
Virus-Based Immuno-Oncology Models



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Oncologic Immunomodulation
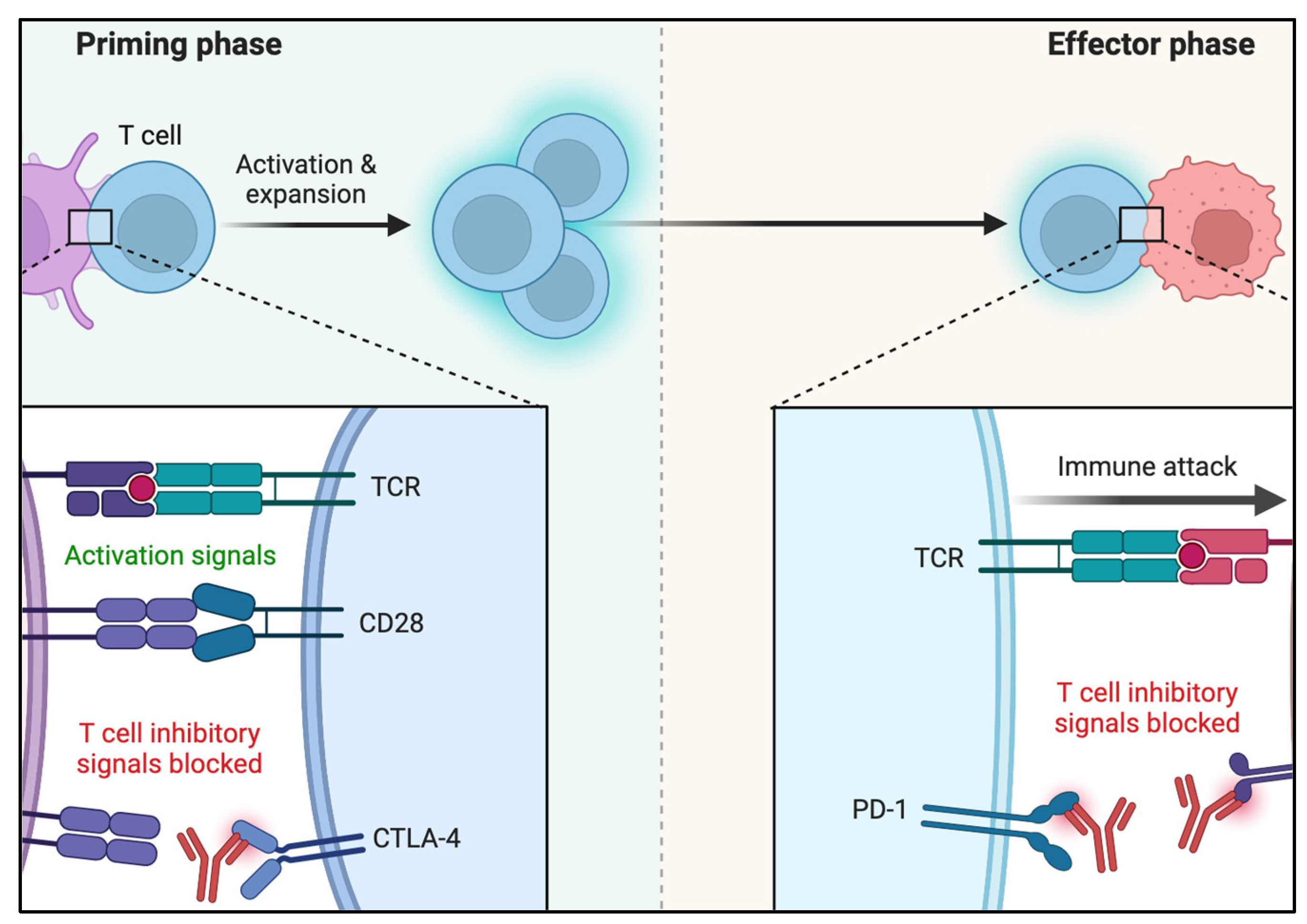
Mechanisms of Tumor Evasion
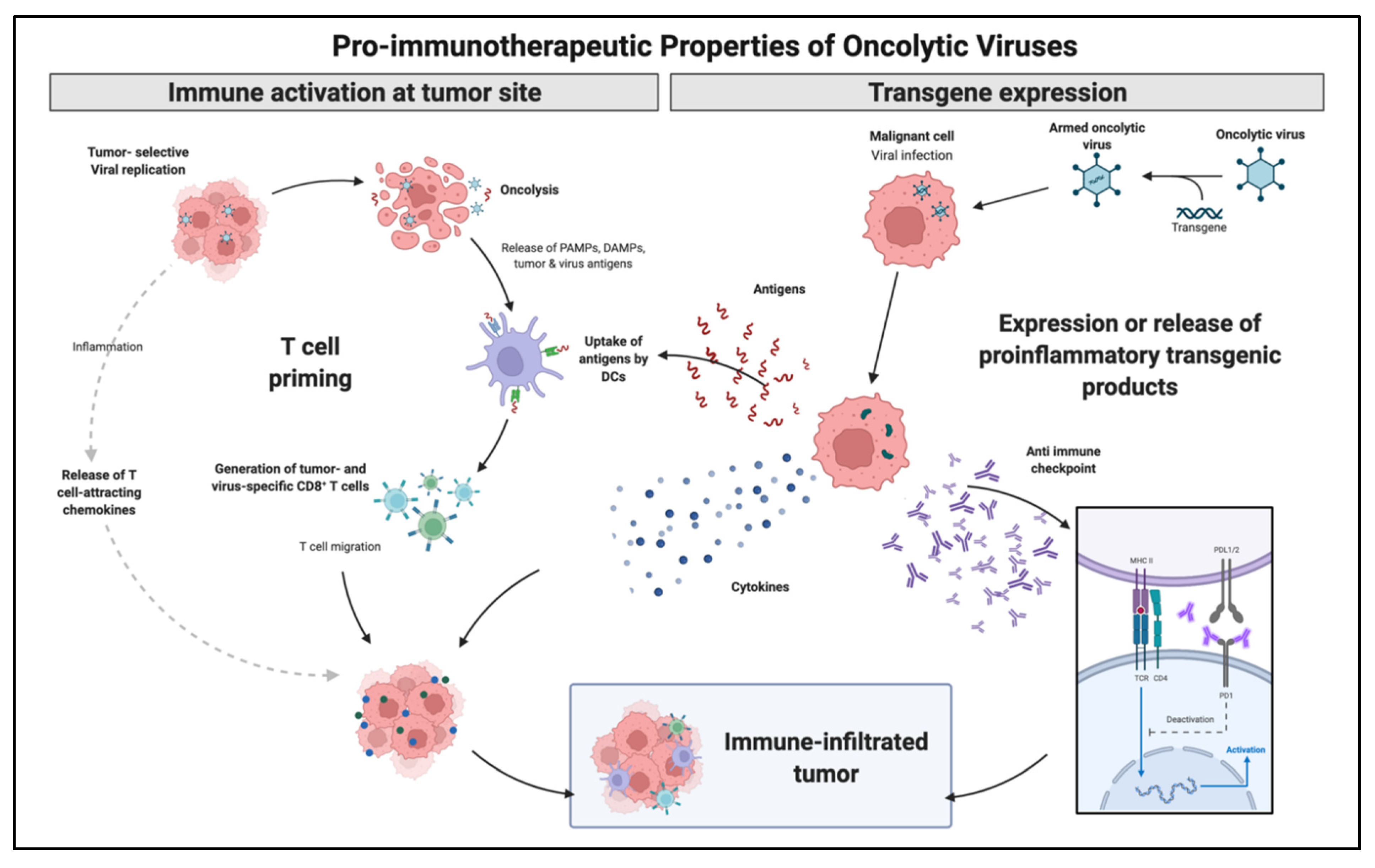
3. Oncolytic Viruses and Immuno-Oncologic Modulation
Oncolytic Viruses and Immunotherapeutic Drugs
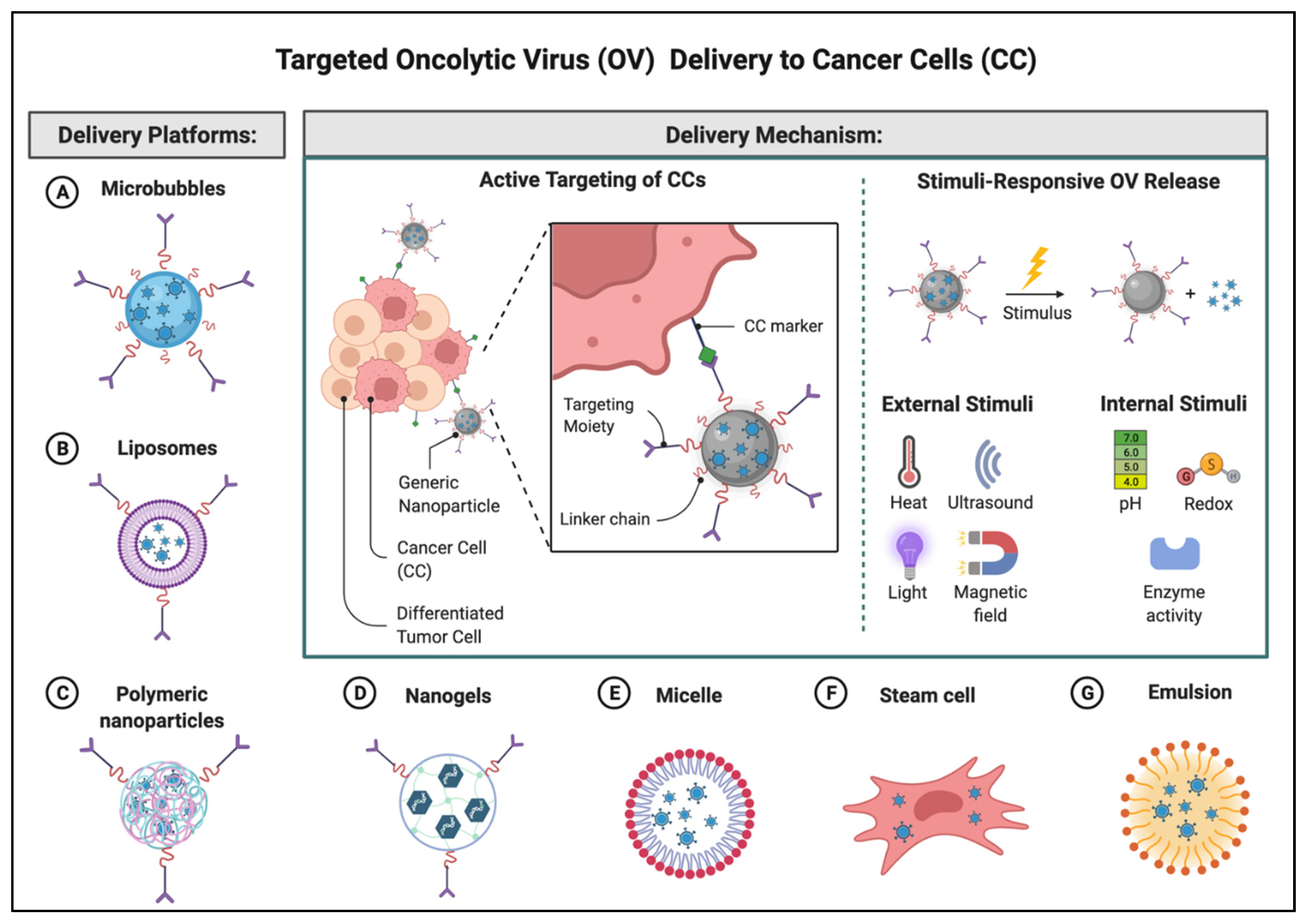
4. Delivery Systems
5. Preclinical Models
Human Immune System Engraftment Approaches
6. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Esfahani, K.; Roudaia, L.; Buhlaiga, N.; del Rincon, S.V.; Papneja, N.; Miller, W.H. A review of cancer immunotherapy: From the past, to the present, to the future. Curr. Oncol. 2020, 27 (Suppl. 2), S87–S97. [Google Scholar] [CrossRef] [PubMed]
- Ventola, C.L. Cancer Immunotherapy, Part 3: Challenges and Future Trends. Pharm. Ther. 2017, 42, 514–521. [Google Scholar]
- Bernadic, M.; Duchon, R.; Aziri, R.; Mladosievicova, B. New principles of cancer therapy give new hope for oncological patients. Bratisl. Med. J. 2019, 120, 15–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Z.S. The 2018 Nobel Prize in medicine goes to cancer immunotherapy (editorial for BMC cancer). BMC Cancer 2018, 18, 1086. [Google Scholar] [CrossRef] [Green Version]
- Hegde, P.S.; Chen, D.S. Top 10 Challenges in Cancer Immunotherapy. Immunity 2020, 52, 17–35. [Google Scholar] [CrossRef]
- Ireson, C.R.; Alavijeh, M.S.; Palmer, A.M.; Fowler, E.R.; Jones, H.J. The role of mouse tumour models in the discovery and development of anticancer drugs. Br. J. Cancer 2019, 121, 101–108. [Google Scholar] [CrossRef]
- Guerin, E.; Man, S.; Xu, P.; Kerbel, R.S. A model of postsurgical advanced metastatic breast cancer more accurately replicates the clinical efficacy of antiangiogenic drugs. Cancer Res. 2013, 73, 2743–2748. [Google Scholar] [CrossRef] [Green Version]
- Adigbli, G.; Ménoret, S.; Cross, A.R.; Hester, J.; Issa, F.; Anegon, I. Humanization of Immunodeficient Animals for the Modeling of Transplantation, Graft Versus Host Disease, and Regenerative Medicine. Transplantation 2020, 104, 2290–2306. [Google Scholar] [CrossRef] [PubMed]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved Survival with Ipilimumab in Patients with Metastatic Melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Grosso, J.F.; Jure-Kunkel, M.N. CTLA-4 blockade in tumor models: An overview of preclinical and translational research. Cancer Immun. 2013, 13, 5. [Google Scholar]
- Ascierto, P.A.; Marincola, F.M.; Ribas, A. Anti-CTLA4 monoclonal antibodies: The past and the future in clinical application. J. Transl. Med. 2011, 9, 196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolchok, J.D.; Chiarion-Sileni, V.; Gonzalez, R.; Rutkowski, P.; Grob, J.-J.; Cowey, C.L.; Lao, C.D.; Wagstaff, J.; Schadendorf, D.; Ferrucci, P.F.; et al. Overall Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2017, 377, 1345–1356. [Google Scholar] [CrossRef] [PubMed]
- Michot, J.; Bigenwald, C.; Champiat, S.; Collins, M.; Carbonnel, F.; Postel-Vinay, S.; Berdelou, A.; Varga, A.; Bahleda, R.; Hollebecque, A.; et al. Immune-related adverse events with immune checkpoint blockade: A comprehensive review. Eur. J. Cancer 2016, 54, 139–148. [Google Scholar] [CrossRef]
- Andrews, A. Treating with Checkpoint Inhibitors-Figure $1 Million per Patient. Am. Health Drug Benefits 2015, 8, 9. [Google Scholar]
- Walunas, T.L.; Lenschow, D.J.; Bakker, C.Y.; Linsley, P.S.; Freeman, G.J.; Green, J.M.; Thompson, C.B.; Bluestone, J.A. CTLA-4 can function as a negative regulator of T cell activation. Immunity 1994, 1, 405–413. [Google Scholar] [CrossRef]
- Brunner, M.C.; Chambers, C.A.; Chan, F.K.; Hanke, J.; Winoto, A.; Allison, J.P. CTLA-4-Mediated inhibition of early events of T cell proliferation. J. Immunol. 1999, 162, 5813–5820. [Google Scholar] [PubMed]
- Linsley, P.S.; Greene, J.L.; Brady, W.; Bajorath, J.; Ledbetter, J.A.; Peach, R. Human B7-1 (CD80) and B7-2 (CD86) bind with similar avidities but distinct kinetics to CD28 and CTLA-4 receptors. Immunity 1994, 1, 793–801. [Google Scholar] [CrossRef]
- van der Merwe, P.A.; Bodian, D.L.; Daenke, S.; Linsley, P.; Davis, S.J. CD80 (B7-1) Binds Both CD28 and CTLA-4 with a Low Affinity and Very Fast Kinetics. J. Exp. Med. 1997, 185, 393–404. [Google Scholar] [CrossRef] [Green Version]
- Buchbinder, E.I.; Desai, A. CTLA-4 and PD-1 Pathways: Similarities, Differences, and Implications of Their Inhibition. Am. J. Clin. Oncol. 2016, 39, 98–106. [Google Scholar] [CrossRef] [Green Version]
- Jain, N.; Nguyen, H.; Chambers, C.; Kang, J. Dual function of CTLA-4 in regulatory T cells and conventional T cells to prevent multiorgan autoimmunity. Proc. Natl. Acad. Sci. USA 2010, 107, 1524–1528. [Google Scholar] [CrossRef] [Green Version]
- Wei, S.C.; Duffy, C.R.; Allison, J.P. Fundamental Mechanisms of Immune Checkpoint Blockade Therapy. Cancer Discov. 2018, 8, 1069–1086. [Google Scholar] [CrossRef] [Green Version]
- Joalland, N.; Scotet, E. Emerging Challenges of Preclinical Models of Anti-tumor Immunotherapeutic Strategies Utilizing Vγ9Vδ2 T Cells. Front. Immunol. 2020, 11, 992. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, R.; Sugiura, D.; Shimizu, K.; Maruhashi, T.; Watada, M.; Okazaki, I.-M.; Okazaki, T. PD-1 Primarily Targets TCR Signal in the Inhibition of Functional T Cell Activation. Front. Immunol. 2019, 10, 630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yokosuka, T.; Takamatsu, M.; Kobayashi-Imanishi, W.; Hashimoto-Tane, A.; Azuma, M.; Saito, T. Programmed cell death 1 forms negative costimulatory microclusters that directly inhibit T cell receptor signaling by recruiting phosphatase SHP2. J. Exp. Med. 2012, 209, 1201–1217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, M.; Huang, Q.; Xie, Y.; Wu, X.; Ma, H.; Zhang, Y.; Xia, Y. Improvement of the anticancer efficacy of PD-1/PD-L1 blockade via combination therapy and PD-L1 regulation. J. Hematol. Oncol. 2022, 15, 24. [Google Scholar] [CrossRef]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef] [Green Version]
- Wherry, E.J.; Kurachi, M. Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 2015, 15, 486–499. [Google Scholar] [CrossRef]
- Patsoukis, N.; Bardhan, K.; Chatterjee, P.; Sari, D.; Liu, B.; Bell, L.N.; Karoly, E.D.; Freeman, G.J.; Petkova, V.; Seth, P.; et al. PD-1 alters T-cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nat. Commun. 2015, 6, 6692. [Google Scholar] [CrossRef] [Green Version]
- Braun, M.Y. The Natural History of T Cell Metabolism. Int. J. Mol. Sci. 2021, 22, 6779. [Google Scholar] [CrossRef] [PubMed]
- Kaminski, H.; Lemoine, M.; Pradeu, T. Immunological exhaustion: How to make a disparate concept operational? PLoS Pathog. 2021, 17, e1009892. [Google Scholar] [CrossRef]
- Nair, V.S.; Saleh, R.; Toor, S.M.; Taha, R.Z.; Ahmed, A.A.; Kurer, M.A.; Murshed, K.; Abu Nada, M.; Elkord, E. Epigenetic regulation of immune checkpoints and T cell exhaustion markers in tumor-infiltrating T cells of colorectal cancer patients. Epigenomics 2020, 12, 1871–1882. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Hakeem, M.S.; Manne, S.; Beltra, J.-C.; Stelekati, E.; Chen, Z.; Nzingha, K.; Ali, M.-A.; Johnson, J.L.; Giles, J.R.; Mathew, D.; et al. Epigenetic scarring of exhausted T cells hinders memory differentiation upon eliminating chronic antigenic stimulation. Nat. Immunol. 2021, 22, 1008–1019. [Google Scholar] [CrossRef] [PubMed]
- Arlauckas, S.P.; Garris, C.S.; Kohler, R.H.; Kitaoka, M.; Cuccarese, M.F.; Yang, K.S.; Miller, M.A.; Carlson, J.C.; Freeman, G.J.; Anthony, R.M.; et al. In vivo imaging reveals a tumor-associated macrophage–mediated resistance pathway in anti–PD-1 therapy. Sci. Transl. Med. 2017, 9, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeNardo, D.G.; Ruffell, B. Macrophages as regulators of tumour immunity and immunotherapy. Nat. Rev. Immunol. 2019, 19, 369–382. [Google Scholar] [CrossRef] [PubMed]
- Peranzoni, E.; Lemoine, J.; Vimeux, L.; Feuillet, V.; Barrin, S.; Kantari-Mimoun, C.; Bercovici, N.; Guérin, M.; Biton, J.; Ouakrim, H.; et al. Macrophages impede CD8 T cells from reaching tumor cells and limit the efficacy of anti–PD-1 treatment. Proc. Natl. Acad. Sci. USA 2018, 115, E4041–E4050. [Google Scholar] [CrossRef] [Green Version]
- Fridman, W.H.; Zitvogel, L.; Sautes-Fridman, C.; Kroemer, G. The immune contexture in cancer prognosis and treatment. Nat. Rev. Clin. Oncol. 2017, 14, 717–734. [Google Scholar] [CrossRef]
- Mantovani, A.; Marchesi, F.; Malesci, A.; Laghi, L.; Allavena, P. Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 2017, 14, 399–416. [Google Scholar] [CrossRef] [PubMed]
- Rora, S.; Dev, K.; Agarwal, B.; Das, P.; Syed, M.A. Macrophages: Their role, activation and polarization in pulmonary diseases. Immunobiology 2017, 223, 383–396. [Google Scholar] [CrossRef]
- Pan, Y.; Yu, Y.; Wang, X.; Zhang, T. Tumor-Associated Macrophages in Tumor Immunity. Front. Immunol. 2020, 11, 583084. [Google Scholar] [CrossRef]
- Tokunaga, R.; Zhang, W.; Naseem, M.; Puccini, A.; Berger, M.D.; Soni, S.; McSkane, M.; Baba, H.; Lenz, H.-J. CXCL9, CXCL10, CXCL11/CXCR3 axis for immune activation—A target for novel cancer therapy. Cancer Treat. Rev. 2018, 63, 40–47. [Google Scholar] [CrossRef]
- Zwirner, N.W.; Ziblat, A. Regulation of NK Cell Activation and Effector Functions by the IL-12 Family of Cytokines: The Case of IL-27. Front. Immunol. 2017, 8, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeda, N.; O’Dea, E.L.; Doedens, A.; Kim, J.-W.; Weidemann, A.; Stockmann, C.; Asagiri, M.; Simon, M.C.; Hoffmann, A.; Johnson, R.S. Differential activation and antagonistic function of HIF-α isoforms in macrophages are essential for NO homeostasis. Genes Dev. 2010, 24, 491–501. [Google Scholar] [CrossRef] [Green Version]
- Franklin, R.A.; Liao, W.; Sarkar, A.; Kim, M.V.; Bivona, M.R.; Liu, K.; Pamer, E.G.; Li, M.O. The cellular and molecular origin of tumor-associated macrophages. Science 2014, 344, 921–925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.; Zheng, M.; Kim, B.; Rouse, B.T. Role of matrix metalloproteinase-9 in angiogenesis caused by ocular infection with herpes simplex virus. J. Clin. Investig. 2002, 110, 1105–1111. [Google Scholar] [CrossRef] [PubMed]
- Asano, K.; Nabeyama, A.; Miyake, Y.; Qiu, C.-H.; Kurita, A.; Tomura, M.; Kanagawa, O.; Fujii, S.-I.; Tanaka, M. CD169-Positive Macrophages Dominate Antitumor Immunity by Crosspresenting Dead Cell-Associated Antigens. Immunity. 2011, 34, 85–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawaguchi, S.; Kawahara, K.; Fujiwara, Y.; Ohnishi, K.; Pan, C.; Yano, H.; Hirosue, A.; Nagata, M.; Hirayama, M.; Sakata, J.; et al. Naringenin potentiates anti-tumor immunity against oral cancer by inducing lymph node CD169-positive macrophage activation and cytotoxic T cell infiltration. Cancer Immunol. Immunother. 2022, 1–13. [Google Scholar] [CrossRef]
- Kim, R.; Emi, M.; Tanabe, K. Cancer immunoediting from immune surveillance to immune escape. Immunology. 2007, 121, 1–14. [Google Scholar] [CrossRef]
- Hashimoto, W.; Osaki, T.; Okamura, H.; Robbins, P.D.; Kurimoto, M.; Nagata, S.; Lotze, M.T.; Tahara, H. Differential antitumor effects of administration of recombinant IL-18 or recombinant IL-12 are mediated primarily by Fas-Fas ligand- and perforin-induced tumor apoptosis, respectively. J. Immunol. 1999, 163, 583–589. [Google Scholar]
- Böttcher, J.P.; Bonavita, E.; Chakravarty, P.; Blees, H.; Cabeza-Cabrerizo, M.; Sammicheli, S.; Rogers, N.C.; Sahai, E.; Zelenay, S.; e Sousa, C.R. NK Cells Stimulate Recruitment of cDC1 into the Tumor Microenvironment Promoting Cancer Immune Control. Cell. 2018, 172, 1022–1037.e14. [Google Scholar] [CrossRef] [Green Version]
- López-Soto, A.; Gonzalez, S.; Smyth, M.J.; Galluzzi, L. Control of Metastasis by NK Cells. Cancer Cell. 2017, 32, 135–154. [Google Scholar] [CrossRef] [PubMed]
- Ribas, A.; Wolchok, J.D. Cancer immunotherapy using checkpoint blockade. Science 2018, 359, 1350–1355. [Google Scholar] [CrossRef] [Green Version]
- O’Donnell, J.S.; Teng, M.W.L.; Smyth, M.J. Cancer immunoediting and resistance to T cell-based immunotherapy. Nat. Rev. Clin. Oncol. 2019, 16, 151–167. [Google Scholar] [CrossRef]
- Bandola-Simon, J.; Roche, P.A. Dysfunction of antigen processing and presentation by dendritic cells in cancer. Mol. Immunol. 2019, 113, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Mohme, M.; Neidert, M.C. Tumor-Specific T Cell Activation in Malignant Brain Tumors. Front. Immunol. 2020, 11, 205. [Google Scholar] [CrossRef] [PubMed]
- Mittal, D.; Gubin, M.M.; Schreiber, R.D.; Smyth, M.J. New insights into cancer immunoediting and its three component phases—elimination, equilibrium and escape. Curr. Opin. Immunol. 2014, 27, 16–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; van der Merwe, P.A.; Sivakumar, S. Biomarkers of response to PD-1 pathway blockade. Br. J. Cancer 2022, 126, 1663–1675. [Google Scholar] [CrossRef]
- Hellmann, M.D.; Nathanson, T.; Rizvi, H.; Creelan, B.C.; Sanchez-Vega, F.; Ahuja, A.; Ni, A.; Novik, J.B.; Mangarin, L.M.; Abu-Akeel, M.; et al. Genomic Features of Response to Combination Immunotherapy in Patients with Advanced Non-Small-Cell Lung Cancer. Cancer Cell 2018, 33, 843–852.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duan, F.; Duitama, J.; Al Seesi, S.; Ayres, C.M.; Corcelli, S.A.; Pawashe, A.P.; Blanchard, T.; McMahon, D.; Sidney, J.; Sette, A.; et al. Genomic and bioinformatic profiling of mutational neoepitopes reveals new rules to predict anticancer immunogenicity. J. Exp. Med. 2014, 211, 2231–2248. [Google Scholar] [CrossRef]
- Kim, S.; Kim, H.S.; Kim, E.; Lee, M.G.; Shin, E.-C.; Paik, S.; Kim, S. Neopepsee: Accurate genome-level prediction of neoantigens by harnessing sequence and amino acid immunogenicity information. Ann. Oncol. 2018, 29, 1030–1036. [Google Scholar] [CrossRef] [PubMed]
- Yarchoan, M.; Hopkins, A.; Jaffee, E.M. Tumor Mutational Burden and Response Rate to PD-1 Inhibition. N. Engl. J. Med. 2017, 377, 2500–2501. [Google Scholar] [CrossRef]
- Zhao, P.; Li, L.; Jiang, X.; Li, Q. Mismatch repair deficiency/microsatellite instability-high as a predictor for anti-PD-1/PD-L1 immunotherapy efficacy. J. Hematol. Oncol. 2019, 12, 54. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Tong, Z.; Zhang, W.; Zhang, W.; Buzdin, A.; Mu, X.; Yan, Q.; Zhao, X.; Chang, H.-H.; Duhon, M.; et al. FDA-Approved and Emerging Next Generation Predictive Biomarkers for Immune Checkpoint Inhibitors in Cancer Patients. Front. Oncol. 2021, 11, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Trujillo, J.A.; Sweis, R.F.; Bao, R.; Luke, J.J. T Cell-Inflamed versus Non-T Cell-Inflamed Tumors: A Conceptual Framework for Cancer Immunotherapy Drug Development and Combination Therapy Selection. Cancer Immunol. Res. 2018, 6, 990–1000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonaventura, P.; Shekarian, T.; Alcazer, V.; Valladeau-Guilemond, J.; Valsesia-Wittmann, S.; Amigorena, S.; Caux, C.; Depil, S. Cold Tumors: A Therapeutic Challenge for Immunotherapy. Front. Immunol. 2019, 10, 168. [Google Scholar] [CrossRef] [Green Version]
- Cerullo, V.; Koski, A.; Vähä-Koskela, M.; Hemminki, A. Oncolytic Adenoviruses for Cancer Immunotherapy. ScienceDirect 2012, 115, 265–318. [Google Scholar] [CrossRef]
- Cerullo, V.; Pesonen, S.; Diaconu, I.; Escutenaire, S.; Arstila, P.T.; Ugolini, M.; Nokisalmi, P.; Raki, M.; Laasonen, L.; Särkioja, M.; et al. Oncolytic Adenovirus Coding for Granulocyte Macrophage Colony-Stimulating Factor Induces Antitumoral Immunity in Cancer Patients. Cancer Res. 2010, 70, 4297–4309. [Google Scholar] [CrossRef] [Green Version]
- Keshavarz-Fathi, M.; Rezaei, N. Candidate Cancers for Vaccination. In Vaccines for Cancer Immunotherapy; Elsevier: Amsterdam, The Netherlands, 2019; pp. 145–152. [Google Scholar] [CrossRef]
- Kantoff, P.W.; Higano, C.S.; Shore, N.D.; Berger, E.R.; Small, E.J.; Penson, D.F.; Redfern, C.H.; Ferrari, A.C.; Dreicer, R.; Sims, R.B.; et al. Sipuleucel-T Immunotherapy for Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2010, 363, 411–422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiocca, E.A.; Rabkin, S.D. Oncolytic Viruses and Their Application to Cancer Immunotherapy. Cancer Immunol. Res. 2014, 2, 295–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ott, P.A.; Hodi, F.S.; Kaufman, H.L.; Wigginton, J.M.; Wolchok, J.D. Combination immunotherapy: A road map. J. ImmunoTherapy Cancer 2017, 5, 16. [Google Scholar] [CrossRef] [Green Version]
- Chesney, J.; Puzanov, I.; Collichio, F.; Singh, P.; Milhem, M.M.; Glaspy, J.; Hamid, O.; Ross, M.; Friedlander, P.; Garbe, C.; et al. Randomized, Open-Label Phase II Study Evaluating the Efficacy and Safety of Talimogene Laherparepvec in Combination With Ipilimumab Versus Ipilimumab Alone in Patients With Advanced, Unresectable Melanoma. J. Clin. Oncol. 2018, 36, 1658–1667. [Google Scholar] [CrossRef]
- Lu, L.; Tao, H.; Chang, A.E.; Jian-Chuan, X.; Shu, G.; Chen, Q.; Egenti, M.; Owen, J.; Moyer, J.S.; Prince, M.E.; et al. Cancer stem cell vaccine inhibits metastases of primary tumors and induces humoral immune responses against cancer stem cells. Oncoimmunology 2015, 4, e990767. [Google Scholar] [CrossRef] [PubMed]
- Pan, Q.; Li, Q.; Liu, S.; Ning, N.; Zhang, X.; Xu, Y.; Chang, A.E.; Wicha, M.S. Concise Review: Targeting Cancer Stem Cells Using Immunologic Approaches. Stem Cells 2015, 33, 2085–2092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dashti, A.; Ebrahimi, M.; Hadjati, J.; Memarnejadian, A.; Moazzeni, S.M. Dendritic cell based immunotherapy using tumor stem cells mediates potent antitumor immune responses. Cancer Lett. 2016, 374, 175–185. [Google Scholar] [CrossRef] [PubMed]
- Naz, F.; Shi, M.; Sajid, S.; Yang, Z.; Yu, C. Cancer stem cells: A major culprit of intra-tumor heterogeneity. Am. J. Cancer Res. 2021, 11, 5782–5811. [Google Scholar]
- Dillman, R.O. Is there a role for therapeutic cancer vaccines in the age of checkpoint inhibitors? Hum. Vaccin Immunother. 2017, 13, 528–532. [Google Scholar] [CrossRef] [Green Version]
- Zheng, F.; Dang, J.; Zhang, H.; Xu, F.; Ba, D.; Zhang, B.; Cheng, F.; Chang, A.E.; Wicha, M.S.; Li, Q. Cancer Stem Cell Vaccination With PD-L1 and CTLA-4 Blockades Enhances the Eradication of Melanoma Stem Cells in a Mouse Tumor Model. J. Immunother. 2018, 41, 361–368. [Google Scholar] [CrossRef]
- Feins, S.; Kong, W.; Williams, E.F.; Milone, M.C.; Fraietta, J.A. An introduction to chimeric antigen receptor (CAR) T-cell immunotherapy for human cancer. Am. J. Hematol. 2019, 94 (Suppl. 1), S3–S9. [Google Scholar] [CrossRef] [Green Version]
- Porter, D.L.; Levine, B.L.; Kalos, M.; Bagg, A.; June, C.H. Chimeric Antigen Receptor–Modified T Cells in Chronic Lymphoid Leukemia. N. Engl. J. Med. 2011, 365, 725–733. [Google Scholar] [CrossRef] [Green Version]
- Kalos, M.; Levine, B.L.; Porter, D.L.; Katz, S.; Grupp, S.A.; Bagg, A.; June, C.H. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci. Transl. Med. 2011, 3, 95ra73. [Google Scholar] [CrossRef] [Green Version]
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.F.; et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 2014, 371, 1507–1517. [Google Scholar] [CrossRef] [Green Version]
- Park, J.H.; Rivière, I.; Gonen, M.; Wang, X.; Sénéchal, B.; Curran, K.J.; Sauter, C.; Wang, Y.; Santomasso, B.; Mead, E.; et al. Long-Term Follow-up of CD19 CAR Therapy in Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 449–459. [Google Scholar] [CrossRef]
- Simula, L.; Corrado, M.; Accordi, B.; Di Rita, A.; Nazio, F.; Antonucci, Y.; Di Daniele, A.; Caicci, F.; Caruana, I.; Soriano, M.E.; et al. JNK1 and ERK1/2 modulate lymphocyte homeostasis via BIM and DRP1 upon AICD induction. Cell Death Differ. 2020, 27, 2749–2767. [Google Scholar] [CrossRef] [PubMed]
- Ivica, N.A.; Young, C.M. Tracking the CAR-T Revolution: Analysis of Clinical Trials of CAR-T and TCR-T Therapies for the Treatment of Cancer (1997–2020). Healthcare 2021, 9, 1062. [Google Scholar] [CrossRef] [PubMed]
- Huan, T.; Chen, D.; Liu, G.; Zhang, H.; Wang, X.; Wu, Z.; Wu, Y.; Xu, Q.; Yu, F. Activation-induced cell death in CAR-T cell therapy. Hum. Cell 2022, 35, 441–447. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Xiao, F.; Zhang, A.; Zhang, D.; Nie, W.; Xu, T.; Han, B.; Seth, P.; Wang, H.; Yang, Y.; et al. Oncolytic adenovirus targeting TGF-β enhances anti-tumor responses of mesothelin-targeted chimeric antigen receptor T cell therapy against breast cancer. Cell. Immunol. 2020, 348, 104041. [Google Scholar] [CrossRef]
- Yang, Y.; Xu, W.; Peng, D.; Wang, H.; Zhang, X.; Wang, H.; Xiao, F.; Zhu, Y.; Ji, Y.; Gulukota, K.; et al. An Oncolytic Adenovirus Targeting Transforming Growth Factor β Inhibits Protumorigenic Signals and Produces Immune Activation: A Novel Approach to Enhance Anti-PD-1 and Anti-CTLA-4 Therapy. Hum. Gene Ther. 2019, 30, 1117–1132. [Google Scholar] [CrossRef]
- Hassan, R.; Bera, T.; Pastan, I. Mesothelin. Clin. Cancer Res. 2004, 10, 3937–3942. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, K.; Luo, Y.; Da, T.; Guedan, S.; Ruella, M.; Scholler, J.; Keith, B.; Young, R.M.; Engels, B.; Sorsa, S.; et al. Pancreatic cancer therapy with combined mesothelin-redirected chimeric antigen receptor T cells and cytokine-armed oncolytic adenoviruses. JCI Insight. 2018, 3, e99573. [Google Scholar] [CrossRef] [Green Version]
- Marchini, A.; Scott, E.; Rommelaere, J. Overcoming Barriers in Oncolytic Virotherapy with HDAC Inhibitors and Immune Checkpoint Blockade. Viruses. 2016, 8, 9. [Google Scholar] [CrossRef] [Green Version]
- Nishikawa, T.; Tung, L.Y.; Kaneda, Y. Systemic Administration of Platelets Incorporating Inactivated Sendai Virus Eradicates Melanoma in Mice. Mol. Ther. 2014, 22, 2046–2055. [Google Scholar] [CrossRef] [Green Version]
- Denton, N.; Chen, C.Y.; Scott, T.; Cripe, T. Tumor-Associated Macrophages in Oncolytic Virotherapy: Friend or Foe? Biomedicines 2016, 4, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burke, S.; Shergold, A.; Elder, M.J.; Whitworth, J.; Cheng, X.; Jin, H.; Wilkinson, R.W.; Harper, J.; Carroll, D.K. Oncolytic Newcastle disease virus activation of the innate immune response and priming of antitumor adaptive responses in vitro. Cancer Immunol. Immunother. 2020, 69, 1015–1027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sitta, J.; Howard, C.M. Applications of Ultrasound-Mediated Drug Delivery and Gene Therapy. Int. J. Mol. Sci. 2021, 22, 11491. [Google Scholar] [CrossRef] [PubMed]
- Nande, R.; Greco, A.; Gossman, M.S.; Lopez, J.P.; Claudio, L.; Salvatore, M.; Brunetti, A.; Denvir, J.; Howard, C.; Claudio, P.P. Microbubble-assisted p53, RB, and p130 gene transfer in combination with radiation therapy in prostate cancer. Curr. Gene Therapy. 2013, 13, 163–174. [Google Scholar] [CrossRef] [Green Version]
- Howard, C.M.; Forsberg, F.; Minimo, C.; Liu, J.B.; Merton, D.A.; Claudio, P.P. Ultrasound guided site specific gene delivery system using adenoviral vectors and commercial ultrasound contrast agents. J. Cell. Physiol. 2006, 209, 413–421. [Google Scholar] [CrossRef]
- Greco, A.; Mancini, M.; Gargiulo, S.; Gramanzini, M.; Claudio, P.P.; Brunetti, A.; Salvatore, M. Ultrasound biomicroscopy in small animal research: Applications in molecular and preclinical imaging. J. Biomed. Biotechnol. 2012, 2012, 1–14. [Google Scholar] [CrossRef]
- Greco, A.; Di Benedetto, A.; Howard, C.; Kelly, S.; Nande, R.; Dementieva, Y.; Miranda, M.; Brunetti, A.; Salvatore, M.; Claudio, L.; et al. Eradication of therapy-resistant human prostate tumors using an ultrasound-guided site-specific cancer terminator virus delivery approach. Mol. Ther. 2010, 18, 295–306. [Google Scholar] [CrossRef]
- Greco, A.; Albanese, S.; Auletta, L.; De Carlo, F.; Salvatore, M.; Howard, C.M.; Claudio, P.P. Advances in molecular preclinical therapy mediated by imaging. Q. J. Nucl. Med. Mol. Imaging 2017, 61, 76–94. [Google Scholar] [CrossRef]
- De Carlo, F.; Thomas, L.; Brooke, B.; Varney, E.T.; Nande, R.; Boskovic, O.; Marshall, G.D.; Claudio, P.P.; Howard, C.M. Microbubble-mediated delivery of human adenoviruses does not elicit innate and adaptive immunity response in an immunocompetent mouse model of prostate cancer. J. Transl. Med. 2019, 17, 19. [Google Scholar] [CrossRef] [Green Version]
- Mahasa, K.J.; De Pillis, L.; Ouifki, R.; Eladdadi, A.; Maini, P.; Yoon, A.-R.; Yun, C.-O. Mesenchymal stem cells used as carrier cells of oncolytic adenovirus results in enhanced oncolytic virotherapy. Sci. Rep. 2020, 10, 425. [Google Scholar] [CrossRef]
- Bosma, G.C.; Custer, R.P.; Bosma, M.J. A severe combined immunodeficiency mutation in the mouse. Nature. 1983, 301, 527–530. [Google Scholar] [CrossRef] [PubMed]
- Gellert, M. V(D)J Recombination: RAG Proteins, Repair Factors, and Regulation. Annu. Rev. Biochem. 2002, 71, 101–132. [Google Scholar] [CrossRef] [PubMed]
- Shultz, L.D.; Schweitzer, P.A.; Christianson, S.W.; Gott, B.; Schweitzer, I.B.; Tennent, B.; McKenna, S.; Mobraaten, L.; Rajan, T.V.; Greiner, D.L. Multiple defects in innate and adaptive immunologic function in NOD/LtSz-scid mice. J. Immunol. 1995, 154, 180–191. [Google Scholar] [PubMed]
- Nagatani, M.; Kodera, T.; Suzuki, D.; Igura, S.; Fukunaga, Y.; Kanemitsu, H.; Nakamura, D.; Mochizuki, M.; Kemi, M.; Tamura, K.; et al. Comparison of biological features between severely immuno-deficient NOD/Shi-scid Il2rgnull and NOD/LtSz-scid Il2rgnull mice. Exp. Anim. 2019, 68, 471–482. [Google Scholar] [CrossRef] [Green Version]
- Marín-Jiménez, J.A.; Capasso, A.; Lewis, M.S.; Bagby, S.M.; Hartman, S.J.; Shulman, J.; Navarro, N.M.; Yu, H.; Rivard, C.J.; Wang, X.; et al. Testing Cancer Immunotherapy in a Human Immune System Mouse Model: Correlating Treatment Responses to Human Chimerism, Therapeutic Variables and Immune Cell Phenotypes. Front. Immunol. 2021, 12, 607282. [Google Scholar] [CrossRef]
- Skelton, J.K.; Ortega-Prieto, A.M.; Dorner, M. A Hitchhiker’s guide to humanized mice: New pathways to studying viral infections. Immunology 2018, 154, 50–61. [Google Scholar] [CrossRef]
- Verbiest, T.; Finnon, R.; Brown, N.; Finnon, P.; Bouffler, S.; Badie, C. NOD Scid Gamma Mice Are Permissive to Allogeneic HSC Transplantation without Prior Conditioning. Int. J. Mol. Sci. 2016, 17, 1850. [Google Scholar] [CrossRef]
- Gimeno, R.; Weijer, K.; Voordouw, A.; Uittenbogaart, C.H.; Legrand, N.; Alves, N.L.; Wijnands, E.; Blom, B.; Spits, H. Monitoring the effect of gene silencing by RNA interference in human CD34+ cells injected into newborn RAG2-/- γc-/- mice: Functional inactivation of p53 in developing T cells. Blood 2004, 104, 3886–3893. [Google Scholar] [CrossRef] [Green Version]
- Shultz, L.D.; Lyons, B.L.; Burzenski, L.M.; Gott, B.; Chen, X.; Chaleff, S.; Kotb, M.; Gillies, S.D.; King, M.; Mangada, J.; et al. Human Lymphoid and Myeloid Cell Development in NOD/LtSz- scid IL2R γ null Mice Engrafted with Mobilized Human Hemopoietic Stem Cells. J. Immunol. 2005, 174, 6477–6489. [Google Scholar] [CrossRef] [Green Version]
- Traggiai, E.; Chicha, L.; Mazzucchelli, L.; Bronz, L.; Piffaretti, J.-C.; Lanzavecchia, A.; Manz, M.G. Development of a Human Adaptive Immune System in Cord Blood Cell-Transplanted Mice. Science 2004, 304, 104–107. [Google Scholar] [CrossRef]
- Walcher, L.; Hilger, N.; Wege, A.K.; Lange, F.; Tretbar, U.S.; Blaudszun, A.; Fricke, S. Humanized mouse model: Hematopoietic stemcell transplantation and tracking using short tandem repeat technology. Immun. Inflamm. Dis. 2020, 8, 363–370. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Beltran, W.F.; Claiborne, D.T.; Maldini, C.R.; Phelps, M.B.; Vrbanac, V.; Karpel, M.E.; Krupp, K.L.; Power, K.A.; Boutwell, C.L.; Balazs, A.B.; et al. Innate Immune Reconstitution in Humanized Bone Marrow-Liver-Thymus (HuBLT) Mice Governs Adaptive Cellular Immune Function and Responses to HIV-1 Infection. Front. Immunol. 2021, 12. [Google Scholar] [CrossRef] [PubMed]
- Willinger, T.; Rongvaux, A.; Takizawa, H.; Yancopoulos, G.D.; Valenzuela, D.M.; Murphy, A.J.; Auerbach, W.; Eynon, E.E.; Stevens, S.; Manz, M.G.; et al. Human IL-3/GM-CSF knock-in mice support human alveolar macrophage development and human immune responses in the lung. Proc. Natl. Acad. Sci. USA 2011, 108, 2390–2395. [Google Scholar] [CrossRef] [PubMed] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sitta, J.; Claudio, P.P.; Howard, C.M. Virus-Based Immuno-Oncology Models. Biomedicines 2022, 10, 1441. https://doi.org/10.3390/biomedicines10061441
Sitta J, Claudio PP, Howard CM. Virus-Based Immuno-Oncology Models. Biomedicines. 2022; 10(6):1441. https://doi.org/10.3390/biomedicines10061441
Chicago/Turabian StyleSitta, Juliana, Pier Paolo Claudio, and Candace M. Howard. 2022. "Virus-Based Immuno-Oncology Models" Biomedicines 10, no. 6: 1441. https://doi.org/10.3390/biomedicines10061441
APA StyleSitta, J., Claudio, P. P., & Howard, C. M. (2022). Virus-Based Immuno-Oncology Models. Biomedicines, 10(6), 1441. https://doi.org/10.3390/biomedicines10061441