
Connexins, Pannexins and Gap Junctions in Perinatal Brain Injury

 , , and
, , and 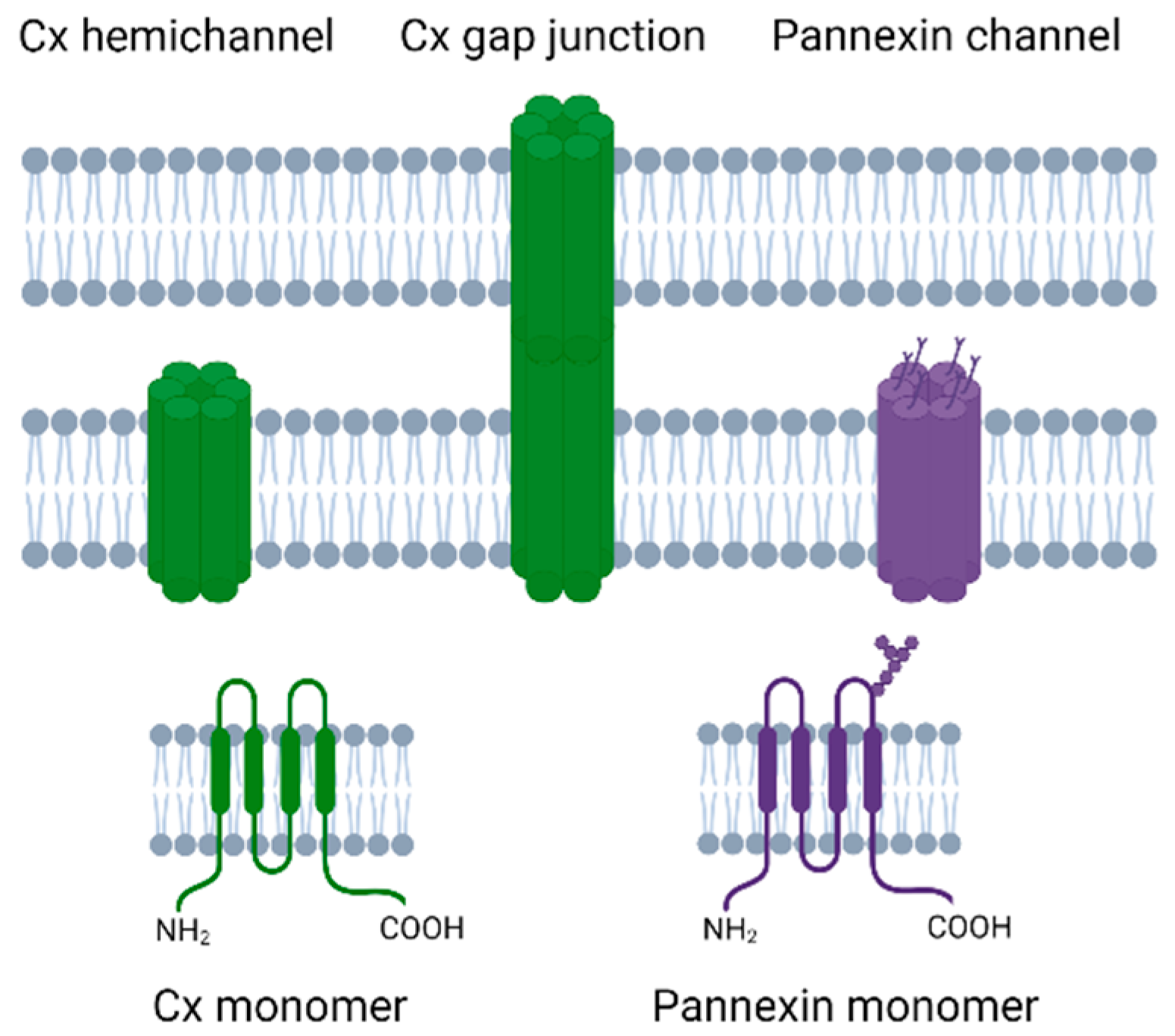{kind=link}
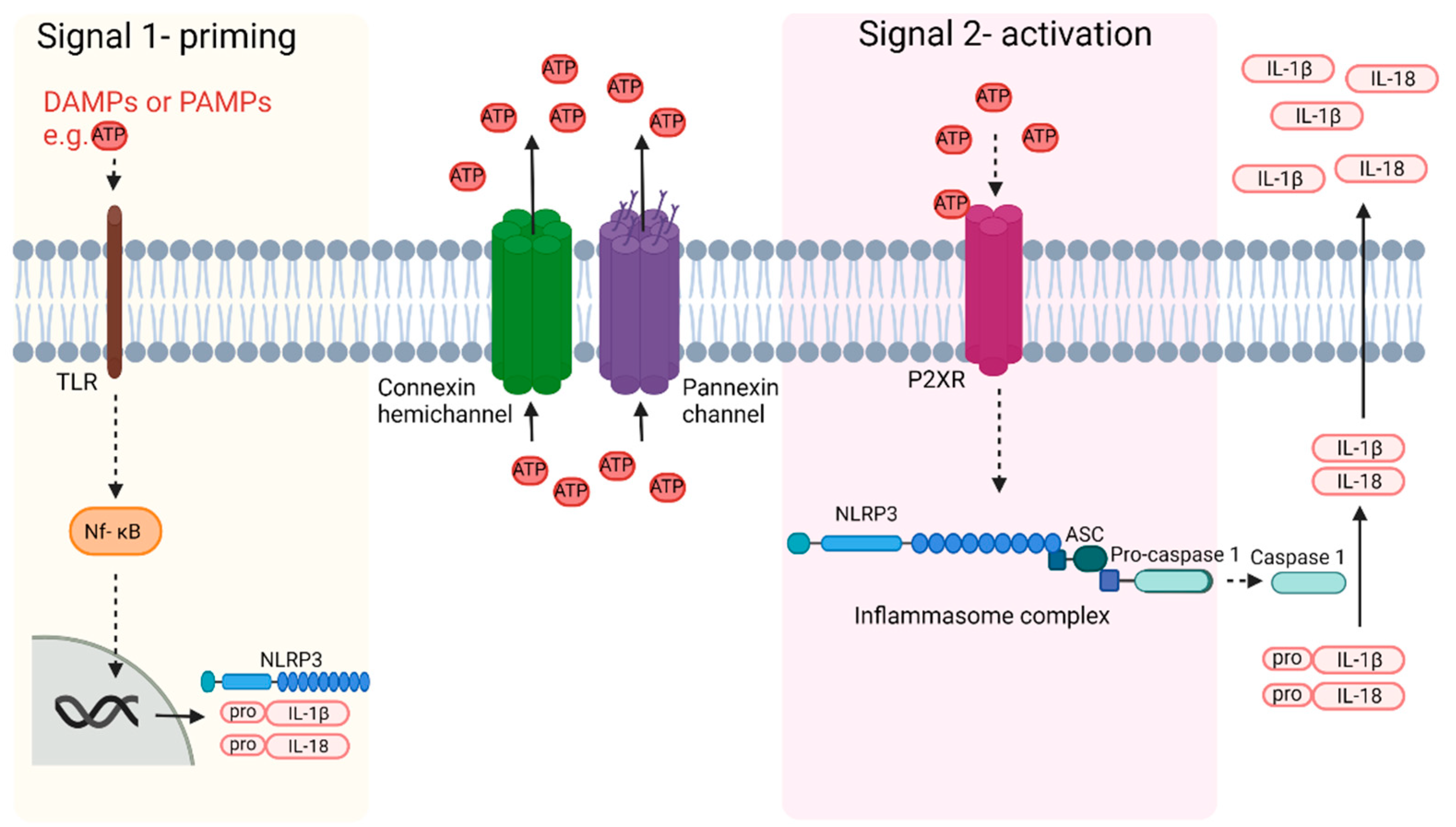{kind=link}
Abstract
:1. Introduction
2. Connexin Proteins and HI
3. Effect of HI on Gap Junctions
4. Effect of HI on Connexin Hemichannels
5. Pannexin Channels
6. Effect of HI on Pannexin Channels
7. Inflammation in Perinatal Brain Injury
8. Astrocytes Involvement in Inflammation
9. Inflammation and the Blood–Brain Barrier
10. Effect of Inflammation in the Absence of HI on Gap Junctions, Hemichannels and Pannexins
11. Role of Connexins and Pannexin Channels in Propagation of Inflammation
12. Inflammasome Activation
13. Therapeutic Potential of Connexin and Pannexin Blockers
13.1. Add on to Therapeutic Hypothermia?
13.2. Treating Perinatal Brain Injury When TH Is Not Recommemded?
14. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Battin, M.; Sadler, L.; Masson, V.; Farquhar, C. Neonatal encephalopathy in New Zealand: Demographics and clinical outcome. J. Paediatr. Child Health 2016, 52, 632–636. [Google Scholar] [CrossRef] [PubMed]
- Jonsson, M.; Agren, J.; Norden-Lindeberg, S.; Ohlin, A.; Hanson, U. Neonatal encephalopathy and the association to asphyxia in labor. Am. J. Obstet. Gynecol. 2014, 211, 667.e1–667.e8. [Google Scholar] [CrossRef] [PubMed]
- Namusoke, H.; Nannyonga, M.M.; Ssebunya, R.; Nakibuuka, V.K.; Mworozi, E. Incidence and short term outcomes of neonates with hypoxic ischemic encephalopathy in a Peri Urban teaching hospital, Uganda: A prospective cohort study. Matern. Health Neonatol. Perinatol. 2018, 4, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simiyu, I.N.; McHaile, D.N.; Katsongeri, K.; Philemon, R.N.; Msuya, S.E. Prevalence, severity and early outcomes of hypoxic ischemic encephalopathy among newborns at a tertiary hospital, in northern Tanzania. BMC Pediatr. 2017, 17, 131. [Google Scholar] [CrossRef] [Green Version]
- Chalak, L.F.; Rollins, N.; Morriss, M.C.; Brion, L.P.; Heyne, R.; Sanchez, P.J. Perinatal acidosis and hypoxic-ischemic encephalopathy in preterm infants of 33 to 35 weeks’ gestation. J. Pediatr. 2012, 160, 388–394. [Google Scholar] [CrossRef] [Green Version]
- Salhab, W.A.; Perlman, J.M. Severe fetal acidemia and subsequent neonatal encephalopathy in the larger premature infant. Pediatr. Neurol. 2005, 32, 25–29. [Google Scholar] [CrossRef]
- Schmidt, J.W.; Walsh, W.F. Hypoxic-ischemic encephalopathy in preterm infants. J. Neonatal Perinatal Med. 2010, 3, 277–284. [Google Scholar] [CrossRef]
- Manuck, T.A.; Rice, M.M.; Bailit, J.L.; Grobman, W.A.; Reddy, U.M.; Wapner, R.J.; Thorp, J.M.; Caritis, S.N.; Prasad, M.; Tita, A.T.; et al. Preterm neonatal morbidity and mortality by gestational age: A contemporary cohort. Am. J. Obstet. Gynecol. 2016, 215, 103.e1–103.e14. [Google Scholar] [CrossRef] [Green Version]
- Galinsky, R.; Polglase, G.R.; Hooper, S.B.; Black, M.J.; Moss, T.J. The consequences of chorioamnionitis: Preterm birth and effects on development. J. Pregnancy 2013, 2013, 412831. [Google Scholar] [CrossRef] [Green Version]
- Jacobs, S.E.; Berg, M.; Hunt, R.; Tarnow-Mordi, W.O.; Inder, T.E.; Davis, P.G. Cooling for newborns with hypoxic ischaemic encephalopathy. Cochrane Database Syst. Rev. 2013, 1, CD003311. [Google Scholar] [CrossRef]
- Gluckman, P.D.; Wyatt, J.S.; Azzopardi, D.; Ballard, R.; Edwards, A.D.; Ferriero, D.M.; Polin, R.A.; Robertson, C.M.; Thoresen, M.; Whitelaw, A.; et al. Selective head cooling with mild systemic hypothermia after neonatal encephalopathy: Multicentre randomised trial. Lancet 2005, 365, 663–670. [Google Scholar] [CrossRef]
- Shankaran, S.; Laptook, A.R.; Ehrenkranz, R.A.; Tyson, J.E.; McDonald, S.A.; Donovan, E.F.; Fanaroff, A.A.; Poole, W.K.; Wright, L.L.; Higgins, R.D.; et al. Whole-body hypothermia for neonates with hypoxic-ischemic encephalopathy. N. Engl. J. Med. 2005, 353, 1574–1584. [Google Scholar] [CrossRef] [PubMed]
- Azzopardi, D.V.; Strohm, B.; Edwards, A.D.; Dyet, L.; Halliday, H.L.; Juszczak, E.; Kapellou, O.; Levene, M.; Marlow, N.; Porter, E.; et al. Moderate hypothermia to treat perinatal asphyxial encephalopathy. N. Engl. J. Med. 2009, 361, 1349–1358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacobs, S.E.; Morley, C.J.; Inder, T.E.; Stewart, M.J.; Smith, K.R.; McNamara, P.J.; Wright, I.M.; Kirpalani, H.M.; Darlow, B.A.; Doyle, L.W. Whole-body hypothermia for term and near-term newborns with hypoxic-ischemic encephalopathy: A randomized controlled trial. Arch. Pediatr. Adolesc. Med. 2011, 165, 692–700. [Google Scholar] [CrossRef] [Green Version]
- McDouall, A.; Wassink, G.; Bennet, L.; Gunn, A.J.; Davidson, J.O. Challenges in developing therapeutic strategies for mild neonatal encephalopathy. Neural Regen. Res. 2022, 17, 277–282. [Google Scholar] [CrossRef] [PubMed]
- Silverman, W.A.; Fertig, J.W.; Berger, A.P. The influence of the thermal environment upon the survival of newly born premature infants. Pediatrics 1958, 22, 876–886. [Google Scholar] [CrossRef] [PubMed]
- Day, R.L.; Caliguiri, L.; Kamenski, C.; Ehrlich, F. Body temperature and survival of premature infants. Pediatrics 1964, 34, 171–181. [Google Scholar] [CrossRef] [PubMed]
- Thayyil, S.; Pant, S.; Montaldo, P.; Shukla, D.; Oliveira, V.; Ivain, P.; Bassett, P.; Swamy, R.; Mendoza, J.; Moreno-Morales, M.; et al. Hypothermia for moderate or severe neonatal encephalopathy in low-income and middle-income countries (HELIX): A randomised controlled trial in India, Sri Lanka, and Bangladesh. Lancet Glob Health 2021, 9, e1273–e1285. [Google Scholar] [CrossRef]
- Wassink, G.; Davidson, J.O.; Dhillon, S.K.; Zhou, K.; Bennet, L.; Thoresen, M.; Gunn, A.J. Therapeutic hypothermia in neonatal hypoxic-ischemic encephalopathy. Curr. Neurol. Neurosci. Rep. 2019, 19, 2. [Google Scholar] [CrossRef] [Green Version]
- Zhou, K.Q.; Davidson, J.O.; Bennet, L.; Gunn, A.J. Combination treatments with therapeutic hypothermia for hypoxic-ischemic neuroprotection. Dev. Med. Child Neurol. 2020, 62, 1131–1137. [Google Scholar] [CrossRef]
- Bennet, L.; Roelfsema, V.; Pathipati, P.; Quaedackers, J.; Gunn, A.J. Relationship between evolving epileptiform activity and delayed loss of mitochondrial activity after asphyxia measured by near-infrared spectroscopy in preterm fetal sheep. J. Physiol. 2006, 572, 141–154. [Google Scholar] [CrossRef]
- Geddes, R.; Vannucci, R.C.; Vannucci, S.J. Delayed cerebral atrophy following moderate hypoxia-ischemia in the immature rat. Dev. Neurosci. 2001, 23, 180–185. [Google Scholar] [CrossRef] [PubMed]
- Davidson, J.O.; Dean, J.M.; Fraser, M.; Wassink, G.; Andelius, T.C.; Dhillon, S.K.; Bennet, L.; Gunn, A.J. Perinatal brain injury: Mechanisms and therapeutic approaches. Front. Biosci. (Landmark Ed.) 2018, 23, 2204–2226. [Google Scholar] [CrossRef] [PubMed]
- Tan, W.K.; Williams, C.E.; During, M.J.; Mallard, C.E.; Gunning, M.I.; Gunn, A.J.; Gluckman, P.D. Accumulation of cytotoxins during the development of seizures and edema after hypoxic-ischemic injury in late gestation fetal sheep. Pediatr. Res. 1996, 39, 791–797. [Google Scholar] [CrossRef] [Green Version]
- Zhou, K.Q.; Green, C.R.; Bennet, L.; Gunn, A.J.; Davidson, J.O. The role of connexin and pannexin channels in perinatal brain injury and inflammation. Front, Physiol. 2019, 10, 141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azzopardi, D.; Wyatt, J.S.; Cady, E.B.; Delpy, D.T.; Baudin, J.; Stewart, A.L.; Hope, P.L.; Hamilton, P.A.; Reynolds, E.O. Prognosis of newborn infants with hypoxic-ischemic brain injury assessed by phosphorus magnetic resonance spectroscopy. Pediatr. Res. 1989, 25, 445–451. [Google Scholar] [CrossRef] [Green Version]
- Gunn, A.J.; Gunn, T.R.; de Haan, H.H.; Williams, C.E.; Gluckman, P.D. Dramatic neuronal rescue with prolonged selective head cooling after ischemia in fetal lambs. J. Clin. Investig. 1997, 99, 248–256. [Google Scholar] [CrossRef]
- Fleiss, B.; Gressens, P. Tertiary mechanisms of brain damage: A new hope for treatment of cerebral palsy? Lancet Neurol. 2012, 11, 556–566. [Google Scholar] [CrossRef]
- Ness, J.K.; Romanko, M.J.; Rothstein, R.P.; Wood, T.L.; Levison, S.W. Perinatal hypoxia-ischemia induces apoptotic and excitotoxic death of periventricular white matter oligodendrocyte progenitors. Dev. Neurosci. 2001, 23, 203–208. [Google Scholar] [CrossRef]
- Romanko, M.J.; Rothstein, R.P.; Levison, S.W. Neural stem cells in the subventricular zone are resilient to hypoxia/ischemia whereas progenitors are vulnerable. J. Cereb. Blood Flow Metab. 2004, 24, 814–825. [Google Scholar] [CrossRef] [Green Version]
- Mugisho, O.O.; Rupenthal, I.D.; Paquet-Durand, F.; Acosta, M.L.; Green, C.R. Targeting connexin hemichannels to control the inflammasome: The correlation between connexin43 and NLRP3 expression in chronic eye disease. Expert Opin. Ther. Targets 2019, 23, 855–863. [Google Scholar] [CrossRef]
- Leybaert, L.; Lampe, P.D.; Dhein, S.; Kwak, B.R.; Ferdinandy, P.; Beyer, E.C.; Laird, D.W.; Naus, C.C.; Green, C.R.; Schulz, R. Connexins in cardiovascular and neurovascular health and disease: Pharmacological implications. Pharmacol. Rev. 2017, 69, 396–478. [Google Scholar] [CrossRef] [PubMed]
- Unger, V.M.; Kumar, N.M.; Gilula, N.B.; Yeager, M. Three-dimensional structure of a recombinant gap junction membrane channel. Science 1999, 283, 1176–1180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theis, M.; Sohl, G.; Eiberger, J.; Willecke, K. Emerging complexities in identity and function of glial connexins. Trends Neurosci. 2005, 28, 188–195. [Google Scholar] [CrossRef] [PubMed]
- Dermietzel, R.; Gao, Y.; Scemes, E.; Vieira, D.; Urban, M.; Kremer, M.; Bennett, M.V.; Spray, D.C. Connexin43 null mice reveal that astrocytes express multiple connexins. Brain Res. Brain Res. Rev. 2000, 32, 45–56. [Google Scholar] [CrossRef]
- Nagy, J.I.; Ionescu, A.V.; Lynn, B.D.; Rash, J.E. Coupling of astrocyte connexins Cx26, Cx30, Cx43 to oligodendrocyte Cx29, Cx32, Cx47: Implications from normal and connexin32 knockout mice. Glia 2003, 44, 205–218. [Google Scholar] [CrossRef] [Green Version]
- Davidson, J.O.; Green, C.R.; Bennet, L.; Nicholson, L.F.; Danesh-Meyer, H.; Carroll, S.J.; Gunn, A.J. A key role for connexin hemichannels in spreading ischemic brain injury. Curr. Drug Targets 2013, 14, 36–46. [Google Scholar] [CrossRef]
- Nakase, T.; Yoshida, Y.; Nagata, K. Enhanced Connexin 43 immunoreactivity in penumbral areas in the human brain following ischemia. Glia 2006, 54, 369–375. [Google Scholar] [CrossRef]
- Nakase, T.; Maeda, T.; Yoshida, Y.; Nagata, K. Ischemia alters the expression of connexins in the aged human brain. J. Biomed. Biotechnol. 2009, 2009, 147946. [Google Scholar] [CrossRef]
- Wang, J.; Ma, A.; Xi, J.; Wang, Y.; Zhao, B. Connexin 43 and its hemichannels mediate hypoxia-ischemia-induced cell death in neonatal rats. Child Neurol. Open 2014, 1, 2329048X14544955. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Zhao, H.; Tan, X.; Kostrzewa, R.M.; Du, G.; Chen, Y.; Zhu, J.; Miao, Z.; Yu, H.; Kong, J.; et al. Inhibition of connexin43 improves functional recovery after ischemic brain injury in neonatal rats. Glia 2015, 63, 1553–1567. [Google Scholar] [CrossRef]
- Davidson, J.O.; Green, C.R.; Nicholson, L.F.B.; O’Carroll, S.J.; Fraser, M.; Bennet, L.; Gunn, A.J. Connexin hemichannel blockade improves outcomes in a model of fetal ischemia. Ann. Neurol. 2012, 71, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Davidson, J.O.; Green, C.R.; Nicholson, L.F.; Bennet, L.; Gunn, A.J. Deleterious effects of high dose connexin 43 mimetic peptide infusion after cerebral ischaemia in near-term fetal sheep. Int. J. Mol. Sci. 2012, 13, 6303–6319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeinieh, M.P.; Talhouk, R.S.; El-Sabban, M.E.; Mikati, M.A. Differential expression of hippocampal connexins after acute hypoxia in the developing brain. Brain Dev. 2010, 32, 810–817. [Google Scholar] [CrossRef] [PubMed]
- Belliveau, D.J.; Kidder, G.M.; Naus, C.C. Expression of gap junction genes during postnatal neural development. Dev. Genet. 1991, 12, 308–317. [Google Scholar] [CrossRef]
- Sadowska, G.B.; Stopa, E.G.; Stonestreet, B.S. Ontogeny of connexin 32 and 43 expression in the cerebral cortices of ovine fetuses, newborns, and adults. Brain Res. 2009, 1255, 51–56. [Google Scholar] [CrossRef] [Green Version]
- Nadarajah, B.; Jones, A.M.; Evans, W.H.; Parnavelas, J.G. Differential expression of connexins during neocortical development and neuronal circuit formation. J. Neurosci. 1997, 17, 3096–3111. [Google Scholar] [CrossRef] [Green Version]
- Nakase, T.; Fushiki, S.; Naus, C.C. Astrocytic gap junctions composed of connexin 43 reduce apoptotic neuronal damage in cerebral ischemia. Stroke 2003, 34, 1987–1993. [Google Scholar] [CrossRef] [Green Version]
- Siushansian, R.; Bechberger, J.F.; Cechetto, D.F.; Hachinski, V.C.; Naus, C.C. Connexin43 null mutation increases infarct size after stroke. J. Comp. Neurol. 2001, 440, 387–394. [Google Scholar] [CrossRef]
- Nakase, T.; Sohl, G.; Theis, M.; Willecke, K.; Naus, C.C. Increased apoptosis and inflammation after focal brain ischemia in mice lacking connexin43 in astrocytes. Am. J. Pathol. 2004, 164, 2067–2075. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Qin, C.; Luo, X.; Wang, J.; Wang, X.; Xie, M.; Hu, J.; Cao, J.; Hu, T.; Goldman, S.A.; et al. Astrocytic connexin 43 potentiates myelin injury in ischemic white matter disease. Theranostics 2019, 9, 4474–4493. [Google Scholar] [CrossRef]
- Krysko, D.V.; Leybaert, L.; Vandenabeele, P.; D’Herde, K. Gap junctions and the propagation of cell survival and cell death signals. Apoptosis 2005, 10, 459–469. [Google Scholar] [CrossRef] [PubMed]
- Cotrina, M.L.; Kang, J.; Lin, J.H.; Bueno, E.; Hansen, T.W.; He, L.; Liu, Y.; Nedergaard, M. Astrocytic gap junctions remain open during ischemic conditions. J. Neurosci. 1998, 18, 2520–2537. [Google Scholar] [CrossRef] [PubMed]
- Li, W.E.; Nagy, J.I. Connexin43 phosphorylation state and intercellular communication in cultured astrocytes following hypoxia and protein phosphatase inhibition. Eur. J. Neurosci. 2000, 12, 2644–2650. [Google Scholar] [CrossRef]
- Rawanduzy, A.; Hansen, A.; Hansen, T.W.; Nedergaard, M. Effective reduction of infarct volume by gap junction blockade in a rodent model of stroke. J. Neurosurg. 1997, 87, 916–920. [Google Scholar] [CrossRef] [PubMed]
- de Pina-Benabou, M.H.; Szostak, V.; Kyrozis, A.; Rempe, D.; Uziel, D.; Urban-Maldonado, M.; Benabou, S.; Spray, D.C.; Federoff, H.J.; Stanton, P.K.; et al. Blockade of gap junctions in vivo provides neuroprotection after perinatal global ischemia. Stroke 2005, 36, 2232–2237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Decrock, E.; Vinken, M.; De Vuyst, E.; Krysko, D.V.; D’Herde, K.; Vanhaecke, T.; Vandenabeele, P.; Rogiers, V.; Leybaert, L. Connexin-related signaling in cell death: To live or let die? Cell Death Differ. 2009, 16, 524–536. [Google Scholar] [CrossRef] [PubMed]
- Ozog, M.A.; Siushansian, R.; Naus, C.C. Blocked gap junctional coupling increases glutamate-induced neurotoxicity in neuron-astrocyte co-cultures. J. Neuropathol. Exp. Neurol. 2002, 61, 132–141. [Google Scholar] [CrossRef] [Green Version]
- O’Carroll, S.J.; Alkadhi, M.; Nicholson, L.F.; Green, C.R. Connexin 43 mimetic peptides reduce swelling, astrogliosis, and neuronal cell death after spinal cord injury. Cell Commun. Adhes. 2008, 15, 27–42. [Google Scholar] [CrossRef] [Green Version]
- Decrock, E.; De Vuyst, E.; Vinken, M.; Van Moorhem, M.; Vranckx, K.; Wang, N.; Van Laeken, L.; De Bock, M.; D’Herde, K.; Lai, C.P.; et al. Connexin 43 hemichannels contribute to the propagation of apoptotic cell death in a rat C6 glioma cell model. Cell Death Differ. 2009, 16, 151–163. [Google Scholar] [CrossRef] [Green Version]
- Contreras, J.E.; Saez, J.C.; Bukauskas, F.F.; Bennett, M.V. Gating and regulation of connexin 43 (Cx43) hemichannels. Proc. Natl. Acad. Sci. USA 2003, 100, 11388–11393. [Google Scholar] [CrossRef] [Green Version]
- Contreras, J.E.; Sanchez, H.A.; Eugenin, E.A.; Speidel, D.; Theis, M.; Willecke, K.; Bukauskas, F.F.; Bennett, M.V.; Saez, J.C. Metabolic inhibition induces opening of unapposed connexin 43 gap junction hemichannels and reduces gap junctional communication in cortical astrocytes in culture. Proc. Natl. Acad. Sci. USA 2002, 99, 495–500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orellana, J.A.; Hernandez, D.E.; Ezan, P.; Velarde, V.; Bennett, M.V.; Giaume, C.; Saez, J.C. Hypoxia in high glucose followed by reoxygenation in normal glucose reduces the viability of cortical astrocytes through increased permeability of connexin 43 hemichannels. Glia 2010, 58, 329–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Retamal, M.A.; Cortes, C.J.; Reuss, L.; Bennett, M.V.; Saez, J.C. S-nitrosylation and permeation through connexin 43 hemichannels in astrocytes: Induction by oxidant stress and reversal by reducing agents. Proc. Natl. Acad. Sci. USA 2006, 103, 4475–4480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lillo, M.A.; Himelman, E.; Shirokova, N.; Xie, L.H.; Fraidenraich, D.; Contreras, J.E. S-nitrosylation of connexin43 hemichannels elicits cardiac stress-induced arrhythmias in Duchenne muscular dystrophy mice. JCI Insight 2019, 4, e130091. [Google Scholar] [CrossRef] [PubMed]
- Evans, W.H.; De Vuyst, E.; Leybaert, L. The gap junction cellular internet: Connexin hemichannels enter the signalling limelight. Biochem. J. 2006, 397, 1–14. [Google Scholar] [CrossRef]
- Paul, D.L.; Ebihara, L.; Takemoto, L.J.; Swenson, K.I.; Goodenough, D.A. Connexin46, a novel lens gap junction protein, induces voltage-gated currents in nonjunctional plasma membrane of Xenopus oocytes. J. Cell Biol. 1991, 115, 1077–1089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomez-Hernandez, J.M.; de Miguel, M.; Larrosa, B.; Gonzalez, D.; Barrio, L.C. Molecular basis of calcium regulation in connexin-32 hemichannels. Proc. Natl. Acad. Sci. USA 2003, 100, 16030–16035. [Google Scholar] [CrossRef] [Green Version]
- Takeuchi, H.; Jin, S.; Wang, J.; Zhang, G.; Kawanokuchi, J.; Kuno, R.; Sonobe, Y.; Mizuno, T.; Suzumura, A. Tumor necrosis factor-alpha induces neurotoxicity via glutamate release from hemichannels of activated microglia in an autocrine manner. J. Biol. Chem. 2006, 281, 21362–21368. [Google Scholar] [CrossRef] [Green Version]
- Gomes, P.; Srinivas, S.P.; Van Driessche, W.; Vereecke, J.; Himpens, B. ATP release through connexin hemichannels in corneal endothelial cells. Invest. Ophthalmol. Vis. Sci. 2005, 46, 1208–1218. [Google Scholar] [CrossRef] [Green Version]
- Ye, Z.C.; Wyeth, M.S.; Baltan-Tekkok, S.; Ransom, B.R. Functional hemichannels in astrocytes: A novel mechanism of glutamate release. J. Neurosci. 2003, 23, 3588–3596. [Google Scholar] [CrossRef] [Green Version]
- Davidson, J.O.; Green, C.R.; Nicholson, L.F.; Bennet, L.; Gunn, A.J. Connexin hemichannel blockade is neuroprotective after, but not during, global cerebral ischemia in near-term fetal sheep. Exp. Neurol. 2013, 248, 301–308. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.; Davidson, J.O.; Fowke, T.M.; Galinsky, R.; Wassink, G.; Karunasinghe, R.N.; Prasad, J.D.; Ranasinghe, S.; Green, C.R.; Bennet, L.; et al. Connexin hemichannel mimetic peptide attenuates cortical interneuron loss and perineuronal net disruption following cerebral ischemia in near-term fetal sheep. Int. J. Mol. Sci. 2020, 21, 6475. [Google Scholar] [CrossRef] [PubMed]
- Galinsky, R.; Davidson, J.O.; Lear, C.A.; Bennet, L.; Green, C.R.; Gunn, A.J. Connexin hemichannel blockade improves survival of striatal GABA-ergic neurons after global cerebral ischaemia in term-equivalent fetal sheep. Sci. Rep. 2017, 7, 6304. [Google Scholar] [CrossRef] [PubMed]
- Davidson, J.O.; Drury, P.P.; Green, C.R.; Nicholson, L.F.; Bennet, L.; Gunn, A.J. Connexin hemichannel blockade is neuroprotective after asphyxia in preterm fetal sheep. PLoS ONE 2014, 9, e96558. [Google Scholar] [CrossRef]
- Davidson, J.O.; Rout, A.L.; Wassink, G.; Yuill, C.A.; Zhang, F.G.; Green, C.R.; Bennet, L.; Gunn, A.J. Non-additive effects of delayed connexin hemichannel blockade and hypothermia after cerebral ischemia in near-term fetal sheep. J. Cereb. Blood Flow Metab. 2015, 35, 2052–2061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phelan, P.; Bacon, J.P.; Davies, J.A.; Stebbings, L.A.; Todman, M.G.; Avery, L.; Baines, R.A.; Barnes, T.M.; Ford, C.; Hekimi, S.; et al. Innexins: A family of invertebrate gap-junction proteins. Trends Genet. 1998, 14, 348–349. [Google Scholar] [CrossRef] [Green Version]
- Baranova, A.; Ivanov, D.; Petrash, N.; Pestova, A.; Skoblov, M.; Kelmanson, I.; Shagin, D.; Nazarenko, S.; Geraymovych, E.; Litvin, O.; et al. The mammalian pannexin family is homologous to the invertebrate innexin gap junction proteins. Genomics 2004, 83, 706–716. [Google Scholar] [CrossRef] [PubMed]
- Panchin, Y.V. Evolution of gap junction proteins—The pannexin alternative. J. Exp. Biol. 2005, 208, 1415–1419. [Google Scholar] [CrossRef] [Green Version]
- Michalski, K.; Syrjanen, J.L.; Henze, E.; Kumpf, J.; Furukawa, H.; Kawate, T. The Cryo-EM structure of pannexin 1 reveals unique motifs for ion selection and inhibition. Elife 2020, 9, e54670. [Google Scholar] [CrossRef] [Green Version]
- Sosinsky, G.E.; Boassa, D.; Dermietzel, R.; Duffy, H.S.; Laird, D.W.; MacVicar, B.; Naus, C.C.; Penuela, S.; Scemes, E.; Spray, D.C.; et al. Pannexin channels are not gap junction hemichannels. Channels (Austin) 2011, 5, 193–197. [Google Scholar] [CrossRef]
- Ishikawa, M.; Iwamoto, T.; Nakamura, T.; Doyle, A.; Fukumoto, S.; Yamada, Y. Pannexin 3 functions as an ER Ca(2+) channel, hemichannel, and gap junction to promote osteoblast differentiation. J. Cell Biol. 2011, 193, 1257–1274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sahu, G.; Sukumaran, S.; Bera, A.K. Pannexins form gap junctions with electrophysiological and pharmacological properties distinct from connexins. Sci. Rep. 2014, 4, 4955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruzzone, R.; Hormuzdi, S.G.; Barbe, M.T.; Herb, A.; Monyer, H. Pannexins, a family of gap junction proteins expressed in brain. Proc. Natl. Acad. Sci. USA 2003, 100, 13644–13649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogt, A.; Hormuzdi, S.G.; Monyer, H. Pannexin1 and Pannexin2 expression in the developing and mature rat brain. Brain Res. Mol. Brain Res. 2005, 141, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Boassa, D.; Ambrosi, C.; Qiu, F.; Dahl, G.; Gaietta, G.; Sosinsky, G. Pannexin1 channels contain a glycosylation site that targets the hexamer to the plasma membrane. J. Biol. Chem. 2007, 282, 31733–31743. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Deng, T.; Sun, Y.; Liu, K.; Yang, Y.; Zheng, X. Role for nitric oxide in permeability of hippocampal neuronal hemichannels during oxygen glucose deprivation. J. Neurosci. Res. 2008, 86, 2281–2291. [Google Scholar] [CrossRef]
- Silverman, W.R.; de Rivero Vaccari, J.P.; Locovei, S.; Qiu, F.; Carlsson, S.K.; Scemes, E.; Keane, R.W.; Dahl, G. The pannexin 1 channel activates the inflammasome in neurons and astrocytes. J. Biol. Chem. 2009, 284, 18143–18151. [Google Scholar] [CrossRef] [Green Version]
- Thompson, R.J.; Jackson, M.F.; Olah, M.E.; Rungta, R.L.; Hines, D.J.; Beazely, M.A.; MacDonald, J.F.; MacVicar, B.A. Activation of pannexin-1 hemichannels augments aberrant bursting in the hippocampus. Science 2008, 322, 1555–1559. [Google Scholar] [CrossRef] [Green Version]
- Kawamura, M., Jr.; Ruskin, D.N.; Masino, S.A. Metabolic autocrine regulation of neurons involves cooperation among pannexin hemichannels, adenosine receptors, and KATP channels. J. Neurosci. 2010, 30, 3886–3895. [Google Scholar] [CrossRef]
- Santiago, M.F.; Veliskova, J.; Patel, N.K.; Lutz, S.E.; Caille, D.; Charollais, A.; Meda, P.; Scemes, E. Targeting pannexin1 improves seizure outcome. PloS ONE 2011, 6, e25178. [Google Scholar] [CrossRef] [Green Version]
- Vanden Abeele, F.; Bidaux, G.; Gordienko, D.; Beck, B.; Panchin, Y.V.; Baranova, A.V.; Ivanov, D.V.; Skryma, R.; Prevarskaya, N. Functional implications of calcium permeability of the channel formed by pannexin 1. J. Cell Biol. 2006, 174, 535–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, L.; Sheng, H.; Chen, L.; Hao, B.; Shi, X.; Chen, Y. Effect of pannexin-1 on the release of glutamate and cytokines in astrocytes. J. Clin. Neurosci. 2016, 23, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Weilinger, N.L.; Tang, P.L.; Thompson, R.J. Anoxia-induced NMDA receptor activation opens pannexin channels via Src family kinases. J. Neurosci. 2012, 32, 12579–12588. [Google Scholar] [CrossRef]
- Cisneros-Mejorado, A.; Gottlieb, M.; Cavaliere, F.; Magnus, T.; Koch-Nolte, F.; Scemes, E.; Perez-Samartin, A.; Matute, C. Blockade of P2X7 receptors or pannexin-1 channels similarly attenuates postischemic damage. J. Cereb. Blood Flow Metab. 2015, 35, 843–850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, R.; Wang, J.; Xu, Y.; Yin, B.; He, F.; Du, Y.; Peng, G.; Luo, B. Probenecid protects against cerebral ischemia/reperfusion injury by inhibiting lysosomal and inflammatory damage in rats. Neuroscience 2015, 301, 168–177. [Google Scholar] [CrossRef]
- Cruikshank, S.J.; Hopperstad, M.; Younger, M.; Connors, B.W.; Spray, D.C.; Srinivas, M. Potent block of Cx36 and Cx50 gap junction channels by mefloquine. Proc. Natl. Acad. Sci. USA 2004, 101, 12364–12369. [Google Scholar] [CrossRef] [Green Version]
- Bargiotas, P.; Krenz, A.; Hormuzdi, S.G.; Ridder, D.A.; Herb, A.; Barakat, W.; Penuela, S.; von Engelhardt, J.; Monyer, H.; Schwaninger, M. Pannexins in ischemia-induced neurodegeneration. Proc. Natl. Acad. Sci. USA 2011, 108, 20772–20777. [Google Scholar] [CrossRef] [Green Version]
- Freitas-Andrade, M.; Bechberger, J.F.; MacVicar, B.A.; Viau, V.; Naus, C.C. Pannexin1 knockout and blockade reduces ischemic stroke injury in female, but not in male mice. Oncotarget 2017, 8, 36973–36983. [Google Scholar] [CrossRef] [Green Version]
- Qiu, F.; Dahl, G. A permeant regulating its permeation pore: Inhibition of pannexin 1 channels by ATP. Am. J. Physiol. Cell Physiol. 2009, 296, C250–C255. [Google Scholar] [CrossRef]
- Kim, Y.; Griffin, J.M.; Nor, M.N.M.; Zhang, J.; Freestone, P.S.; Danesh-Meyer, H.V.; Rupenthal, I.D.; Acosta, M.; Nicholson, L.F.B.; O’Carroll, S.J.; et al. Tonabersat prevents inflammatory damage in the central nervous system by blocking connexin43 hemichannels. Neurotherapeutics 2017, 14, 1148–1165. [Google Scholar] [CrossRef] [Green Version]
- Hagberg, H.; Mallard, C.; Ferriero, D.M.; Vannucci, S.J.; Levison, S.W.; Vexler, Z.S.; Gressens, P. The role of inflammation in perinatal brain injury. Nat. Rev. Neurol. 2015, 11, 192–208. [Google Scholar] [CrossRef] [PubMed]
- Bartha, A.I.; Foster-Barber, A.; Miller, S.P.; Vigneron, D.B.; Glidden, D.V.; Barkovich, A.J.; Ferriero, D.M. Neonatal encephalopathy: Association of cytokines with MR spectroscopy and outcome. Pediatr. Res. 2004, 56, 960–966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Shea, T.M.; Allred, E.N.; Kuban, K.C.; Dammann, O.; Paneth, N.; Fichorova, R.; Hirtz, D.; Leviton, A. Elevated concentrations of inflammation-related proteins in postnatal blood predict severe developmental delay at 2 years of age in extremely preterm infants. J. Pediatr. 2012, 160, 395–401 e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuban, K.C.; Joseph, R.M.; O’Shea, T.M.; Heeren, T.; Fichorova, R.N.; Douglass, L.; Jara, H.; Frazier, J.A.; Hirtz, D.; Rollins, J.V.; et al. Circulating inflammatory-associated proteins in the first month of life and cognitive impairment at age 10 years in children born extremely preterm. J. Pediatr. 2017, 180, 116–123 e1. [Google Scholar] [CrossRef] [Green Version]
- Markiewicz, I.; Lukomska, B. The role of astrocytes in the physiology and pathology of the central nervous system. Acta Neurobiol. Exp. (Warsz) 2006, 66, 343–358. [Google Scholar]
- Nagy, J.I.; Ochalski, P.A.; Li, J.; Hertzberg, E.L. Evidence for the co-localization of another connexin with connexin-43 at astrocytic gap junctions in rat brain. Neuroscience 1997, 78, 533–548. [Google Scholar] [CrossRef]
- Nedergaard, M. Direct signaling from astrocytes to neurons in cultures of mammalian brain cells. Science 1994, 263, 1768–1771. [Google Scholar] [CrossRef]
- Wallraff, A.; Kohling, R.; Heinemann, U.; Theis, M.; Willecke, K.; Steinhauser, C. The impact of astrocytic gap junctional coupling on potassium buffering in the hippocampus. J. Neurosci. 2006, 26, 5438–5447. [Google Scholar] [CrossRef] [Green Version]
- Rothstein, J.D.; Martin, L.; Levey, A.I.; Dykes-Hoberg, M.; Jin, L.; Wu, D.; Nash, N.; Kuncl, R.W. Localization of neuronal and glial glutamate transporters. Neuron 1994, 13, 713–725. [Google Scholar] [CrossRef]
- Tsacopoulos, M.; Magistretti, P.J. Metabolic coupling between glia and neurons. J. Neurosci. 1996, 16, 877–885. [Google Scholar] [CrossRef]
- Liang, Z.; Wang, X.; Hao, Y.; Qiu, L.; Lou, Y.; Zhang, Y.; Ma, D.; Feng, J. The multifaceted role of astrocyte connexin 43 in ischemic stroke through forming hemichannels and gap junctions. Front. Neurol. 2020, 11, 703. [Google Scholar] [CrossRef] [PubMed]
- Farina, C.; Aloisi, F.; Meinl, E. Astrocytes are active players in cerebral innate immunity. Trends Immunol. 2007, 28, 138–145. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Feng, L.; Ma, D.; Yin, P.; Wang, X.; Hou, S.; Hao, Y.; Zhang, J.; Xin, M.; Feng, J. Roles of astrocytic connexin-43, hemichannels, and gap junctions in oxygen-glucose deprivation/reperfusion injury induced neuroinflammation and the possible regulatory mechanisms of salvianolic acid B and carbenoxolone. J. Neuroinflamm. 2018, 15, 97. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, B.T.; Davis, T.P. The blood-brain barrier/neurovascular unit in health and disease. Pharmacol. Rev. 2005, 57, 173–185. [Google Scholar] [CrossRef] [PubMed]
- Shestopalov, V.I.; Panchin, Y. Pannexins and gap junction protein diversity. Cell. Mol. Life Sci. 2008, 65, 376–394. [Google Scholar] [CrossRef]
- Ezan, P.; Andre, P.; Cisternino, S.; Saubamea, B.; Boulay, A.C.; Doutremer, S.; Thomas, M.A.; Quenech’du, N.; Giaume, C.; Cohen-Salmon, M. Deletion of astroglial connexins weakens the blood-brain barrier. J. Cereb. Blood Flow Metab. 2012, 32, 1457–1467. [Google Scholar] [CrossRef] [Green Version]
- Moretti, R.; Pansiot, J.; Bettati, D.; Strazielle, N.; Ghersi-Egea, J.F.; Damante, G.; Fleiss, B.; Titomanlio, L.; Gressens, P. Blood-brain barrier dysfunction in disorders of the developing brain. Front. Neurosci. 2015, 9, 40. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Rousset, C.I.; Hagberg, H.; Mallard, C. Lipopolysaccharide-induced inflammation and perinatal brain injury. Semin. Fetal Neonatal Med. 2006, 11, 343–353. [Google Scholar] [CrossRef]
- Prasad, J.D.; Gunn, K.C.; Davidson, J.O.; Galinsky, R.; Graham, S.E.; Berry, M.J.; Bennet, L.; Gunn, A.J.; Dean, J.M. Anti-inflammatory therapies for treatment of inflammation-related Preterm brain injury. Int. J. Mol. Sci. 2021, 22, 4008. [Google Scholar] [CrossRef]
- Kim, Y.; Davidson, J.O.; Green, C.R.; Nicholson, L.F.; O’Carroll, S.J.; Zhang, J. Connexins and Pannexins in cerebral ischemia. Biochim. Biophys. Acta 2018, 1860, 224–236. [Google Scholar] [CrossRef]
- Abbott, N.J. Inflammatory mediators and modulation of blood-brain barrier permeability. Cell. Mol. Neurobiol. 2000, 20, 131–147. [Google Scholar] [CrossRef] [PubMed]
- De Bock, M.; Culot, M.; Wang, N.; Bol, M.; Decrock, E.; De Vuyst, E.; da Costa, A.; Dauwe, I.; Vinken, M.; Simon, A.M.; et al. Connexin channels provide a target to manipulate brain endothelial calcium dynamics and blood-brain barrier permeability. J. Cereb. Blood Flow Metab. 2011, 31, 1942–1957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lohman, A.W.; Billaud, M.; Isakson, B.E. Mechanisms of ATP release and signalling in the blood vessel wall. Cardiovasc. Res. 2012, 95, 269–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danesh-Meyer, H.V.; Kerr, N.M.; Zhang, J.; Eady, E.K.; O’Carroll, S.J.; Nicholson, L.F.; Johnson, C.S.; Green, C.R. Connexin43 mimetic peptide reduces vascular leak and retinal ganglion cell death following retinal ischaemia. Brain 2012, 135, 506–520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- John, G.R.; Scemes, E.; Suadicani, S.O.; Liu, J.S.; Charles, P.C.; Lee, S.C.; Spray, D.C.; Brosnan, C.F. IL-1beta differentially regulates calcium wave propagation between primary human fetal astrocytes via pathways involving P2 receptors and gap junction channels. Proc. Natl. Acad. Sci. USA 1999, 96, 11613–11618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duffy, H.S.; John, G.R.; Lee, S.C.; Brosnan, C.F.; Spray, D.C. Reciprocal regulation of the junctional proteins claudin-1 and connexin43 by interleukin-1beta in primary human fetal astrocytes. J. Neurosci. 2000, 20, RC114. [Google Scholar] [CrossRef] [Green Version]
- Sayyah, M.; Kaviani, B.; Khoshkholgh-Sima, B.; Bagheri, M.; Olad, M.; Choopani, S.; Mahdian, R. Effect of chronic intracerebroventricluar administration of lipopolysaccharide on connexin43 protein expression in rat hippocampus. Iran Biomed. J. 2012, 16, 25–32. [Google Scholar] [CrossRef]
- Zvalova, D.; Cordier, J.; Mesnil, M.; Junier, M.P.; Chneiweiss, H. p38/SAPK2 controls gap junction closure in astrocytes. Glia 2004, 46, 323–333. [Google Scholar] [CrossRef]
- Haghikia, A.; Ladage, K.; Lafênetre, P.; Haghikia, A.; Hinkerohe, D.; Smikalla, D.; Haase, C.G.; Dermietzel, R.; Faustmann, P.M. Intracellular application of TNF-alpha impairs cell to cell communication via gap junctions in glioma cells. J. Neurooncol. 2008, 86, 143–152. [Google Scholar] [CrossRef]
- Avendaño, B.C.; Montero, T.D.; Chávez, C.E.; von Bernhardi, R.; Orellana, J.A. Prenatal exposure to inflammatory conditions increases Cx43 and Panx1 unopposed channel opening and activation of astrocytes in the offspring effect on neuronal survival. Glia 2015, 63, 2058–2072. [Google Scholar] [CrossRef]
- Willebrords, J.; Crespo Yanguas, S.; Maes, M.; Decrock, E.; Wang, N.; Leybaert, L.; Kwak, B.R.; Green, C.R.; Cogliati, B.; Vinken, M. Connexins and their channels in inflammation. Crit. Rev. Biochem. Mol. Biol. 2016, 51, 413–439. [Google Scholar] [CrossRef] [PubMed]
- De Vuyst, E.; Decrock, E.; De Bock, M.; Yamasaki, H.; Naus, C.C.; Evans, W.H.; Leybaert, L. Connexin hemichannels and gap junction channels are differentially influenced by lipopolysaccharide and basic fibroblast growth factor. Mol. Biol. Cell 2007, 18, 34–46. [Google Scholar] [CrossRef] [PubMed]
- Liao, C.K.; Wang, S.M.; Chen, Y.L.; Wang, H.S.; Wu, J.C. Lipopolysaccharide-induced inhibition of connexin43 gap junction communication in astrocytes is mediated by downregulation of caveolin-3. Int. J. Biochem. Cell Biol. 2010, 42, 762–770. [Google Scholar] [CrossRef] [PubMed]
- Retamal, M.A.; Froger, N.; Palacios-Prado, N.; Ezan, P.; Saez, P.J.; Saez, J.C.; Giaume, C. Cx43 hemichannels and gap junction channels in astrocytes are regulated oppositely by proinflammatory cytokines released from activated microglia. J. Neurosci. 2007, 27, 13781–13792. [Google Scholar] [CrossRef]
- Karpuk, N.; Burkovetskaya, M.; Fritz, T.; Angle, A.; Kielian, T. Neuroinflammation leads to region-dependent alterations in astrocyte gap junction communication and hemichannel activity. J. Neurosci. 2011, 31, 414–425. [Google Scholar] [CrossRef] [Green Version]
- Chávez, C.E.; Oyarzún, J.E.; Avendaño, B.C.; Mellado, L.A.; Inostroza, C.A.; Alvear, T.F.; Orellana, J.A. The Opening of Connexin 43 Hemichannels Alters Hippocampal Astrocyte Function and Neuronal Survival in Prenatally LPS-Exposed Adult Offspring. Front. Cell. Neurosci. 2019, 13, 460. [Google Scholar] [CrossRef] [Green Version]
- Yang, D.; He, Y.; Muñoz-Planillo, R.; Liu, Q.; Núñez, G. Caspase-11 Requires the Pannexin-1 Channel and the Purinergic P2X7 Pore to Mediate Pyroptosis and Endotoxic Shock. Immunity 2015, 43, 923–932. [Google Scholar] [CrossRef] [Green Version]
- Da Silva, J.; Pierrat, B.; Mary, J.L.; Lesslauer, W. Blockade of p38 mitogen-activated protein kinase pathway inhibits inducible nitric-oxide synthase expression in mouse astrocytes. J. Biol. Chem. 1997, 272, 28373–28380. [Google Scholar] [CrossRef] [Green Version]
- Lohman, A.W.; Weaver, J.L.; Billaud, M.; Sandilos, J.K.; Griffiths, R.; Straub, A.C.; Penuela, S.; Leitinger, N.; Laird, D.W.; Bayliss, D.A.; et al. S-nitrosylation inhibits pannexin 1 channel function. J. Biol. Chem. 2012, 287, 39602–39612. [Google Scholar] [CrossRef] [Green Version]
- Prieto-Villalobos, J.; Alvear, T.F.; Liberona, A.; Lucero, C.M.; Martínez-Araya, C.J.; Balmazabal, J.; Inostroza, C.A.; Ramírez, G.; Gómez, G.I.; Orellana, J.A. Astroglial hemichannels and pannexons: The hidden link between maternal inflammation and neurological disorders. Int. J. Mol. Sci. 2021, 22, 9503. [Google Scholar] [CrossRef]
- Melani, A.; Turchi, D.; Vannucchi, M.G.; Cipriani, S.; Gianfriddo, M.; Pedata, F. ATP extracellular concentrations are increased in the rat striatum during in vivo ischemia. Neurochem. Int. 2005, 47, 442–448. [Google Scholar] [CrossRef] [PubMed]
- Sperlagh, B.; Vizi, E.S.; Wirkner, K.; Illes, P. P2X7 receptors in the nervous system. Prog. Neurobiol. 2006, 78, 327–346. [Google Scholar] [CrossRef] [PubMed]
- Bo, X.; Kim, M.; Nori, S.L.; Schoepfer, R.; Burnstock, G.; North, R.A. Tissue distribution of P2X4 receptors studied with an ectodomain antibody. Cell Tissue Res. 2003, 313, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Hattori, M.; Gouaux, E. Molecular mechanism of ATP binding and ion channel activation in P2X receptors. Nature 2012, 485, 207–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stout, C.E.; Costantin, J.L.; Naus, C.C.; Charles, A.C. Intercellular calcium signaling in astrocytes via ATP release through connexin hemichannels. J. Biol. Chem. 2002, 277, 10482–10488. [Google Scholar] [CrossRef] [Green Version]
- Baroja-Mazo, A.; Barbera-Cremades, M.; Pelegrin, P. The participation of plasma membrane hemichannels to purinergic signaling. Biochim. Biophys. Acta 2013, 1828, 79–93. [Google Scholar] [CrossRef] [Green Version]
- Iwabuchi, S.; Kawahara, K. Functional significance of the negative-feedback regulation of ATP release via pannexin-1 hemichannels under ischemic stress in astrocytes. Neurochem. Int. 2011, 58, 376–384. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Piskuric, N.A.; Vollmer, C.; Nurse, C.A. P2Y2 receptor activation opens pannexin-1 channels in rat carotid body type II cells: Potential role in amplifying the neurotransmitter ATP. J. Physiol. 2012, 590, 4335–4350. [Google Scholar] [CrossRef]
- Ceriani, F.; Pozzan, T.; Mammano, F. Critical role of ATP-induced ATP release for Ca2+ signaling in nonsensory cell networks of the developing cochlea. Proc. Natl. Acad. Sci. USA 2016, 113, E7194–E7201. [Google Scholar] [CrossRef] [Green Version]
- Cavaliere, F.; Florenzano, F.; Amadio, S.; Fusco, F.R.; Viscomi, M.T.; D’Ambrosi, N.; Vacca, F.; Sancesario, G.; Bernardi, G.; Molinari, M.; et al. Up-regulation of P2X2, P2X4 receptor and ischemic cell death: Prevention by P2 antagonists. Neuroscience 2003, 120, 85–98. [Google Scholar] [CrossRef]
- Li, F.; Wang, L.; Li, J.W.; Gong, M.; He, L.; Feng, R.; Dai, Z.; Li, S.Q. Hypoxia induced amoeboid microglial cell activation in postnatal rat brain is mediated by ATP receptor P2X4. BMC Neurosci. 2011, 12, 111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wixey, J.A.; Reinebrant, H.E.; Carty, M.L.; Buller, K.M. Delayed P2X4R expression after hypoxia-ischemia is associated with microglia in the immature rat brain. J. Neuroimmunol. 2009, 212, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Melani, A.; Amadio, S.; Gianfriddo, M.; Vannucchi, M.G.; Volonte, C.; Bernardi, G.; Pedata, F.; Sancesario, G. P2X7 receptor modulation on microglial cells and reduction of brain infarct caused by middle cerebral artery occlusion in rat. J. Cereb. Blood Flow Metab. 2006, 26, 974–982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frizzo, J.K.; Cardoso, M.P.; de Assis, A.M.; Perry, M.L.; Volonte, C.; Frizzo, M.E. Effects of acute perinatal asphyxia in the rat hippocampus. Cell. Mol. Neurobiol. 2010, 30, 683–692. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, P.; Cronin, C.G.; Scranton, V.L.; Jacobson, K.A.; Liang, B.T.; Verma, R. Neuroprotective and neuro-rehabilitative effects of acute purinergic receptor P2X4 (P2X4R) blockade after ischemic stroke. Exp. Neurol. 2020, 329, 113308. [Google Scholar] [CrossRef]
- Franke, H.; Gunther, A.; Grosche, J.; Schmidt, R.; Rossner, S.; Reinhardt, R.; Faber-Zuschratter, H.; Schneider, D.; Illes, P. P2X7 receptor expression after ischemia in the cerebral cortex of rats. J. Neuropathol. Exp. Neurol. 2004, 63, 686–699. [Google Scholar] [CrossRef] [Green Version]
- Kaiser, M.; Penk, A.; Franke, H.; Krugel, U.; Norenberg, W.; Huster, D.; Schaefer, M. Lack of functional P2X7 receptor aggravates brain edema development after middle cerebral artery occlusion. Purinergic signal. 2016, 12, 453–463. [Google Scholar] [CrossRef] [Green Version]
- Ozaki, E.; Campbell, M.; Doyle, S.L. Targeting the NLRP3 inflammasome in chronic inflammatory diseases: Current perspectives. J. Inflamm. Res. 2015, 8, 15–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Omer, M.; Melo, A.M.; Kelly, L.; Mac Dermott, E.J.; Leahy, T.R.; Killeen, O.; Saugstad, O.D.; Savani, R.C.; Molloy, E.J. Emerging Role of the NLRP3 Inflammasome and Interleukin-1β in Neonates. Neonatology 2020, 117, 545–554. [Google Scholar] [CrossRef]
- Jun, H.K.; Lee, S.H.; Lee, H.R.; Choi, B.K. Integrin alpha5beta1 activates the NLRP3 inflammasome by direct interaction with a bacterial surface protein. Immunity 2012, 36, 755–768. [Google Scholar] [CrossRef] [Green Version]
- Chakrabarti, A.; Banerjee, S.; Franchi, L.; Loo, Y.M.; Gale, M., Jr.; Nunez, G.; Silverman, R.H. RNase L activates the NLRP3 inflammasome during viral infections. Cell Host Microbe 2015, 17, 466–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, T.; Yang, Y.; Jin, T.; Jiang, W.; Zhou, R. Orchestration of NLRP3 inflammasome activation by ion fluxes. Trends Immunol. 2018, 39, 393–406. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F.; Mayor, A.; Tschopp, J. The inflammasomes: Guardians of the body. Annu. Rev. Immunol. 2009, 27, 229–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, Y.; Hara, H.; Nunez, G. Mechanism and regulation of NLRP3 inflammasome activation. Trends Biochem. Sci. 2016, 41, 1012–1021. [Google Scholar] [CrossRef] [Green Version]
- Chan, A.H.; Schroder, K. Inflammasome signaling and regulation of interleukin-1 family cytokines. J. Exp. Med. 2020, 217, e20190314. [Google Scholar] [CrossRef] [Green Version]
- Simões, A.P.; Duarte, J.A.; Agasse, F.; Canas, P.M.; Tomé, A.R.; Agostinho, P.; Cunha, R.A. Blockade of adenosine A2A receptors prevents interleukin-1β-induced exacerbation of neuronal toxicity through a p38 mitogen-activated protein kinase pathway. J. Neuroinflamm. 2012, 9, 204. [Google Scholar] [CrossRef] [Green Version]
- Savard, A.; Brochu, M.E.; Chevin, M.; Guiraut, C.; Grbic, D.; Sébire, G. Neuronal self-injury mediated by IL-1β and MMP-9 in a cerebral palsy model of severe neonatal encephalopathy induced by immune activation plus hypoxia-ischemia. J. Neuroinflamm. 2015, 12, 111. [Google Scholar] [CrossRef] [Green Version]
- Li, N.; Liu, C.; Wang, C.; Chen, R.; Li, X.; Wang, Y.; Wang, C. Early changes of NLRP3 inflammasome activation after hypoxic-ischemic brain injury in neonatal rats. Int. J. Clin. Exp. Pathol. 2021, 14, 209–220. [Google Scholar]
- Ystgaard, M.B.; Sejersted, Y.; Loberg, E.M.; Lien, E.; Yndestad, A.; Saugstad, O.D. Early upregulation of NLRP3 in the brain of neonatal mice exposed to hypoxia-ischemia: No early neuroprotective effects of NLRP3 deficiency. Neonatology 2015, 108, 211–219. [Google Scholar] [CrossRef] [Green Version]
- Kelly, L.A.; O’Dea, M.I.; Zareen, Z.; Melo, A.M.; McKenna, E.; Strickland, T.; McEneaney, V.; Donoghue, V.; Boylan, G.; Sweetman, D.; et al. Altered inflammasome activation in neonatal encephalopathy persists in childhood. Clin. Exp. Immunol. 2021, 205, 89–97. [Google Scholar] [CrossRef]
- Acosta, M.L.; Mat Nor, M.N.; Guo, C.X.; Mugisho, O.O.; Coutinho, F.P.; Rupenthal, I.D.; Green, C.R. Connexin therapeutics: Blocking connexin hemichannel pores is distinct from blocking pannexin channels or gap junctions. Neural Regen. Res. 2021, 16, 482–488. [Google Scholar] [CrossRef] [PubMed]
- Ystgaard, M.B.; Scheffler, K.; Suganthan, R.; Bjørås, M.; Ranheim, T.; Sagen, E.L.; Halvorsen, B.; Saugstad, O.D.; Yndestad, A. Neuromodulatory Effect of NLRP3 and ASC in Neonatal Hypoxic Ischemic Encephalopathy. Neonatology 2019, 115, 355–362. [Google Scholar] [CrossRef] [PubMed]
- Louie, H.H.; Shome, A.; Kuo, C.Y.; Rupenthal, I.D.; Green, C.R.; Mugisho, O.O. Connexin43 hemichannel block inhibits NLRP3 inflammasome activation in a human retinal explant model of diabetic retinopathy. Exp. Eye Res. 2021, 202, 108384. [Google Scholar] [CrossRef] [PubMed]
- Davidson, J.O.; Green, C.R.; Bennet, L.; Gunn, A.J. Battle of the hemichannels—Connexins and Pannexins in ischemic brain injury. Int. J. Dev. Neurosci. 2015, 45, 66–74. [Google Scholar] [CrossRef] [PubMed]
- Green, C.R.; Law, L.Y.; Lin, J.S.; Becker, D.L. Spatiotemporal depletion of connexins using antisense oligonucleotides. Methods Mol. Biol. 2001, 154, 175–185. [Google Scholar] [CrossRef]
- Cronin, M.; Anderson, P.N.; Cook, J.E.; Green, C.R.; Becker, D.L. Blocking connexin43 expression reduces inflammation and improves functional recovery after spinal cord injury. Mol. Cell. Neurosci. 2008, 39, 152–160. [Google Scholar] [CrossRef]
- Grupcheva, C.N.; Laux, W.T.; Rupenthal, I.D.; McGhee, J.; McGhee, C.N.; Green, C.R. Improved corneal wound healing through modulation of gap junction communication using connexin43-specific antisense oligodeoxynucleotides. Invest. Ophthalmol. Vis. Sci. 2012, 53, 1130–1138. [Google Scholar] [CrossRef] [Green Version]
- Agrawal, S.; Temsamani, J.; Galbraith, W.; Tang, J. Pharmacokinetics of antisense oligonucleotides. Clin. Pharmacokinet. 1995, 28, 7–16. [Google Scholar] [CrossRef]
- Falanga, A.; Galdiero, M.; Galdiero, S. Membranotropic Cell Penetrating Peptides: The Outstanding Journey. Int. J. Mol. Sci. 2015, 16, 25323–25337. [Google Scholar] [CrossRef]
- Coutinho, F.P.; Green, C.R.; Rupenthal, I.D. Intracellular oligonucleotide delivery using the cell penetrating peptide Xentry. Sci. Rep. 2018, 8, 11256. [Google Scholar] [CrossRef] [Green Version]
- Wang, N.; De Bock, M.; Decrock, E.; Bol, M.; Gadicherla, A.; Bultynck, G.; Leybaert, L. Connexin targeting peptides as inhibitors of voltage- and intracellular Ca2+-triggered Cx43 hemichannel opening. Neuropharmacology 2013, 75, 506–516. [Google Scholar] [CrossRef] [PubMed]
- Abudara, V.; Bechberger, J.; Freitas-Andrade, M.; De Bock, M.; Wang, N.; Bultynck, G.; Naus, C.C.; Leybaert, L.; Giaume, C. The connexin43 mimetic peptide Gap19 inhibits hemichannels without altering gap junctional communication in astrocytes. Front. Cell. Neurosci. 2014, 8, 306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, B.; Yang, L.; Chen, J.; Chen, Y.; Zhang, L.; Wang, L.; Li, X.; Li, Y.; Yu, H. Inhibition of Connexin43 hemichannels with Gap19 protects cerebral ischemia/reperfusion injury via the JAK2/STAT3 pathway in mice. Brain Res. Bull. 2019, 146, 124–135. [Google Scholar] [CrossRef] [PubMed]
- Ponsaerts, R.; De Vuyst, E.; Retamal, M.; D’Hondt, C.; Vermeire, D.; Wang, N.; De Smedt, H.; Zimmermann, P.; Himpens, B.; Vereecke, J.; et al. Intramolecular loop/tail interactions are essential for connexin 43-hemichannel activity. FASEB J. 2010, 24, 4378–4395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walrave, L.; Pierre, A.; Albertini, G.; Aourz, N.; De Bundel, D.; Van Eeckhaut, A.; Vinken, M.; Giaume, C.; Leybaert, L.; Smolders, I. Inhibition of astroglial connexin43 hemichannels with TAT-Gap19 exerts anticonvulsant effects in rodents. Glia 2018, 66, 1788–1804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crespo Yanguas, S.; da Silva, T.C.; Pereira, I.V.A.; Willebrords, J.; Maes, M.; Sayuri Nogueira, M.; Alves de Castro, I.; Leclercq, I.; Romualdo, G.R.; Barbisan, L.F.; et al. TAT-Gap19 and carbenoxolone alleviate liver fibrosis in mice. Int. J. Mol. Sci. 2018, 19, 817. [Google Scholar] [CrossRef] [Green Version]
- Coutinho, F.P.; Green, C.R.; Acosta, M.L.; Rupenthal, I.D. Xentry-Gap19 inhibits Connexin43 hemichannel opening especially during hypoxic injury. Drug Deliv. Transl. Res. 2020, 10, 751–765. [Google Scholar] [CrossRef]
- Mat Nor, M.N.; Rupenthal, I.D.; Green, C.R.; Acosta, M.L. Connexin Hemichannel Block Using Orally Delivered Tonabersat Improves Outcomes in Animal Models of Retinal Disease. Neurotherapeutics 2020, 17, 371–387. [Google Scholar] [CrossRef]
- Silberstein, S.D.; Schoenen, J.; Gobel, H.; Diener, H.C.; Elkind, A.H.; Klapper, J.A.; Howard, R.A. Tonabersat, a gap-junction modulator: Efficacy and safety in two randomized, placebo-controlled, dose-ranging studies of acute migraine. Cephalalgia 2009, 29 (Suppl. S2), 17–27. [Google Scholar] [CrossRef]
- Hauge, A.W.; Asghar, M.S.; Schytz, H.W.; Christensen, K.; Olesen, J. Effects of tonabersat on migraine with aura: A randomised, double-blind, placebo-controlled crossover study. Lancet Neurol. 2009, 8, 718–723. [Google Scholar] [CrossRef]
- Kursun, O.; Yemisci, M.; van den Maagdenberg, A.; Karatas, H. Migraine and neuroinflammation: The inflammasome perspective. J. Headache Pain 2021, 22, 55. [Google Scholar] [CrossRef]
- Lyon, H.; Shome, A.; Rupenthal, I.D.; Green, C.R.; Mugisho, O.O. Tonabersat Inhibits Connexin43 Hemichannel Opening and Inflammasome Activation in an In Vitro Retinal Epithelial Cell Model of Diabetic Retinopathy. Int. J. Mol. Sci. 2020, 22, 298. [Google Scholar] [CrossRef]
- Almad, A.A.; Taga, A.; Joseph, J.; Gross, S.K.; Welsh, C.; Patankar, A.; Richard, J.P.; Rust, K.; Pokharel, A.; Plott, C.; et al. Cx43 hemichannels contribute to astrocyte-mediated toxicity in sporadic and familial ALS. Proc. Natl. Acad. Sci. USA 2022, 119, e2107391119. [Google Scholar] [CrossRef] [PubMed]
- Sarrouilhe, D.; Dejean, C.; Mesnil, M. Involvement of gap junction channels in the pathophysiology of migraine with aura. Front. Physiol. 2014, 5, 78. [Google Scholar] [CrossRef] [Green Version]
- Peroutka, S.J. 2008: The year in review. Headache 2009, 49, 796–802. [Google Scholar] [CrossRef] [PubMed]
- Silverman, W.; Locovei, S.; Dahl, G. Probenecid, a gout remedy, inhibits pannexin 1 channels. Am. J. Physiol. Cell Physiol. 2008, 295, C761–C767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lodygensky, G.A.; Battin, M.R.; Gunn, A.J. Mild Neonatal Encephalopathy—How, When, and How Much to Treat? JAMA Pediatrics 2018, 172, 3–4. [Google Scholar] [CrossRef] [PubMed]
- Davidson, J.O.; Battin, M.R.; Gunn, A.J. Implications of the HELIX trial for treating infants with hypoxic-ischaemic encephalopathy in low-to-middle-income countries. Arch. Dis. Child. Fetal Neonatal Ed. 2022. Epub Feb 21. [Google Scholar] [CrossRef]
- El-Dib, M.; Inder, T.E.; Chalak, L.F.; Massaro, A.N.; Thoresen, M.; Gunn, A.J. Should therapeutic hypothermia be offered to babies with mild neonatal encephalopathy in the first 6 h after birth? Pediatr. Res. 2019, 85, 442–448. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
McDouall, A.; Zhou, K.Q.; Bennet, L.; Green, C.R.; Gunn, A.J.; Davidson, J.O. Connexins, Pannexins and Gap Junctions in Perinatal Brain Injury. Biomedicines 2022, 10, 1445. https://doi.org/10.3390/biomedicines10061445
McDouall A, Zhou KQ, Bennet L, Green CR, Gunn AJ, Davidson JO. Connexins, Pannexins and Gap Junctions in Perinatal Brain Injury. Biomedicines. 2022; 10(6):1445. https://doi.org/10.3390/biomedicines10061445
Chicago/Turabian StyleMcDouall, Alice, Kelly Q. Zhou, Laura Bennet, Colin R. Green, Alistair J. Gunn, and Joanne O. Davidson. 2022. "Connexins, Pannexins and Gap Junctions in Perinatal Brain Injury" Biomedicines 10, no. 6: 1445. https://doi.org/10.3390/biomedicines10061445
APA StyleMcDouall, A., Zhou, K. Q., Bennet, L., Green, C. R., Gunn, A. J., & Davidson, J. O. (2022). Connexins, Pannexins and Gap Junctions in Perinatal Brain Injury. Biomedicines, 10(6), 1445. https://doi.org/10.3390/biomedicines10061445