chronic Hepatitis B Patients. Biomedicines. 2022; 10(5):1194. https://doi.org/10.3390/biomedicines10051194
Chicago/Turabian StyleGarcía-García, Selene, Andrea Caballero-Garralda, David Tabernero, Maria Francesca Cortese, Josep Gregori, Francisco Rodriguez-Algarra, Josep Quer, Mar Riveiro-Barciela, Maria Homs, Ariadna Rando-Segura, and et al. 2022. "Hepatitis B Virus Variants with Multiple Insertions and/or Deletions in the X Open Reading Frame 3′ End: Common Members of Viral Quasispecies in Chronic Hepatitis B Patients" Biomedicines 10, no. 5: 1194. https://doi.org/10.3390/biomedicines10051194
APA StyleGarcía-García, S., Caballero-Garralda, A., Tabernero, D., Cortese, M. F., Gregori, J., Rodriguez-Algarra, F., Quer, J., Riveiro-Barciela, M., Homs, M., Rando-Segura, A., Pacin-Ruiz, B., Vila, M., Ferrer-Costa, R., Pumarola, T., Buti, M., & Rodriguez-Frias, F. (2022). Hepatitis B Virus Variants with Multiple Insertions and/or Deletions in the X Open Reading Frame 3′ End: Common Members of Viral Quasispecies in Chronic Hepatitis B Patients. Biomedicines, 10(5), 1194. https://doi.org/10.3390/biomedicines10051194