
Telomere Dysfunction Is Associated with Altered DNA Organization in Trichoplein/Tchp/Mitostatin (TpMs) Depleted Cells

 , ,
, ,  , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Reagents
2.2. Lentivirus Production and Infection
2.3. Three-Dimensional Quantitative Fluorescent In Situ Hybridization (3D Q-FISH)
2.4. Widefield 3D Imaging and Analysis
2.5. SIM Images Acquisition and Analysis
2.6. 3D-SIM Imaging and Analysis
2.7. Western Blot
2.8. Statistical Analysis
3. Results
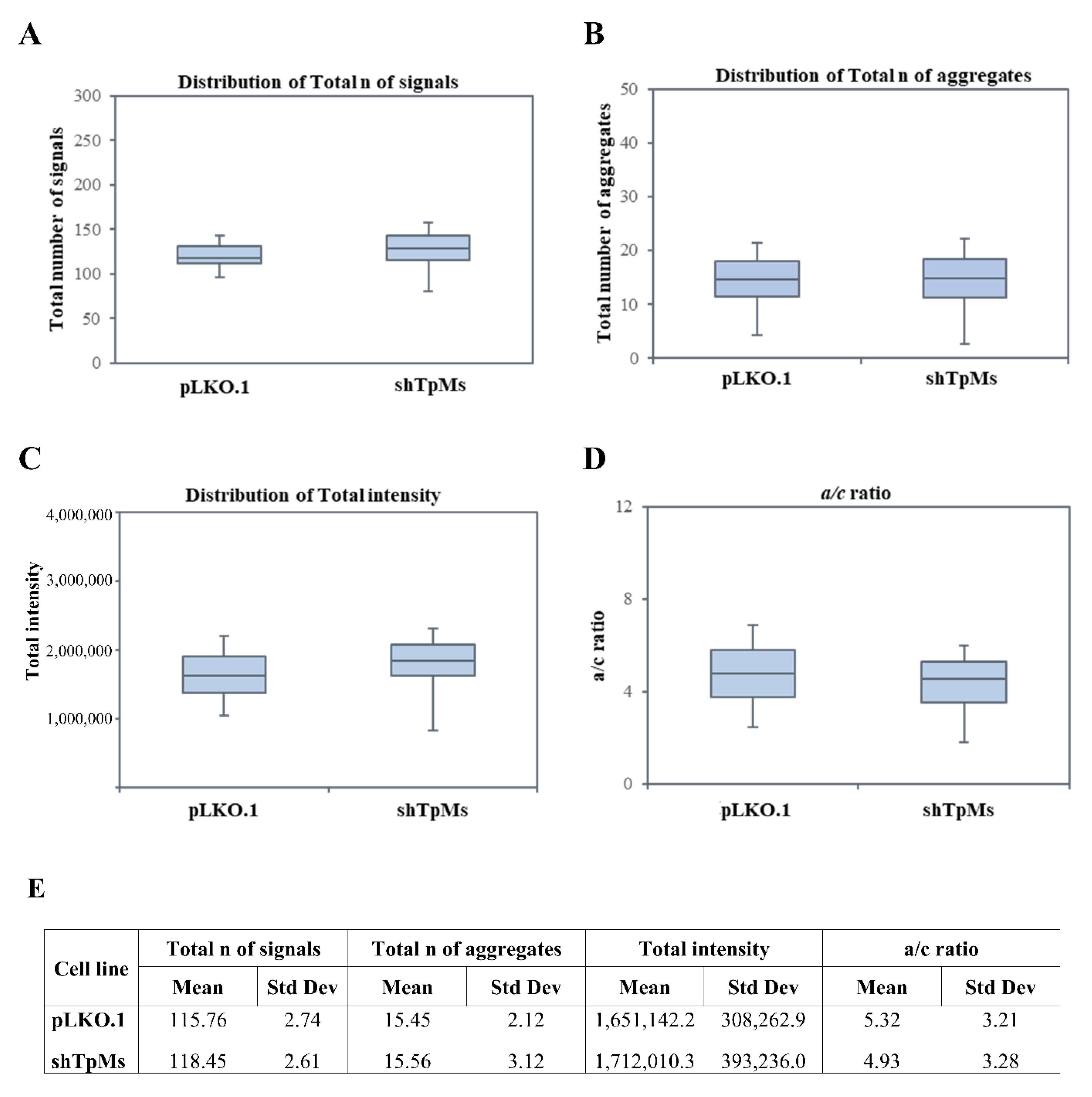
3.1. Telomere Alteration Increases in Tpms-Depleted Colon Cancer HCT116 Cells but Not in Tpms-Depleted Normal Prostatic Epidermal pnt1a Cell Line
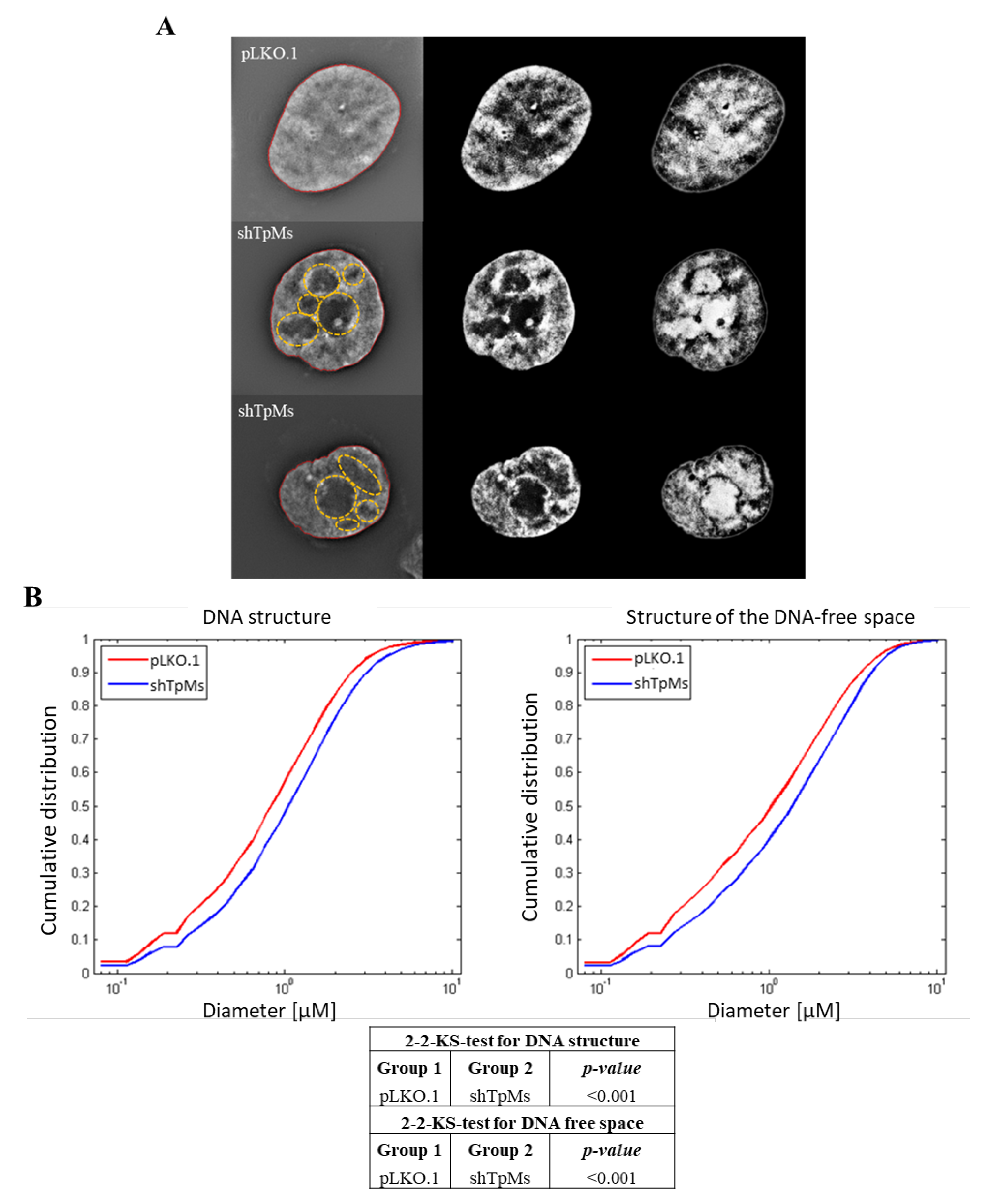
3.2. TpMs-Depleted HCT116 Cells Display Differences in Nuclear DNA Organization
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nishizawa, M.; Izawa, I.; Inoko, A.; Hayashi, Y.; Nagata, K.; Yokoyama, T.; Usukura, J.; Inagaki, M. Identification of Trichoplein, a Novel Keratin Filament-Binding Protein. J. Cell Sci. 2005, 118, 1081–1090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vecchione, A.; Fassan, M.; Anesti, V.; Morrione, A.; Goldoni, S.; Baldassarre, G.; Byrne, D.; D’Arca, D.; Palazzo, J.P.; Lloyd, J.; et al. MITOSTATIN, a Putative Tumor Suppressor on Chromosome 12q24.1, Is Downregulated in Human Bladder and Breast Cancer. Oncogene 2009, 28, 257–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ibi, M.; Zou, P.; Inoko, A.; Shiromizu, T.; Matsuyama, M.; Hayashi, Y.; Enomoto, M.; Mori, D.; Hirotsune, S.; Kiyono, T.; et al. Trichoplein Controls Microtubule Anchoring at the Centrosome by Binding to Odf2 and Ninein. J. Cell Sci. 2011, 124, 857–864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishimura, Y.; Kasahara, K.; Inagaki, M. Intermediate Filaments and IF-Associated Proteins: From Cell Architecture to Cell Proliferation. Proc. Jpn. Acad., Ser. B 2019, 95, 479–493. [Google Scholar] [CrossRef] [Green Version]
- Inoko, A.; Matsuyama, M.; Goto, H.; Ohmuro-Matsuyama, Y.; Hayashi, Y.; Enomoto, M.; Ibi, M.; Urano, T.; Yonemura, S.; Kiyono, T.; et al. Trichoplein and Aurora A Block Aberrant Primary Cilia Assembly in Proliferating Cells. J. Cell Biol. 2012, 197, 391–405. [Google Scholar] [CrossRef] [Green Version]
- Cerqua, C.; Anesti, V.; Pyakurel, A.; Liu, D.; Naon, D.; Wiche, G.; Baffa, R.; Dimmer, K.S.; Scorrano, L. Trichoplein/Mitostatin Regulates Endoplasmic Reticulum–Mitochondria Juxtaposition. EMBO Rep. 2010, 11, 854–860. [Google Scholar] [CrossRef] [PubMed]
- Buraschi, S.; Neill, T.; Iozzo, R.V. Decorin Is a Devouring Proteoglycan: Remodeling of Intracellular Catabolism via Autophagy and Mitophagy. Matrix Biol. 2019, 75–76, 260–270. [Google Scholar] [CrossRef] [PubMed]
- Martello, A.; Lauriola, A.; Mellis, D.; Parish, E.; Dawson, J.C.; Imrie, L.; Vidmar, M.; Gammoh, N.; Mitić, T.; Brittan, M.; et al. Trichoplein Binds PCM 1 and Controls Endothelial Cell Function by Regulating Autophagy. EMBO Rep. 2020, 21. [Google Scholar] [CrossRef] [PubMed]
- Lauriola, A.; Martello, A.; Fantini, S.; Marverti, G.; Zanocco-Marani, T.; Davalli, P.; Guardavaccaro, D.; Mai, S.; Caporali, A.; D’Arca, D. Depletion of Trichoplein (TpMs) Causes Chromosome Mis-Segregation, DNA Damage and Chromosome Instability in Cancer Cells. Cancers 2020, 12, 993. [Google Scholar] [CrossRef] [Green Version]
- Bach, D.-H.; Zhang, W.; Sood, A.K. Chromosomal Instability in Tumor Initiation and Development. Cancer Res. 2019, 79, 3995–4002. [Google Scholar] [CrossRef] [Green Version]
- Bakhoum, S.F.; Cantley, L.C. The Multifaceted Role of Chromosomal Instability in Cancer and Its Microenvironment. Cell 2018, 174, 1347–1360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pino, M.S.; Chung, D.C. The Chromosomal Instability Pathway in Colon Cancer. Gastroenterology 2010, 138, 2059–2072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shay, J.W. Telomeres and Aging. Curr. Opin. Cell Biol. 2018, 52, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Rossiello, F.; Jurk, D.; Passos, J.F.; d’Adda di Fagagna, F. Telomere Dysfunction in Ageing and Age-Related Diseases. Nat. Cell Biol. 2022, 24, 135–147. [Google Scholar] [CrossRef] [PubMed]
- Bär, C.; Blasco, M.A. Telomeres and Telomerase as Therapeutic Targets to Prevent and Treat Age-Related Diseases. F1000Res 2016, 5, 89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernal, A.; Tusell, L. Telomeres: Implications for Cancer Development. IJMS 2018, 19, 294. [Google Scholar] [CrossRef] [Green Version]
- Robinson, N.J.; Schiemann, W.P. Telomerase in Cancer: Function, Regulation, and Clinical Translation. Cancers 2022, 14, 808. [Google Scholar] [CrossRef]
- Stewénius, Y.; Gorunova, L.; Jonson, T.; Larsson, N.; Höglund, M.; Mandahl, N.; Mertens, F.; Mitelman, F.; Gisselsson, D. Structural and Numerical Chromosome Changes in Colon Cancer Develop through Telomere-Mediated Anaphase Bridges, Not through Mitotic Multipolarity. Proc. Natl. Acad. Sci. USA. 2005, 102, 5541–5546. [Google Scholar] [CrossRef] [Green Version]
- Smith, E.M.; Pendlebury, D.F.; Nandakumar, J. Structural Biology of Telomeres and Telomerase. Cell. Mol. Life Sci. 2020, 77, 61–79. [Google Scholar] [CrossRef]
- Roake, C.M.; Artandi, S.E. Regulation of Human Telomerase in Homeostasis and Disease. Nat. Rev. Mol. Cell Biol. 2020, 21, 384–397. [Google Scholar] [CrossRef]
- Okamoto, K.; Seimiya, H. Revisiting Telomere Shortening in Cancer. Cells 2019, 8, 107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maciejowski, J.; de Lange, T. Telomeres in Cancer: Tumour Suppression and Genome Instability. Nat. Rev. Mol. Cell Biol. 2017, 18, 175–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertorelle, R. Telomeres, Telomerase and Colorectal Cancer. WJG 2014, 20, 1940. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Peng, M.; Song, Q. The Co-Expression of Telomerase and ALT Pathway in Human Breast Cancer Tissues. Tumor Biol. 2014, 35, 4087–4093. [Google Scholar] [CrossRef] [PubMed]
- M’kacher, R.; Cuceu, C.; Al Jawhari, M.; Morat, L.; Frenzel, M.; Shim, G.; Lenain, A.; Hempel, W.; Junker, S.; Girinsky, T.; et al. The Transition between Telomerase and ALT Mechanisms in Hodgkin Lymphoma and Its Predictive Value in Clinical Outcomes. Cancers 2018, 10, 169. [Google Scholar] [CrossRef] [Green Version]
- Artandi, S.E.; DePinho, R.A. Telomeres and Telomerase in Cancer. Carcinogenesis 2010, 31, 9–18. [Google Scholar] [CrossRef]
- Bailey, S.M. Telomeres, Chromosome Instability and Cancer. Nucleic Acids Res. 2006, 34, 2408–2417. [Google Scholar] [CrossRef] [Green Version]
- Desmaze, C.; Soria, J.-C.; Freulet-Marrière, M.-A.; Mathieu, N.; Sabatier, L. Telomere-Driven Genomic Instability in Cancer Cells. Cancer Lett. 2003, 194, 173–182. [Google Scholar] [CrossRef]
- Chuang, T.C.Y.; Moshir, S.; Garini, Y.; Chuang, A.Y.-C.; Young, I.T.; Vermolen, B.; van den Doel, R.; Mougey, V.; Perrin, M.; Braun, M.; et al. The Three-Dimensional Organization of Telomeres in the Nucleus of Mammalian Cells. BMC Biol. 2004, 2, 12. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Rangel-Pozzo, A.; Yu, P.; LaL, S.; Asbaghi, Y.; Sisdelli, L.; Tammur, P.; Tamm, A.; Punab, M.; Klewes, L.; Louis, S.; et al. Telomere Architecture Correlates with Aggressiveness in Multiple Myeloma. Cancers 2021, 13, 1969. [Google Scholar] [CrossRef] [PubMed]
- Mai, S. The Three-dimensional Cancer Nucleus. Genes Chromosomes Cancer 2019, 58, 462–473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Righolt, C.H.; Guffei, A.; Knecht, H.; Young, I.T.; Stallinga, S.; van Vliet, L.J.; Mai, S. Differences in Nuclear DNA Organization Between Lymphocytes, Hodgkin and Reed–Sternberg Cells Revealed by Structured Illumination Microscopy. J. Cell. Biochem. 2014, 115, 1441–1448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Counter, C.M.; Avilion, A.A.; LeFeuvre, C.E.; Stewart, N.G.; Greider, C.W.; Harley, C.B.; Bacchetti, S. Telomere Shortening Associated with Chromosome Instability Is Arrested in Immortal Cells Which Express Telomerase Activity. EMBO J. 1992, 11, 1921–1929. [Google Scholar] [CrossRef]
- D’Souza, Y.; Chu, T.W.; Autexier, C. A Translocation-Defective Telomerase with Low Levels of Activity and Processivity Stabilizes Short Telomeres and Confers Immortalization. MBoC 2013, 24, 1469–1479. [Google Scholar] [CrossRef]
- Louis, S.F.; Vermolen, B.J.; Garini, Y.; Young, I.T.; Guffei, A.; Lichtensztejn, Z.; Kuttler, F.; Chuang, T.C.Y.; Moshir, S.; Mougey, V.; et al. C-Myc Induces Chromosomal Rearrangements through Telomere and Chromosome Remodeling in the Interphase Nucleus. Proc. Natl. Acad. Sci. USA. 2005, 102, 9613–9618. [Google Scholar] [CrossRef] [Green Version]
- Poon, S.S.S.; Martens, U.M.; Ward, R.K.; Lansdorp, P.M. Telomere Length Measurements Using Digital Fluorescence Microscopy. Cytometry 1999, 36, 267–278. [Google Scholar] [CrossRef]
- Pienta, K.J.; Partin, A.W.; Coffey, D.S. Cancer as a Disease of DNA Organization and Dynamic Cell Structure. Cancer Res. 1989, 49, 2525–2532. [Google Scholar]
- Cragg, G.E.; So, P.T.C. Lateral Resolution Enhancement with Standing Evanescent Waves. Opt. Lett. 2000, 25, 46. [Google Scholar] [CrossRef]
- Frohn, J.T.; Knapp, H.F.; Stemmer, A. True Optical Resolution beyond the Rayleigh Limit Achieved by Standing Wave Illumination. Proc. Natl. Acad. Sci. USA 2000, 97, 7232–7236. [Google Scholar] [CrossRef] [Green Version]
- Gustafsson, M.G.L. Surpassing the Lateral Resolution Limit by a Factor of Two Using Structured Illumination Microscopy. SHORT COMMUNICATION. J. Microsc. 2000, 198, 82–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D.; Weinberg, R.A. The Hallmarks of Cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [Green Version]
- Holdgaard, S.G.; Cianfanelli, V.; Pupo, E.; Lambrughi, M.; Lubas, M.; Nielsen, J.C.; Eibes, S.; Maiani, E.; Harder, L.M.; Wesch, N.; et al. Selective Autophagy Maintains Centrosome Integrity and Accurate Mitosis by Turnover of Centriolar Satellites. Nat. Commun. 2019, 10, 4176. [Google Scholar] [CrossRef] [PubMed]
- Guardavaccaro, D.; Kudo, Y.; Boulaire, J.; Barchi, M.; Busino, L.; Donzelli, M.; Margottin-Goguet, F.; Jackson, P.K.; Yamasaki, L.; Pagano, M. Control of Meiotic and Mitotic Progression by the F Box Protein β-Trcp1 In Vivo. Dev. Cell 2003, 4, 799–812. [Google Scholar] [CrossRef] [Green Version]
- Meraldi, P.; Draviam, V.M.; Sorger, P.K. Timing and Checkpoints in the Regulation of Mitotic Progression. Dev. Cell 2004, 7, 45–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Lange, T. Shelterin-Mediated Telomere Protection. Annu. Rev. Genet. 2018, 52, 223–247. [Google Scholar] [CrossRef] [PubMed]
- Janssen, A.; van der Burg, M.; Szuhai, K.; Kops, G.J.P.L.; Medema, R.H. Chromosome Segregation Errors as a Cause of DNA Damage and Structural Chromosome Aberrations. Science 2011, 333, 1895–1898. [Google Scholar] [CrossRef] [PubMed]
- Michel, L.S.; Liberal, V.; Chatterjee, A.; Kirchwegger, R.; Pasche, B.; Gerald, W.; Dobles, M.; Sorger, P.K.; Murty, V.V.V.S.; Benezra, R. MAD2 Haplo-Insufficiency Causes Premature Anaphase and Chromosome Instability in Mammalian Cells. Nature 2001, 409, 355–359. [Google Scholar] [CrossRef] [Green Version]
- Baker, D.J.; Jin, F.; Jeganathan, K.B.; van Deursen, J.M. Whole Chromosome Instability Caused by Bub1 Insufficiency Drives Tumorigenesis through Tumor Suppressor Gene Loss of Heterozygosity. Cancer Cell 2009, 16, 475–486. [Google Scholar] [CrossRef] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lauriola, A.; Davalli, P.; Marverti, G.; Caporali, A.; Mai, S.; D’Arca, D. Telomere Dysfunction Is Associated with Altered DNA Organization in Trichoplein/Tchp/Mitostatin (TpMs) Depleted Cells. Biomedicines 2022, 10, 1602. https://doi.org/10.3390/biomedicines10071602
Lauriola A, Davalli P, Marverti G, Caporali A, Mai S, D’Arca D. Telomere Dysfunction Is Associated with Altered DNA Organization in Trichoplein/Tchp/Mitostatin (TpMs) Depleted Cells. Biomedicines. 2022; 10(7):1602. https://doi.org/10.3390/biomedicines10071602
Chicago/Turabian StyleLauriola, Angela, Pierpaola Davalli, Gaetano Marverti, Andrea Caporali, Sabine Mai, and Domenico D’Arca. 2022. "Telomere Dysfunction Is Associated with Altered DNA Organization in Trichoplein/Tchp/Mitostatin (TpMs) Depleted Cells" Biomedicines 10, no. 7: 1602. https://doi.org/10.3390/biomedicines10071602
APA StyleLauriola, A., Davalli, P., Marverti, G., Caporali, A., Mai, S., & D’Arca, D. (2022). Telomere Dysfunction Is Associated with Altered DNA Organization in Trichoplein/Tchp/Mitostatin (TpMs) Depleted Cells. Biomedicines, 10(7), 1602. https://doi.org/10.3390/biomedicines10071602