
Identification of the Potential Molecular Mechanisms Linking RUNX1 Activity with Nonalcoholic Fatty Liver Disease, by Means of Systems Biology

 ,
,  , , ,
, , ,
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Bibliographic and Metadata Analysis in Databases
2.2. Mechanistic Model Generation
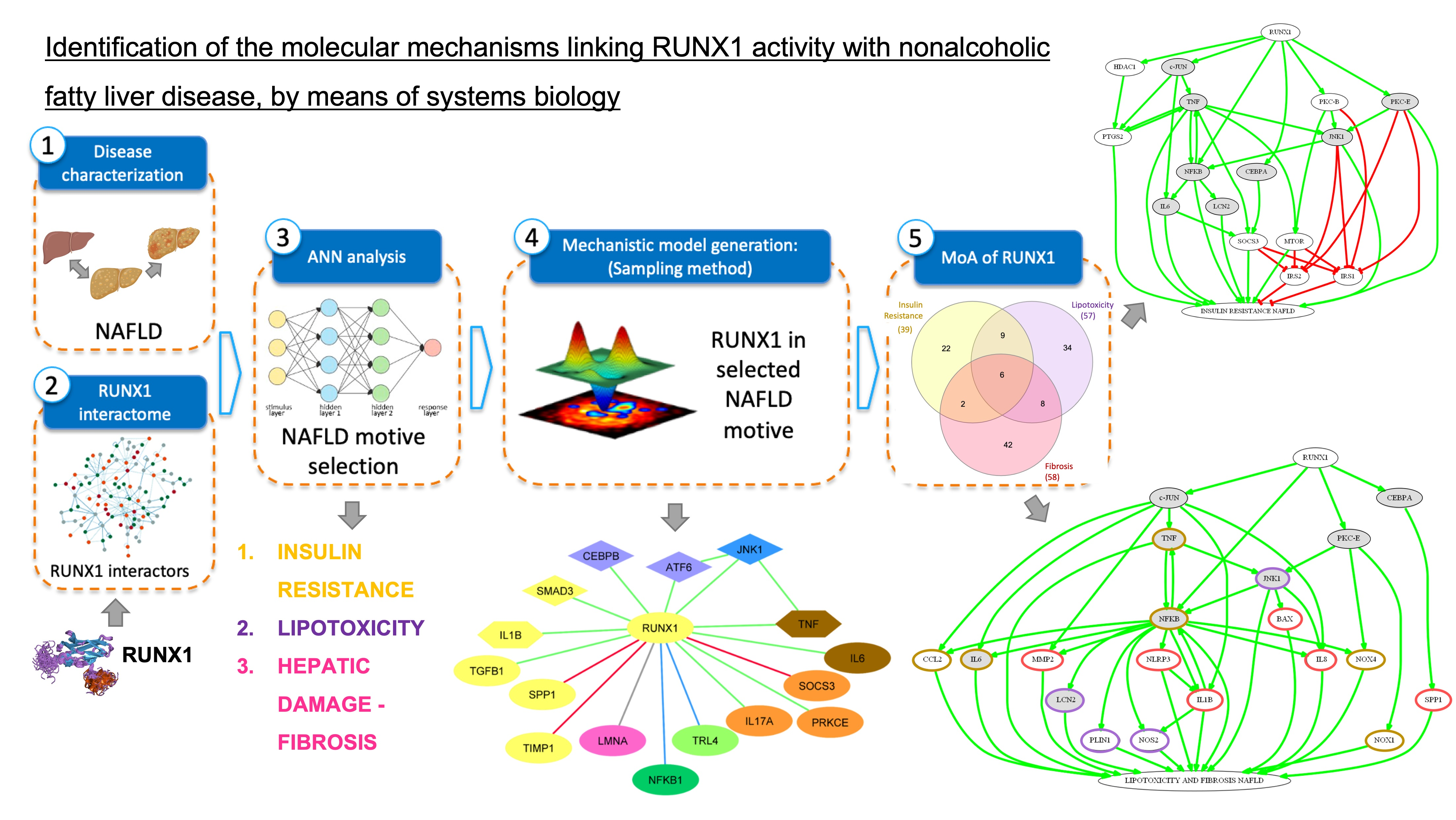
- Activation of RUNX1 promoting insulin resistance (IR).
- Activation of RUNX1 promoting lipotoxicity and hepatic injury and liver fibrosis.
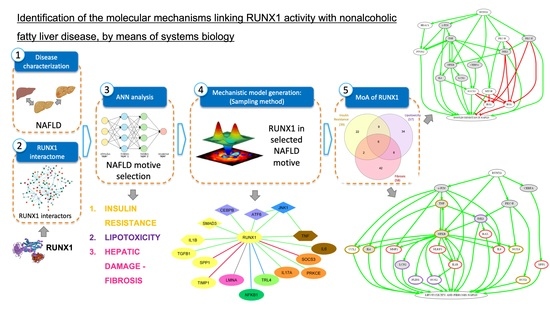
3. Results
3.1. Functional Relationship between RUNX1 and NAFLD: ANNs Analysis
3.2. Mechanisms of Action of RUNX1
3.2.1. Mechanism of Action of RUNX1 Promoting IR
3.2.2. Mechanism of Action of RUNX1 Promoting Lipotoxicity and Hepatic Injury-Liver Fibrosis
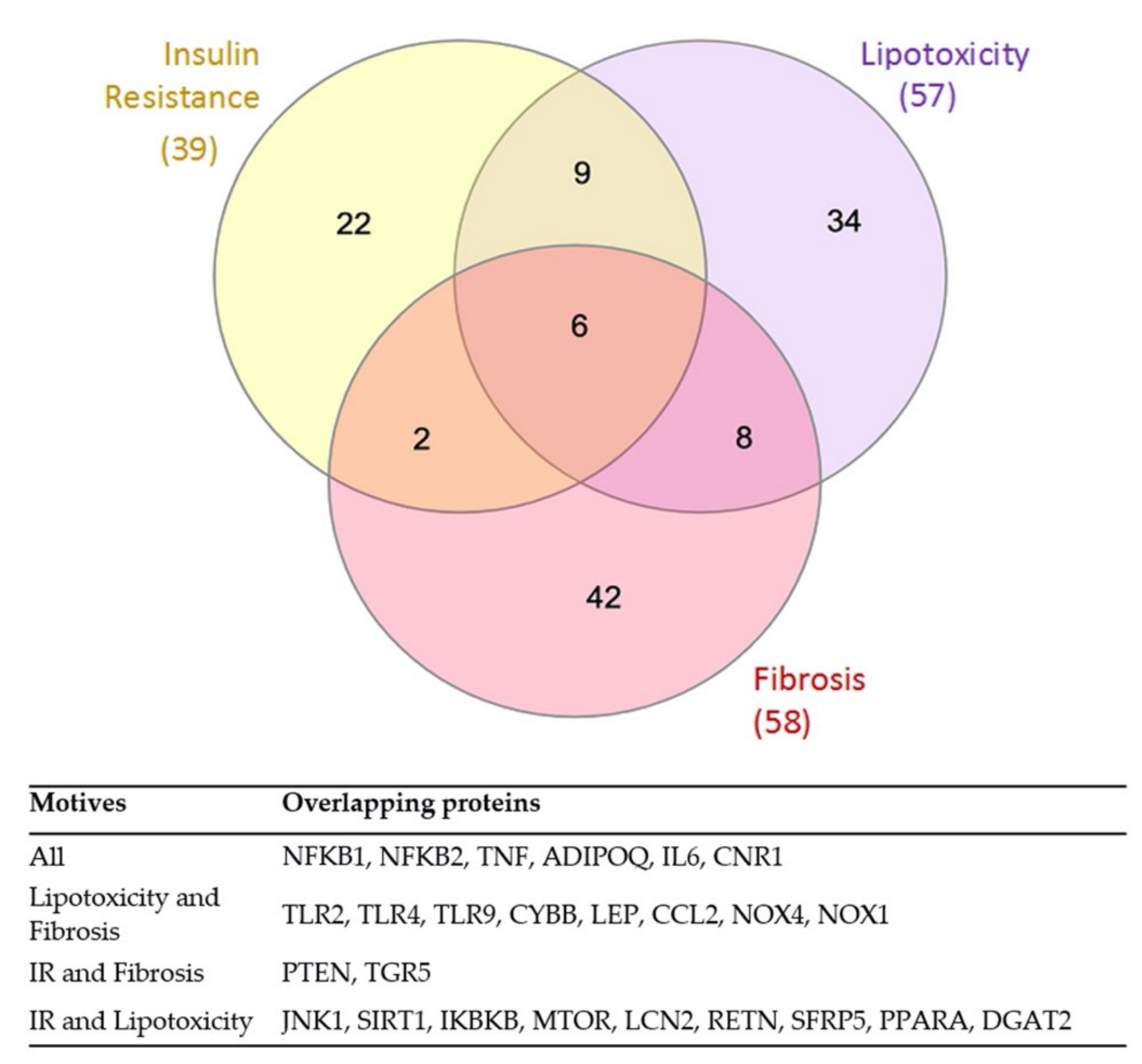
3.3. Overlap between the Mechanistic Pathways Modulated by RUNX1 Activation in IR and Lipotoxicity & Fibrosis Stimulation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A
The Sampling Methods
References
- Arciello, M.; Gori, M.; Maggio, R.; Barbaro, B.; Tarocchi, M.; Galli, A.; Balsano, C. Environmental Pollution: A Tangible Risk for NAFLD Pathogenesis. Int. J. Mol. Sci. 2013, 14, 22052–22066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarwar, R.; Pierce, N.; Koppe, S. Obesity and Nonalcoholic Fatty Liver Disease: Current Perspectives. DMSO 2018, 11, 533–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larter, C.Z.; Chitturi, S.; Heydet, D.; Farrell, G.C. A Fresh Look at NASH Pathogenesis. Part 1: The Metabolic Movers. J. Gastroenterol. Hepatol. 2010, 25, 672–690. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.C.; Horton, J.D.; Hobbs, H.H. Human Fatty Liver Disease: Old Questions and New Insights. Science 2011, 332, 1519–1523. [Google Scholar] [CrossRef] [Green Version]
- Cotter, T.G.; Rinella, M. Nonalcoholic Fatty Liver Disease 2020: The State of the Disease. Gastroenterology 2020, 158, 1851–1864. [Google Scholar] [CrossRef]
- Lazarus, J.V.; Mark, H.E.; Anstee, Q.M.; Arab, J.P.; Batterham, R.L.; Castera, L.; Cortez-Pinto, H.; Crespo, J.; Cusi, K.; Dirac, M.A.; et al. Advancing the Global Public Health Agenda for NAFLD: A Consensus Statement. Nat. Rev. Gastroenterol. Hepatol. 2022, 19, 60–78. [Google Scholar] [CrossRef]
- Brunner, K.T.; Henneberg, C.J.; Wilechansky, R.M.; Long, M.T. Nonalcoholic Fatty Liver Disease and Obesity Treatment. Curr. Obes. Rep. 2019, 8, 220–228. [Google Scholar] [CrossRef]
- Okuda, T.; Nishimura, M.; Nakao, M.; Fujitaa, Y. RUNX1/AML1: A Central Player in Hematopoiesis. Int. J. Hematol. 2001, 74, 252–257. [Google Scholar] [CrossRef]
- Ito, Y. Oncogenic Potential of the RUNX Gene Family: ‘Overview’. Oncogene 2004, 23, 4198–4208. [Google Scholar] [CrossRef] [Green Version]
- Ichikawa, M.; Yoshimi, A.; Nakagawa, M.; Nishimoto, N.; Watanabe-Okochi, N.; Kurokawa, M. A Role for RUNX1 in Hematopoiesis and Myeloid Leukemia. Int. J. Hematol. 2013, 97, 726–734. [Google Scholar] [CrossRef]
- Chen, C.-L.; Broom, D.C.; Liu, Y.; de Nooij, J.C.; Li, Z.; Cen, C.; Samad, O.A.; Jessell, T.M.; Woolf, C.J.; Ma, Q. Runx1 Determines Nociceptive Sensory Neuron Phenotype and Is Required for Thermal and Neuropathic Pain. Neuron 2006, 49, 365–377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwatsuki, K.; Tanaka, K.; Kaneko, T.; Kazama, R.; Okamoto, S.; Nakayama, Y.; Ito, Y.; Satake, M.; Takahashi, S.-I.; Miyajima, A.; et al. Runx1 Promotes Angiogenesis by Downregulation of Insulin-like Growth Factor-Binding Protein-3. Oncogene 2005, 24, 1129–1137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, M.; He, J.; Li, D.; Chu, Y.; Pu, J.; Tong, Q.; Joshi, H.C.; Tang, S.; Li, S. Runt-Related Transcription Factor 1 Promotes Apoptosis and Inhibits Neuroblastoma Progression in Vitro and in Vivo. J. Exp. Clin. Cancer Res. 2020, 39, 52. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Xing, Y.; Lu, W.; Li, S.; Tian, Z.; Xing, H.; Tang, K.; Xu, Y.; Rao, Q.; Wang, M.; et al. RUNX1 Inhibits Proliferation and Induces Apoptosis of t(8;21) Leukemia Cells via KLF4-Mediated Transactivation of P57. Haematologica 2019, 104, 1597–1607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doffo, J.; Bamopoulos, S.A.; Köse, H.; Orben, F.; Zang, C.; Pons, M.; den Dekker, A.T.; Brouwer, R.W.W.; Baluapuri, A.; Habringer, S.; et al. NOXA Expression Drives Synthetic Lethality to RUNX1 Inhibition in Pancreatic Cancer. Proc. Natl. Acad. Sci. USA 2022, 119, e2105691119. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.; Peng, Z.; Zhu, A.; Xue, R.; Lu, R.; Mi, J.; Xi, S.; Chen, W.; Jiang, S. Inhibition of RUNX1 Promotes Cisplatin-Induced Apoptosis in Ovarian Cancer Cells. Biochem. Pharmacol. 2020, 180, 114116. [Google Scholar] [CrossRef]
- Schlegelberger, B.; Heller, P.G. RUNX1 Deficiency (Familial Platelet Disorder with Predisposition to Myeloid Leukemia, FPDMM). Semin. Hematol. 2017, 54, 75–80. [Google Scholar] [CrossRef] [Green Version]
- Xing, X.; Wang, H.; Niu, T.; Jiang, Y.; Shi, X.; Liu, K. RUNX1 Can Mediate the Glucose and O-GlcNAc-Driven Proliferation and Migration of Human Retinal Microvascular Endothelial Cells. BMJ Open Diabetes Res. Care 2021, 9, e001898. [Google Scholar] [CrossRef]
- RUNX1 Gene-RUNX Family Transcription Factor 1. GeneCards. Available online: https://www.genecards.org/cgi-bin/carddisp.pl?gene=RUNX1 (accessed on 2 April 2022).
- Kaur, S.; Rawal, P.; Siddiqui, H.; Rohilla, S.; Sharma, S.; Tripathi, D.M.; Baweja, S.; Hassan, M.; Vlaic, S.; Guthke, R.; et al. Increased Expression of RUNX1 in Liver Correlates with NASH Activity Score in Patients with Non-Alcoholic Steatohepatitis (NASH). Cells 2019, 8, 1277. [Google Scholar] [CrossRef] [Green Version]
- Bertran, L.; Pastor, A.; Portillo-Carrasquer, M.; Binetti, J.; Aguilar, C.; Martínez, S.; Vives, M.; Sabench, F.; Porras, J.A.; Riesco, D.; et al. The Potential Protective Role of RUNX1 in Nonalcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2021, 22, 5239. [Google Scholar] [CrossRef]
- Han, H.; Cho, J.-W.; Lee, S.; Yun, A.; Kim, H.; Bae, D.; Yang, S.; Kim, C.Y.; Lee, M.; Kim, E.; et al. TRRUST v2: An Expanded Reference Database of Human and Mouse Transcriptional Regulatory Interactions. Nucleic Acids Res. 2018, 46, D380–D386. [Google Scholar] [CrossRef]
- The Biological General Repository for Interaction. BioGRID. Available online: https://thebiogrid.org/ (accessed on 23 March 2022).
- Peri, S.; Navarro, J.D.; Amanchy, R.; Kristiansen, T.Z.; Jonnalagadda, C.K.; Surendranath, V.; Niranjan, V.; Muthusamy, B.; Gandhi, T.K.B.; Gronborg, M.; et al. Development of Human Protein Reference Database as an Initial Platform for Approaching Systems Biology in Humans. Genome Res. 2003, 13, 2363–2371. [Google Scholar] [CrossRef] [Green Version]
- Prasad, T.S.K.; Goel, R.; Kandasamy, K.; Keerthikumar, S.; Kumar, S.; Mathivanan, S.; Telikicherla, D.; Raju, R.; Shafreen, B.; Venugopal, A.; et al. Human Protein Reference Database—2009 Update. Nucleic Acids Res. 2009, 37, D767–D772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orchard, S.; Ammari, M.; Aranda, B.; Breuza, L.; Briganti, L.; Broackes-Carter, F.; Campbell, N.H.; Chavali, G.; Chen, C.; del-Toro, N.; et al. The MIntAct Project—IntAct as a Common Curation Platform for 11 Molecular Interaction Databases. Nucl. Acids Res. 2014, 42, D358–D363. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Griss, J.; Viteri, G.; Sidiropoulos, K.; Nguyen, V.; Fabregat, A.; Hermjakob, H. ReactomeGSA–Efficient Multi-Omics Comparative Pathway Analysis. Mol. Cell. Proteom. 2020, 19, 2115–2125. [Google Scholar] [CrossRef] [PubMed]
- Jorba, G.; Aguirre-Plans, J.; Junet, V.; Segú-Vergés, C.; Ruiz, J.L.; Pujol, A.; Fernández-Fuentes, N.; Mas, J.M.; Oliva, B. In-Silico Simulated Prototype-Patients Using TPMS Technology to Study a Potential Adverse Effect of Sacubitril and Valsartan. PLoS ONE 2020, 15, e0228926. [Google Scholar] [CrossRef] [Green Version]
- Bishop, C.M. Pattern Recognition and Machine Learning, 1st ed.; Information Science and Statistics; Springer: New York, NY, USA, 2006; Volume 738. [Google Scholar]
- Artigas, L.; Coma, M.; Matos-Filipe, P.; Aguirre-Plans, J.; Farrés, J.; Valls, R.; Fernandez-Fuentes, N.; de la Haba-Rodriguez, J.; Olvera, A.; Barbera, J.; et al. In-Silico Drug Repurposing Study Predicts the Combination of Pirfenidone and Melatonin as a Promising Candidate Therapy to Reduce SARS-CoV-2 Infection Progression and Respiratory Distress Caused by Cytokine Storm. PLoS ONE 2020, 15, e0240149. [Google Scholar] [CrossRef] [PubMed]
- Segú-Vergés, C.; Coma, M.; Kessel, C.; Smeets, S.; Foell, D.; Aldea, A. Application of Systems Biology-Based in Silico Tools to Optimize Treatment Strategy Identification in Still’s Disease. Arthritis Res. Ther. 2021, 23, 126. [Google Scholar] [CrossRef]
- Liu, C.; Xu, D.; Xue, B.; Liu, B.; Li, J.; Huang, J. Upregulation of RUNX1 Suppresses Proliferation and Migration through Repressing VEGFA Expression in Hepatocellular Carcinoma. Pathol. Oncol. Res. 2020, 26, 1301–1311. [Google Scholar] [CrossRef]
- Michaud, J.; Simpson, K.M.; Escher, R.; Buchet-Poyau, K.; Beissbarth, T.; Carmichael, C.; Ritchie, M.E.; Schütz, F.; Cannon, P.; Liu, M.; et al. Integrative Analysis of RUNX1 Downstream Pathways and Target Genes. BMC Genom. 2008, 9, 363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, A.L.; Marsman, J.; Antony, J.; Schierding, W.; O’Sullivan, J.M.; Horsfield, J.A. Transcriptional Regulation of RUNX1: An Informatics Analysis. Genes 2021, 12, 1175. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zhang, L.; Liu, L.; Sun, S.; Zhao, X.; Wang, Y.; Zhang, Y.; Du, J.; Gu, L. Rasip1 Is a RUNX1 Target Gene and Promotes Migration of NSCLC Cells. CMAR 2018, 10, 4537–4552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riddell, A.; McBride, M.; Braun, T.; Nicklin, S.A.; Cameron, E.; Loughrey, C.M.; Martin, T.P. RUNX1: An Emerging Therapeutic Target for Cardiovascular Disease. Cardiovasc. Res. 2020, 116, 1410–1423. [Google Scholar] [CrossRef]
- McCarroll, C.S.; He, W.; Foote, K.; Bradley, A.; Mcglynn, K.; Vidler, F.; Nixon, C.; Nather, K.; Fattah, C.; Riddell, A.; et al. Runx1 Deficiency Protects Against Adverse Cardiac Remodeling After Myocardial Infarction. Circulation 2018, 137, 57–70. [Google Scholar] [CrossRef]
- Shilpi, S.; Shivvedi, R.; Gurnany, E.; Dixit, S.; Khatri, K.; Dwivedi, D.K. Drug Targeting Strategies for Liver Cancer and Other Liver Diseases. MOJ Drug Des. Dev. Ther. 2018, 2, 171–177. [Google Scholar] [CrossRef] [Green Version]
- Byrne, C.D.; Olufadi, R.; Bruce, K.D.; Cagampang, F.R.; Ahmed, M.H. Metabolic Disturbances in Non-Alcoholic Fatty Liver Disease. Clin. Sci. 2009, 116, 539–564. [Google Scholar] [CrossRef]
- Girija, S.M. The Blind Men see the Elephant-the Many Faces of Fatty Liver Disease. World J. Gastroenterol. 2008, 14, 831–844. [Google Scholar] [CrossRef] [Green Version]
- Tacer, K.F.; Rozman, D. Nonalcoholic Fatty Liver Disease: Focus on Lipoprotein and Lipid Deregulation. J. Lipids 2011, 2011, 783976. [Google Scholar] [CrossRef] [Green Version]
- Jornayvaz, F.R.; Shulman, G.I. Diacylglycerol Activation of Protein Kinase Cε and Hepatic Insulin Resistance. Cell Metab. 2012, 15, 574–584. [Google Scholar] [CrossRef] [Green Version]
- Kumashiro, N.; Erion, D.M.; Zhang, D.; Kahn, M.; Beddow, S.A.; Chu, X.; Still, C.D.; Gerhard, G.S.; Han, X.; Dziura, J.; et al. Cellular Mechanism of Insulin Resistance in Nonalcoholic Fatty Liver Disease. Proc. Natl. Acad. Sci. USA 2011, 108, 16381–16385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, F.; Meng, G.; Strober, W. Interactions among the Transcription Factors Runx1, RORγt and Foxp3 Regulate the Differentiation of Interleukin 17–Producing T Cells. Nat. Immunol. 2008, 9, 1297–1306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukherji, A.; Dachraoui, M.; Baumert, T.F. Perturbation of the Circadian Clock and Pathogenesis of NAFLD. Metabolism 2020, 111, 154337. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.-C.; Zhou, S.-Y.; Feng, D.-Y.; Xiao, J.; Li, W.-Y.; Xu, C.-D.; Wang, H.-Y.; Zhou, T. Runt-Related Transcription Factor 1 (RUNX1) Binds to P50 in Macrophages and Enhances TLR4-Triggered Inflammation and Septic Shock. J. Biol. Chem. 2016, 291, 22011–22020. [Google Scholar] [CrossRef] [Green Version]
- Tilg, H.; Moschen, A.R. Insulin Resistance, Inflammation, and Non-Alcoholic Fatty Liver Disease. Trends Endocrinol. Metab. 2008, 19, 371–379. [Google Scholar] [CrossRef]
- Rada, P.; González-Rodríguez, Á.; García-Monzón, C.; Valverde, Á.M. Understanding Lipotoxicity in NAFLD Pathogenesis: Is CD36 a Key Driver? Cell Death Dis. 2020, 11, 802. [Google Scholar] [CrossRef]
- Farrell, G.C.; Teoh, N.C.; Mccuskey, R.S. Hepatic Microcirculation in Fatty Liver Disease. Anat. Rec. 2008, 291, 684–692. [Google Scholar] [CrossRef]
- Lemoinne, S.; Thabut, D.; Housset, C. Portal Myofibroblasts Connect Angiogenesis and Fibrosis in Liver. Cell Tissue Res. 2016, 365, 583–589. [Google Scholar] [CrossRef]
- Coulon, S.; Legry, V.; Heindryckx, F.; Van Steenkiste, C.; Casteleyn, C.; Olievier, K.; Libbrecht, L.; Carmeliet, P.; Jonckx, B.; Stassen, J.-M.; et al. Role of Vascular Endothelial Growth Factor in the Pathophysiology of Nonalcoholic Steatohepatitis in Two Rodent Models. Hepatology 2013, 57, 1793–1805. [Google Scholar] [CrossRef]
- North, T.E.; de Bruijn, M.F.T.R.; Stacy, T.; Talebian, L.; Lind, E.; Robin, C.; Binder, M.; Dzierzak, E.; Speck, N.A. Runx1 Expression Marks Long-Term Repopulating Hematopoietic Stem Cells in the Midgestation Mouse Embryo. Immunity 2002, 16, 661–672. [Google Scholar] [CrossRef] [Green Version]
- Coulon, S.; Francque, S.; Colle, I.; Verrijken, A.; Blomme, B.; Heindryckx, F.; De Munter, S.; Prawitt, J.; Caron, S.; Staels, B.; et al. Evaluation of Inflammatory and Angiogenic Factors in Patients with Non-Alcoholic Fatty Liver Disease. Cytokine 2012, 59, 442–449. [Google Scholar] [CrossRef]
- Rahman, S.M.; Janssen, R.C.; Choudhury, M.; Baquero, K.C.; Aikens, R.M.; de la Houssaye, B.A.; Friedman, J.E. CCAAT/Enhancer-Binding Protein β (C/EBPβ) Expression Regulates Dietary-Induced Inflammation in Macrophages and Adipose Tissue in Mice. J. Biol. Chem. 2012, 287, 34349–34360. [Google Scholar] [CrossRef] [Green Version]
- Amacher, D.E. The Mechanistic Basis for the Induction of Hepatic Steatosis by Xenobiotics. Expert Opin. Drug Metab. Toxicol. 2011, 7, 949–965. [Google Scholar] [CrossRef]
- Masarone, M.; Rosato, V.; Dallio, M.; Gravina, A.G.; Aglitti, A.; Loguercio, C.; Federico, A.; Persico, M. Role of Oxidative Stress in Pathophysiology of Nonalcoholic Fatty Liver Disease. Oxidative Med. Cell. Longev. 2018, 2018, 9547613. [Google Scholar] [CrossRef] [PubMed]
- Chan, V.; Chan, T.K.; Liu, V.W.S.; Wong, A.C.K. Restriction Fragment Length Polymorphisms Associated with Factor VIII: C Gene in Chinese. Hum. Genet. 1988, 79, 128–131. [Google Scholar] [CrossRef] [PubMed]
- Kantner, H.-P.; Warsch, W.; Delogu, A.; Bauer, E.; Esterbauer, H.; Casanova, E.; Sexl, V.; Stoiber, D. ETV6/RUNX1 Induces Reactive Oxygen Species and Drives the Accumulation of DNA Damage in B Cells. Neoplasia 2013, 15, 1292–1300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whitmore, H.A.B.; Amarnani, D.; O’Hare, M.; Delgado-Tirado, S.; Gonzalez-Buendia, L.; An, M.; Pedron, J.; Bushweller, J.H.; Arboleda-Velasquez, J.F.; Kim, L.A. TNF-α Signaling Regulates RUNX1 Function in Endothelial Cells. FASEB J. 2020, 35, e21155. [Google Scholar] [CrossRef] [PubMed]
- Kanda, T.; Matsuoka, S.; Yamazaki, M.; Shibata, T.; Nirei, K.; Takahashi, H.; Kaneko, T.; Fujisawa, M.; Higuchi, T.; Nakamura, H.; et al. Apoptosis and Non-Alcoholic Fatty Liver Diseases. World J. Gastroenterol. 2018, 24, 2661–2672. [Google Scholar] [CrossRef]
- Petta, S.; Gastaldelli, A.; Rebelos, E.; Bugianesi, E.; Messa, P.; Miele, L.; Svegliati-Baroni, G.; Valenti, L.; Bonino, F. Pathophysiology of Non Alcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2016, 17, 2082. [Google Scholar] [CrossRef] [PubMed]
- O’Hare, M.; Amarnani, D.; Whitmore, H.A.B.; An, M.; Marino, C.; Ramos, L.; Delgado-Tirado, S.; Hu, X.; Chmielewska, N.; Chandrahas, A.; et al. Targeting Runt-Related Transcription Factor 1 Prevents Pulmonary Fibrosis and Reduces Expression of Severe Acute Respiratory Syndrome Coronavirus 2 Host Mediators. Am. J. Pathol. 2021, 191, 1193–1208. [Google Scholar] [CrossRef]
- Bertrand-Philippe, M.; Ruddell, R.G.; Arthur, M.J.P.; Thomas, J.; Mungalsingh, N.; Mann, D.A. Regulation of Tissue Inhibitor of Metalloproteinase 1 Gene Transcription by RUNX1 and RUNX2. J. Biol. Chem. 2004, 279, 24530–24539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, K.; Hu, H.; Jiang, H.; Zhang, H.; Gong, S.; Wei, D.; Yu, Z. RUNX1 Promotes MAPK Signaling to Increase Tumor Progression and Metastasis via OPN in Head and Neck Cancer. Carcinogenesis 2021, 42, 414–422. [Google Scholar] [CrossRef] [PubMed]
- Bellissimo, D.C.; Chen, C.; Zhu, Q.; Bagga, S.; Lee, C.-T.; He, B.; Wertheim, G.B.; Jordan, M.; Tan, K.; Worthen, G.S.; et al. Runx1 Negatively Regulates Inflammatory Cytokine Production by Neutrophils in Response to Toll-like Receptor Signaling. Blood Adv. 2020, 4, 1145–1158. [Google Scholar] [CrossRef] [PubMed]
- Manco, M. Insulin Resistance and NAFLD: A Dangerous Liaison beyond the Genetics. Children 2017, 4, 74. [Google Scholar] [CrossRef] [Green Version]
- Guo, H.; Friedman, A.D. Phosphorylation of RUNX1 by Cyclin-Dependent Kinase Reduces Direct Interaction with HDAC1 and HDAC3. J. Biol. Chem. 2011, 286, 208–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, W.; Hong, S.H.; Choe, J. IL-4 and HDAC Inhibitors Suppress Cyclooxygenase-2 Expression in Human Follicular Dendritic Cells. Immune Netw. 2013, 13, 75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsieh, P.-S.; Jin, J.-S.; Chiang, C.-F.; Chan, P.-C.; Chen, C.-H.; Shih, K.-C. COX-2-Mediated Inflammation in Fat Is Crucial for Obesity-Linked Insulin Resistance and Fatty Liver. Obesity 2009, 17, 1150–1157. [Google Scholar] [CrossRef]
- Chan, P.-C.; Liao, M.-T.; Hsieh, P.-S. The Dualistic Effect of COX-2-Mediated Signaling in Obesity and Insulin Resistance. Int. J. Mol. Sci. 2019, 20, 3115. [Google Scholar] [CrossRef] [Green Version]
- Qiao, Y.; He, H.; Jonsson, P.; Sinha, I.; Zhao, C.; Dahlman-Wright, K. AP-1 Is a Key Regulator of Proinflammatory Cytokine TNFα-Mediated Triple-Negative Breast Cancer Progression. J. Biol. Chem. 2016, 291, 5068–5079. [Google Scholar] [CrossRef] [Green Version]
- Monaco, C.; Andreakos, E.; Kiriakidis, S.; Mauri, C.; Bicknell, C.; Foxwell, B.; Cheshire, N.; Paleolog, E.; Feldmann, M. Canonical Pathway of Nuclear Factor ΚB Activation Selectively Regulates Proinflammatory and Prothrombotic Responses in Human Atherosclerosis. Proc. Natl. Acad. Sci. USA 2004, 101, 5634–5639. [Google Scholar] [CrossRef] [Green Version]
- Scheidereit, C. IκB Kinase Complexes: Gateways to NF-ΚB Activation and Transcription. Oncogene 2006, 25, 6685–6705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhingra, S.; Sharma, A.K.; Arora, R.C.; Slezak, J.; Singal, P.K. IL-10 Attenuates TNF-α-Induced NFκB Pathway Activation and Cardiomyocyte Apoptosis. Cardiovasc. Res. 2009, 82, 59–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tien, Y.-C.; Lin, J.-Y.; Lai, C.-H.; Kuo, C.-H.; Lin, W.-Y.; Tsai, C.-H.; Tsai, F.-J.; Cheng, Y.-C.; Peng, W.-H.; Huang, C.-Y. Carthamus Tinctorius L. Prevents LPS-Induced TNFα Signaling Activation and Cell Apoptosis through JNK1/2–NFκB Pathway Inhibition in H9c2 Cardiomyoblast Cells. J. Ethnopharmacol. 2010, 130, 505–513. [Google Scholar] [CrossRef] [PubMed]
- Cronin, J.G.; Turner, M.L.; Goetze, L.; Bryant, C.E.; Sheldon, I.M. Toll-Like Receptor 4 and MYD88-Dependent Signaling Mechanisms of the Innate Immune System Are Essential for the Response to Lipopolysaccharide by Epithelial and Stromal Cells of the Bovine Endometrium. Biol. Reprod. 2012, 86, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Candido, S.; Maestro, R.; Polesel, J.; Catania, A.; Maira, F.; Signorelli, S.S.; McCubrey, J.A.; Libra, M. Roles of Neutrophil Gelatinase-Associated Lipocalin (NGAL) in Human Cancer. Oncotarget 2014, 5, 1576–1594. [Google Scholar] [CrossRef] [Green Version]
- Schaefer, F.M.; Peng, J.; Hu, W.; Drvarov, O.; Nevzorova, Y.A.; Zhao, G.; Masaoudi, M.A.; Davis, R.J.; Trautwein, C.; Cubero, F.J. Bone Marrow-Derived c-Jun N-Terminal Kinase-1 (JNK1) Mediates Liver Regeneration. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2015, 1852, 137–145. [Google Scholar] [CrossRef] [Green Version]
- Faggioli, L.; Costanzo, C.; Donadelli, M.; Palmieri, M. Activation of the Interleukin-6 Promoter by a Dominant Negative Mutant of c-Jun. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2004, 1692, 17–24. [Google Scholar] [CrossRef]
- Torisu, T.; Sato, N.; Yoshiga, D.; Kobayashi, T.; Yoshioka, T.; Mori, H.; Iida, M.; Yoshimura, A. The Dual Function of Hepatic SOCS3 in Insulin Resistance in Vivo. Genes Cells 2007, 12, 143–154. [Google Scholar] [CrossRef]
- Kim, K.; Kim, K.H.; Cheong, J. Hepatitis B Virus X Protein Impairs Hepatic Insulin Signaling Through Degradation of IRS1 and Induction of SOCS3. PLoS ONE 2010, 5, e8649. [Google Scholar] [CrossRef] [Green Version]
- Rui, L.; Yuan, M.; Frantz, D.; Shoelson, S.; White, M.F. SOCS-1 and SOCS-3 Block Insulin Signaling by Ubiquitin-Mediated Degradation of IRS1 and IRS2. J. Biol. Chem. 2002, 277, 42394–42398. [Google Scholar] [CrossRef] [Green Version]
- Laplante, M.; Sabatini, D.M. MTOR Signaling at a Glance. J. Cell Sci. 2009, 122, 3589–3594. [Google Scholar] [CrossRef] [Green Version]
- Tai, H.; Wang, X.; Zhou, J.; Han, X.; Fang, T.; Gong, H.; Huang, N.; Chen, H.; Qin, J.; Yang, M.; et al. Protein Kinase Cβ Activates Fat Mass and Obesity-associated Protein by Influencing Its Ubiquitin/Proteasome Degradation. FASEB J. 2017, 31, 4396–4406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aguirre, V.; Uchida, T.; Yenush, L.; Davis, R.; White, M.F. The C-Jun NH2-Terminal Kinase Promotes Insulin Resistance during Association with Insulin Receptor Substrate-1 and Phosphorylation of Ser307. J. Biol. Chem. 2000, 275, 9047–9054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, K.; Li, Q.; Rask-Madsen, C.; Mima, A.; Mizutani, K.; Winnay, J.; Maeda, Y.; D’Aquino, K.; White, M.F.; Feener, E.P.; et al. Serine Phosphorylation Sites on IRS2 Activated by Angiotensin II and Protein Kinase C To Induce Selective Insulin Resistance in Endothelial Cells. Mol. Cell Biol. 2013, 33, 3227–3241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López-Bergami, P.; Habelhah, H.; Bhoumik, A.; Zhang, W.; Wang, L.-H.; Ronai, Z. Receptor for RACK1 Mediates Activation of JNK by Protein Kinase C. Mol. Cell 2005, 19, 309–320. [Google Scholar] [CrossRef] [PubMed]
- Comalada, M.; Xaus, J.; Valledor, A.F.; López-López, C.; Pennington, D.J.; Celada, A. PKCϵ Is Involved in JNK Activation That Mediates LPS-Induced TNF-α, Which Induces Apoptosis in Macrophages. Am. J. Physiol.-Cell Physiol. 2003, 285, C1235–C1245. [Google Scholar] [CrossRef] [Green Version]
- Kolter, T.; Uphues, I.; Eckel, J. Molecular Analysis of Insulin Resistance in Isolated Ventricular Cardiomyocytes of Obese Zucker Rats. Am. J. Physiol.-Endocrinol. Metab. 1997, 273, E59–E67. [Google Scholar] [CrossRef]
- Naruse, K.; Rask-Madsen, C.; Takahara, N.; Ha, S.; Suzuma, K.; Way, K.J.; Jacobs, J.R.C.; Clermont, A.C.; Ueki, K.; Ohshiro, Y.; et al. Activation of Vascular Protein Kinase C-β Inhibits Akt-Dependent Endothelial Nitric Oxide Synthase Function in Obesity-Associated Insulin Resistance. Diabetes 2006, 55, 691–698. [Google Scholar] [CrossRef] [Green Version]
- Sampson, S.; Cooper, D. Specific Protein Kinase C Isoforms as Transducers and Modulators of Insulin Signaling. Mol. Genet. Metab. 2006, 89, 32–47. [Google Scholar] [CrossRef] [Green Version]
- Matsuda, S.; Kobayashi, M.; Kitagishi, Y. Roles for PI3K/AKT/PTEN Pathway in Cell Signaling of Nonalcoholic Fatty Liver Disease. Int. Sch. Res. Not. 2013, 2013, 472432. [Google Scholar] [CrossRef]
- Huang, X.; Liu, G.; Guo, J.; Su, Z. The PI3K/AKT Pathway in Obesity and Type 2 Diabetes. Int. J. Biol. Sci. 2018, 14, 1483–1496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peyrou, M.; Bourgoin, L.; Foti, M. PTEN in Non-Alcoholic Fatty Liver Disease/Non-Alcoholic Steatohepatitis and Cancer. Dig. Dis 2010, 28, 236–246. [Google Scholar] [CrossRef] [PubMed]
- Ding, R.-B.; Bao, J.; Deng, C.-X. Emerging Roles of SIRT1 in Fatty Liver Diseases. Int. J. Biol. Sci. 2017, 13, 852–867. [Google Scholar] [CrossRef] [PubMed]
- Wasilewska, N.; Lebensztejn, D. Non-Alcoholic Fatty Liver Disease and Lipotoxicity. Clin. Exp. Hepatol. 2021, 7, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Haslinger, B.; Mandl-Weber, S.; Sellmayer, A.; Sitter, T. Hyaluronan Fragments Induce the Synthesis of MCP-1 and IL-8 in Cultured Human Peritoneal Mesothelial Cells. Cell Tissue Res. 2001, 305, 79–86. [Google Scholar] [CrossRef]
- Curran, J.E.; Weinstein, S.R.; Griffiths, L.R. Polymorphic Variants of NFKB1 and Its Inhibitory Protein NFKBIA, and Their Involvement in Sporadic Breast Cancer. Cancer Lett. 2002, 188, 103–107. [Google Scholar] [CrossRef]
- Petta, S.; Muratore, C.; Craxì, A. Non-Alcoholic Fatty Liver Disease Pathogenesis: The Present and the Future. Dig. Liver Dis. 2009, 41, 615–625. [Google Scholar] [CrossRef]
- Feldstein, A.E.; Werneburg, N.W.; Canbay, A.; Guicciardi, M.E.; Bronk, S.F.; Rydzewski, R.; Burgart, L.J.; Gores, G.J. Free Fatty Acids Promote Hepatic Lipotoxicity by Stimulating TNF-α Expression via a Lysosomal Pathway. Hepatology 2004, 40, 185–194. [Google Scholar] [CrossRef]
- Ferraris, S.E.; Isoniemi, K.; Torvaldson, E.; Anckar, J.; Westermarck, J.; Eriksson, J.E. Nucleolar AATF Regulates C-Jun–Mediated Apoptosis. Mol. Biol. Cell 2012, 23, 4323–4332. [Google Scholar] [CrossRef]
- Kagoya, Y.; Yoshimi, A.; Kataoka, K.; Nakagawa, M.; Kumano, K.; Arai, S.; Kobayashi, H.; Saito, T.; Iwakura, Y.; Kurokawa, M. Positive Feedback between NF-ΚB and TNF-α Promotes Leukemia-Initiating Cell Capacity. J. Clin. Investig. 2014, 124, 528–542. [Google Scholar] [CrossRef]
- Lee, I.-T.; Liu, S.-W.; Chi, P.-L.; Lin, C.-C.; Hsiao, L.-D.; Yang, C.-M. TNF-α Mediates PKCδ/JNK1/2/c-Jun-Dependent Monocyte Adhesion via ICAM-1 Induction in Human Retinal Pigment Epithelial Cells. PLoS ONE 2015, 10, e0117911. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.-J.; Lo, J.-F.; Kuo, C.-H.; Chu, C.-H.; Chen, L.-M.; Tsai, F.-J.; Tsai, C.-H.; Tzang, B.-S.; Kuo, W.-W.; Huang, C.-Y. Akt Mediates 17β-Estradiol and/or Estrogen Receptor-α Inhibition of LPS-Induced Tumor Necresis Factor-α Expression and Myocardial Cell Apoptosis by Suppressing the JNK1/2-NFκB Pathway. J. Cell. Mol. Med. 2009, 13, 3655–3667. [Google Scholar] [CrossRef] [PubMed]
- Tiniakos, D.G.; Vos, M.B.; Brunt, E.M. Nonalcoholic Fatty Liver Disease: Pathology and Pathogenesis. Annu. Rev. Pathol. Mech. Dis. 2010, 5, 145–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luedde, T.; Schwabe, R.F. NF-ΚB in the Liver—Linking Injury, Fibrosis and Hepatocellular Carcinoma. Nat. Rev. Gastroenterol. Hepatol 2011, 8, 108–118. [Google Scholar] [CrossRef] [Green Version]
- Ferro, D.; Baratta, F.; Pastori, D.; Cocomello, N.; Colantoni, A.; Angelico, F.; Del Ben, M. New Insights into the Pathogenesis of Non-Alcoholic Fatty Liver Disease: Gut-Derived Lipopolysaccharides and Oxidative Stress. Nutrients 2020, 12, 2762. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Tian, R.; She, Z.; Cai, J.; Li, H. Role of Oxidative Stress in the Pathogenesis of Nonalcoholic Fatty Liver Disease. Free Radic. Biol. Med. 2020, 152, 116–141. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Goettsch, C.; Xia, N.; Horke, S.; Morawietz, H.; Förstermann, U.; Li, H. Differential Roles of PKCα and PKCɛ in Controlling the Gene Expression of Nox4 in Human Endothelial Cells. Free. Radic. Biol. Med. 2008, 44, 1656–1667. [Google Scholar] [CrossRef] [PubMed]
- Sattayakhom, A.; Chunglok, W.; Ittarat, W.; Chamulitrat, W. Study Designs to Investigate Nox1 Acceleration of Neoplastic Progression in Immortalized Human Epithelial Cells by Selection of Differentiation Resistant Cells. Redox Biol. 2014, 2, 140–147. [Google Scholar] [CrossRef] [Green Version]
- Manea, A.; Tanase, L.I.; Raicu, M.; Simionescu, M. Transcriptional Regulation of NADPH Oxidase Isoforms, Nox1 and Nox4, by Nuclear Factor-ΚB in Human Aortic Smooth Muscle Cells. Biochem. Biophys. Res. Commun. 2010, 396, 901–907. [Google Scholar] [CrossRef]
- Milacic, M.; Haw, R.; Rothfels, K.; Wu, G.; Croft, D.; Hermjakob, H.; D’Eustachio, P.; Stein, L. Annotating Cancer Variants and Anti-Cancer Therapeutics in Reactome. Cancers 2012, 4, 1180–1211. [Google Scholar] [CrossRef]
- Valls, R.; (Anaxomics Biotech, Barcelona, Spain); Pujol, A.; (Anaxomics Biotech; Institute for Research in Biomedicine and Barcelona Supercomputing Center, Barcelona, Spain); Farrés, J.; (Anaxomics Biotech, Barcelona, Spain); Artigas, L.; (Anaxomics Biotech, Barcelona, Spain); Mas, J.M.; (Anaxomics Biotech, Barcelona, Spain). Anaxomics’ Methodologies–Understanding the Complexity of Biological Processes. Personal communication (Protocol), 2013. [Google Scholar]
- Croft, D.; Mundo, A.F.; Haw, R.; Milacic, M.; Weiser, J.; Wu, G.; Caudy, M.; Garapati, P.; Gillespie, M.; Kamdar, M.R.; et al. The Reactome Pathway Knowledgebase. Nucl. Acids Res. 2014, 42, D472–D477. [Google Scholar] [CrossRef] [PubMed]
- Stark, C.; Breitkreutz, B.-J.; Reguly, T.; Boucher, L.; Breitkreutz, A.; Tyers, M. BioGRID: A General Repository for Interaction Datasets. Nucleic Acids Res. 2006, 34, D535–D539. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NAFLD | SS/NASH | SS/NASH | SS/NASH | NASH | NASH | |
|---|---|---|---|---|---|---|
| Increased Body Fat | Hepatic Insulin Resistance | Altered Fatty Acid Metabolism | Lipotoxicity | Hepatic Injury and Liver Fibrosis | ||
| RUNX1 | MEDIUM (67%) | LOW (37%) | MEDIUM (67%) | LOW (22%) | MEDIUM (61%) | HIGH (78%) |
| Gene Name | Protein Name | Causative Effect in NAFLD | MoA Activation by RUNX1 |
|---|---|---|---|
| NFKB1 | Nuclear factor NF-kappa-B p105 subunit | 1 | 1.000 |
| JNK1 | c-Jun N-terminal kinase 1 | 1 | 0.992 |
| PKC-E | Protein kinase C epsilon type | 1 | 0.883 |
| TNF | Tumor necrosis factor | 1 | 0.875 |
| IKBKB | Inhibitor of nuclear factor kB kinase subunit beta | 1 | 0.859 |
| PTGS2 | Prostaglandin G/H synthase 2 | 1 | 0.842 |
| IL17A | Interleukin 17A | 1 | 0.688 |
| MTOR | Serine/threonine-protein kinase mTOR | 1 | 0.684 |
| APOC3 | Apolipoprotein C-III | 1 | 0.605 |
| NFKB2 | Nuclear factor NF-kappa-B p100 subunit | 1 | 0.543 |
| LCN2 | Neutrophil gelatinase-associated lipocalin | 1 | 0.457 |
| SOCS3 | Suppressor of cytokine signaling 3 | 1 | 0.436 |
| INS | Insulin | 1 | 0.422 |
| NT | Neurotensin | 1 | 0.362 |
| IL6 | Interleukin-6 | 1 | 0.230 |
| CNR1 | Cannabinoid receptor 1 | 1 | 0.102 |
| ADIPOQ | Adiponectin | −1 | −0.184 |
| NRG4 | Pro-neuregulin-4, membrane-bound isoform | −1 | −0.305 |
| AKT2 | RAC-beta serine/threonine-protein kinase | −1 | −0.375 |
| PTPN1 | Tyrosine-protein phosphatase non-receptor type 1 | −1 | −0.436 |
| GSK3 | Glycogen synthase kinase-3 alpha | −1 | −0.504 |
| SIRT1 | Sirtuin 1 | −1 | −0.868 |
| IRS2 | Insulin receptor substrate 2 | −1 | −0.916 |
| PTEN | Phosphatase and tensin homolog | −1 | −0.930 |
| IRS1 | Insulin receptor substrate 1 | −1 | −0.996 |
| Gene Name | Protein Name | Causative Effect in NAFLD | Activation by RUNX1 |
|---|---|---|---|
| LIPOTOXICITY | |||
| JNK1 | c-Jun N-terminal kinase 1 | 1 | 0.999 |
| CEBPB | CCAAT/enhancer-binding protein beta | 1 | 0.825 |
| IKBKB | Inhibitor of nuclear factor kappa-B kinase subunit beta | 1 | 0.819 |
| MAP3K7 | Transforming growth factor beta-activated kinase 1/Mitogen-activated protein kinase 7 | 1 | 0.722 |
| NOS2 | Nitric oxide synthase, inducible | 1 | 0.667 |
| MTOR | Serine/threonine-protein kinase mTOR | 1 | 0.617 |
| LCN2 | Neutrophil gelatinase-associated lipocalin | 1 | 0.609 |
| PLIN1 | Perilipin-1 | 1 | 0.563 |
| HMOX1 | Heme oxygenase 1 | 1 | 0.490 |
| MAP3K5 | Apoptosis signal-regulating kinase 1/mitogen-activated protein kinase 5 | 1 | 0.460 |
| PPARG | Peroxisome proliferator-activated receptor gamma | 1 | 0.402 |
| XBP1 | X-box-binding protein 1 | 1 | 0.327 |
| UCP2 | Mitochondrial uncoupling protein 2 | 1 | 0.183 |
| ACC1 | Acetyl-CoA carboxylase 1 | 1 | 0.113 |
| ADIPOR2 | Adiponectin receptor protein 2 | −1 | −0.600 |
| ADIPOR1 | Adiponectin receptor protein 1 | −1 | −0.633 |
| SIRT1 | Sirtuin 1 | −1 | −0.780 |
| FIBROSIS | |||
| SMAD3 | Mothers against decapentaplegic homolog 3 | 1 | 0.859 |
| ANGPT2 | Angiopoietin-2 | 1 | 0.854 |
| BAX | Apoptosis regulator BAX | 1 | 0.839 |
| AGTR1 | Type-1 angiotensin II receptor | 1 | 0.839 |
| TGFB1 | Transforming growth factor beta-1 | 1 | 0.827 |
| IL1B | Interleukin-1 beta | 1 | 0.693 |
| IL8 | Interleukin-8 | 1 | 0.683 |
| MMP9 | Matrix metalloproteinase-9 | 1 | 0.614 |
| FAS | Tumor necrosis factor receptor superfamily member 6 | 1 | 0.518 |
| MMP2 | 72 kDa type IV collagenase | 1 | 0.508 |
| CTGF | Connective tissue growth factor | 1 | 0.497 |
| CASP1 | Caspase-1 | 1 | 0.472 |
| NLRP3 | NACHT, LRR and PYD domains-containing protein 3 | 1 | 0.352 |
| AGT | Angiotensinogen | 1 | 0.340 |
| SPP1 | Osteopontin | 1 | 0.330 |
| TIMP1 | Metalloproteinase inhibitor 1 | 1 | 0.282 |
| MYD88 | Myeloid differentiation primary response protein MyD88 | 1 | 0.277 |
| PDGFA | Platelet-derived growth factor subunit A | 1 | 0.248 |
| LY96 | Lymphocyte antigen 96 | 1 | 0.224 |
| COL1A1 | Collagen alpha-1(I) chain | 1 | 0.209 |
| COL1A2 | Collagen alpha-2(I) chain | 1 | 0.124 |
| NR1H4 | Bile acid receptor | −1 | −0.441 |
| PTEN | Phosphatase and tensin homolog | −1 | −0.958 |
| LIPOTOXICITY AND FIBROSIS | |||
| NFKB1 | Nuclear factor NF-kappa-B p105 subunit | 1 | 0.999 |
| NOX1 | NADPH oxidase 1 | 1 | 0.894 |
| NOX4 | NADPH oxidase 4 | 1 | 0.822 |
| CCL2 | C-C motif chemokine 2 | 1 | 0.813 |
| TNF | Tumor necrosis factor | 1 | 0.812 |
| CYBB | Cytochrome b-245 heavy chain | 1 | 0.640 |
| NFKB2 | Nuclear factor NF-kappa-B p100 subunit | 1 | 0.535 |
| TLR4 | Toll-like receptor 4 | 1 | 0.501 |
| IL6 | Interleukin-6 | 1 | 0.342 |
| TLR2 | Toll-like receptor 2 | 1 | 0.292 |
| TLR9 | Toll-like receptor 9 | 1 | 0.200 |
| ADIPOQ | Adiponectin | −1 | −0.142 |
| Gene Name | Protein Code | Causative Effect in NAFLD | Activity in IR MoA | Activity in L&F MoA | Present in the Most Represented MoA | |
|---|---|---|---|---|---|---|
| IR (Figure 2) | L&F (Figure 3) | |||||
| Common RUNX1-modulated effector proteins in three motives | ||||||
| NFKB1 | P19838 | 1 | 1.000 | 0.999 | Yes | Yes |
| TNF | P01375 | 1 | 0.875 | 0.812 | Yes | Yes |
| NFKB2 | Q00653 | 1 | 0.543 | 0.535 | - | - |
| IL6 | P05231 | 1 | 0.230 | 0.342 | Yes | Yes |
| ADIPOQ | Q15848 | −1 | −0.184 | −0.142 | - | - |
| Common RUNX1-modulated effector proteins in lipotoxicity and fibrosis | ||||||
| NOX1 | Q9Y5S8 | 1 | - | 0.894 | - | Yes |
| NOX4 | Q9NPH5 | 1 | - | 0.822 | - | Yes |
| CCL2 | P13500 | 1 | - | 0.813 | - | Yes |
| CYBB | P04839 | 1 | - | 0.640 | - | - |
| TLR4 | O00206 | 1 | - | 0.501 | - | - |
| TLR2 | O60603 | 1 | - | 0.292 | - | - |
| TLR9 | Q9NR96 | 1 | - | 0.200 | - | - |
| Common RUNX1-modulated effector proteins in IR and lipotoxicity | ||||||
| JNK1 | P45983 | 1 | 0.992 | 0.999 | Yes | Yes |
| IKBKB | O14920 | 1 | 0.859 | 0.819 | - | - |
| MTOR | P42345 | 1 | 0.684 | 0.617 | Yes | - |
| LCN2 | P80188 | 1 | 0.457 | 0.609 | Yes | Yes |
| SIRT1 | Q96EB6 | −1 | −0.868 | −0.780 | - | - |
| Common RUNX1-modulated effector proteins in IR and fibrosis | ||||||
| PTEN | P60484 | −1 | −0.930 | −0.958 | - | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bertran, L.; Eigbefoh-Addeh, A.; Portillo-Carrasquer, M.; Barrientos-Riosalido, A.; Binetti, J.; Aguilar, C.; Ugarte Chicote, J.; Bartra, H.; Artigas, L.; Coma, M.; et al. Identification of the Potential Molecular Mechanisms Linking RUNX1 Activity with Nonalcoholic Fatty Liver Disease, by Means of Systems Biology. Biomedicines 2022, 10, 1315. https://doi.org/10.3390/biomedicines10061315
Bertran L, Eigbefoh-Addeh A, Portillo-Carrasquer M, Barrientos-Riosalido A, Binetti J, Aguilar C, Ugarte Chicote J, Bartra H, Artigas L, Coma M, et al. Identification of the Potential Molecular Mechanisms Linking RUNX1 Activity with Nonalcoholic Fatty Liver Disease, by Means of Systems Biology. Biomedicines. 2022; 10(6):1315. https://doi.org/10.3390/biomedicines10061315
Chicago/Turabian StyleBertran, Laia, Ailende Eigbefoh-Addeh, Marta Portillo-Carrasquer, Andrea Barrientos-Riosalido, Jessica Binetti, Carmen Aguilar, Javier Ugarte Chicote, Helena Bartra, Laura Artigas, Mireia Coma, and et al. 2022. "Identification of the Potential Molecular Mechanisms Linking RUNX1 Activity with Nonalcoholic Fatty Liver Disease, by Means of Systems Biology" Biomedicines 10, no. 6: 1315. https://doi.org/10.3390/biomedicines10061315
APA StyleBertran, L., Eigbefoh-Addeh, A., Portillo-Carrasquer, M., Barrientos-Riosalido, A., Binetti, J., Aguilar, C., Ugarte Chicote, J., Bartra, H., Artigas, L., Coma, M., Richart, C., & Auguet, T. (2022). Identification of the Potential Molecular Mechanisms Linking RUNX1 Activity with Nonalcoholic Fatty Liver Disease, by Means of Systems Biology. Biomedicines, 10(6), 1315. https://doi.org/10.3390/biomedicines10061315