
Molecular Threat of Splicing Factor Mutations to Myeloid Malignancies and Potential Therapeutic Modulations


Abstract
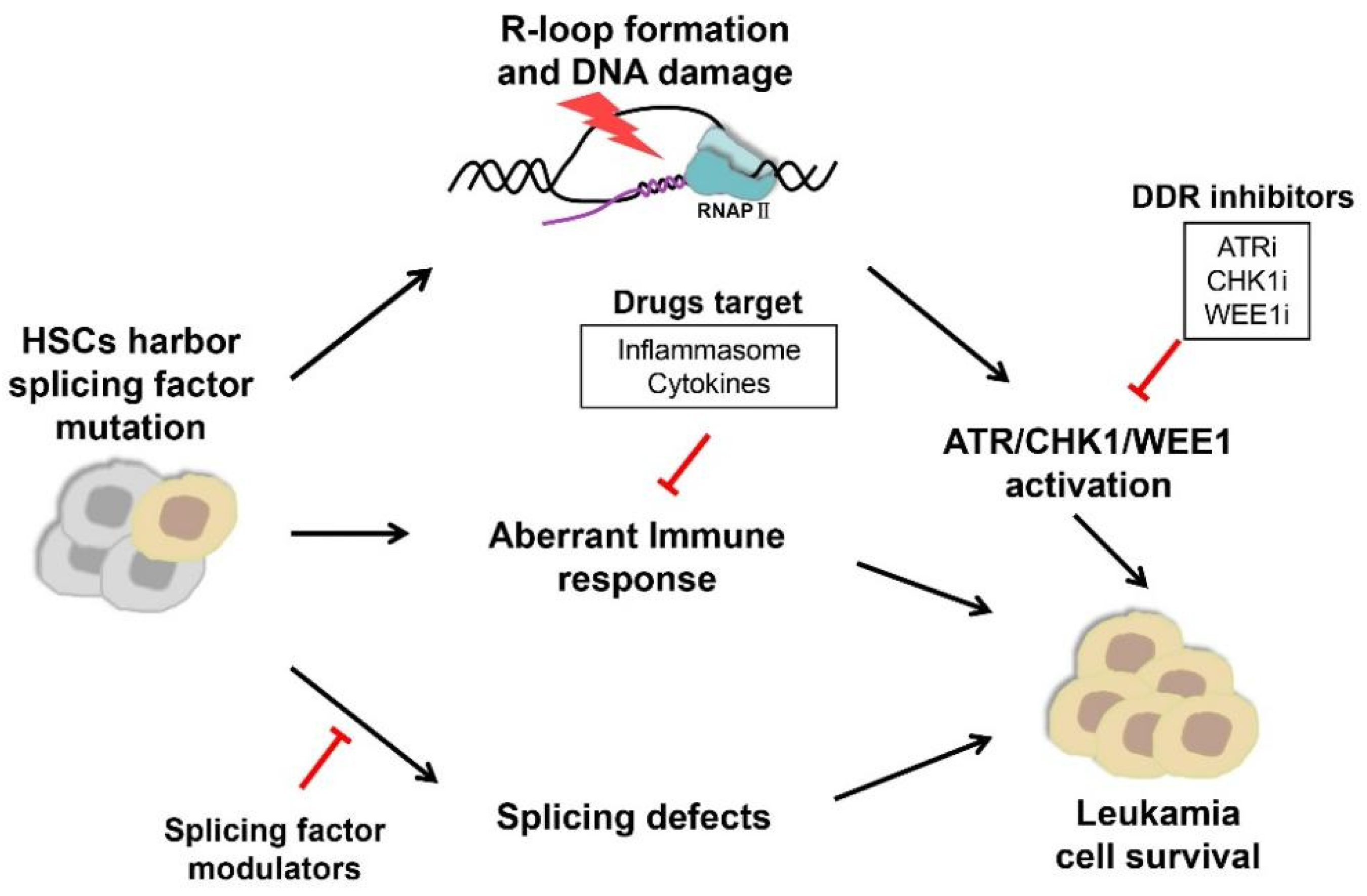
:1. Introduction
2. The Discovery and Pathogenic Roles of Splicing Factor Mutations during the Development of MDS/AML
3. Mechanisms That Contribute to Leukemogenesis beyond Splicing Defects
4. Development of Drugs Targeting Cancer Cells with Splicing Factor Mutations
4.1. Direct Targeting of the Spliceosome with Splicing Factor Modulators
4.2. Targeting DDR with Small Molecule Inhibitors
4.3. Targeting Immune Responses and Inflammation Pathways
5. Concluding Remarks and Future Perspectives
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Saez, B.; Walter, M.J.; Graubert, T.A. Splicing factor gene mutations in hematologic malignancies. Blood J. Am. Soc. Hematol. 2017, 129, 1260–1269. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, K.; Sanada, M.; Shiraishi, Y.; Nowak, D.; Nagata, Y.; Yamamoto, R.; Sato, Y.; Sato-Otsubo, A.; Kon, A.; Nagasaki, M. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature 2011, 478, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Seiler, M.; Peng, S.; Agrawal, A.A.; Palacino, J.; Teng, T.; Zhu, P.; Smith, P.G.; Caesar-Johnson, S.J.; Demchok, J.A.; Felau, I. Somatic mutational landscape of splicing factor genes and their functional consequences across 33 cancer types. Cell Rep. 2018, 23, 282–296.e4. [Google Scholar] [CrossRef]
- Papaemmanuil, E.; Gerstung, M.; Malcovati, L.; Tauro, S.; Gundem, G.; Van Loo, P.; Yoon, C.J.; Ellis, P.; Wedge, D.C.; Pellagatti, A. Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood J. Am. Soc. Hematol. 2013, 122, 3616–3627. [Google Scholar] [CrossRef]
- Papaemmanuil, E.; Gerstung, M.; Bullinger, L.; Gaidzik, V.I.; Paschka, P.; Roberts, N.D.; Potter, N.E.; Heuser, M.; Thol, F.; Bolli, N. Genomic classification and prognosis in acute myeloid leukemia. N. Engl. J. Med. 2016, 374, 2209–2221. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, S. Genetics of MDS. Blood. J. Am. Soc. Hematol. 2019, 133, 1049–1059. [Google Scholar]
- Haferlach, T.; Nagata, Y.; Grossmann, V.; Okuno, Y.; Bacher, U.; Nagae, G.; Schnittger, S.; Sanada, M.; Kon, A.; Alpermann, T. Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia 2014, 28, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Darman, R.B.; Seiler, M.; Agrawal, A.A.; Lim, K.H.; Peng, S.; Aird, D.; Bailey, S.L.; Bhavsar, E.B.; Chan, B.; Colla, S. Cancer-associated SF3B1 hotspot mutations induce cryptic 3′ splice site selection through use of a different branch point. Cell Rep. 2015, 13, 1033–1045. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Ilagan, J.O.; Liang, Y.; Daubner, G.M.; Lee, S.C.-W.; Ramakrishnan, A.; Li, Y.; Chung, Y.R.; Micol, J.-B.; Murphy, M.E. SRSF2 mutations contribute to myelodysplasia by mutant-specific effects on exon recognition. Cancer Cell 2015, 27, 617–630. [Google Scholar] [CrossRef]
- Obeng, E.A.; Chappell, R.J.; Seiler, M.; Chen, M.C.; Campagna, D.R.; Schmidt, P.J.; Schneider, R.K.; Lord, A.M.; Wang, L.; Gambe, R.G. Physiologic expression of Sf3b1K700E causes impaired erythropoiesis, aberrant splicing, and sensitivity to therapeutic spliceosome modulation. Cancer Cell 2016, 30, 404–417. [Google Scholar] [CrossRef]
- Shirai, C.L.; Ley, J.N.; White, B.S.; Kim, S.; Tibbitts, J.; Shao, J.; Ndonwi, M.; Wadugu, B.; Duncavage, E.J.; Okeyo-Owuor, T. Mutant U2AF1 expression alters hematopoiesis and pre-mRNA splicing in vivo. Cancer Cell 2015, 27, 631–643. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.C.-W.; Dvinge, H.; Kim, E.; Cho, H.; Micol, J.-B.; Chung, Y.R.; Durham, B.H.; Yoshimi, A.; Kim, Y.J.; Thomas, M. Modulation of splicing catalysis for therapeutic targeting of leukemia with mutations in genes encoding spliceosomal proteins. Nat. Med. 2016, 22, 672–678. [Google Scholar] [CrossRef] [PubMed]
- Fei, D.L.; Motowski, H.; Chatrikhi, R.; Prasad, S.; Yu, J.; Gao, S.; Kielkopf, C.L.; Bradley, R.K.; Varmus, H. Wild-type U2AF1 antagonizes the splicing program characteristic of U2AF1-mutant tumors and is required for cell survival. PLoS Genet. 2016, 12, e1006384. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Ahmed, D.; Dolatshad, H.; Tatwavedi, D.; Schulze, U.; Sanchi, A.; Ryley, S.; Dhir, A.; Carpenter, L.; Watt, S.M. SF3B1 mutations induce R-loop accumulation and DNA damage in MDS and leukemia cells with therapeutic implications. Leukemia 2020, 34, 2525–2530. [Google Scholar] [CrossRef]
- Chen, L.; Chen, J.-Y.; Huang, Y.-J.; Gu, Y.; Qiu, J.; Qian, H.; Shao, C.; Zhang, X.; Hu, J.; Li, H. The augmented R-loop is a unifying mechanism for myelodysplastic syndromes induced by high-risk splicing factor mutations. Mol. Cell 2018, 69, 412–425.e6. [Google Scholar] [CrossRef]
- Nguyen, H.D.; Yadav, T.; Giri, S.; Saez, B.; Graubert, T.A.; Zou, L. Functions of replication protein A as a sensor of R loops and a regulator of RNaseH1. Mol. Cell 2017, 65, 832–847.e4. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.A.; Choudhary, G.S.; Pellagatti, A.; Choi, K.; Bolanos, L.C.; Bhagat, T.D.; Gordon-Mitchell, S.; Von Ahrens, D.; Pradhan, K.; Steeples, V. U2AF1 mutations induce oncogenic IRAK4 isoforms and activate innate immune pathways in myeloid malignancies. Nat. Cell Biol. 2019, 21, 640–650. [Google Scholar] [CrossRef]
- Pollyea, D.A.; Harris, C.; Rabe, J.L.; Hedin, B.R.; De Arras, L.; Katz, S.; Wheeler, E.; Bejar, R.; Walter, M.J.; Jordan, C.T. Myelodysplastic syndrome-associated spliceosome gene mutations enhance innate immune signaling. Haematologica 2019, 104, e388–e392. [Google Scholar] [CrossRef]
- Wang, L.; Lawrence, M.S.; Wan, Y.; Stojanov, P.; Sougnez, C.; Stevenson, K.; Werner, L.; Sivachenko, A.; DeLuca, D.S.; Zhang, L. SF3B1 and other novel cancer genes in chronic lymphocytic leukemia. N. Engl. J. Med. 2011, 365, 2497–2506. [Google Scholar] [CrossRef]
- Ferrando, A.A.; Lopez-Otin, C. Clonal evolution in leukemia. Nat. Med. 2017, 23, 1135–1145. [Google Scholar] [CrossRef] [PubMed]
- Pararajalingam, P.; Coyle, K.M.; Arthur, S.E.; Thomas, N.; Alcaide, M.; Meissner, B.; Boyle, M.; Qureshi, Q.; Grande, B.M.; Rushton, C. Coding and noncoding drivers of mantle cell lymphoma identified through exome and genome sequencing. Blood 2020, 136, 572–584. [Google Scholar] [CrossRef] [PubMed]
- Morrison, S.J.; Spradling, A.C. Stem cells and niches: Mechanisms that promote stem cell maintenance throughout life. Cell 2008, 132, 598–611. [Google Scholar] [CrossRef]
- de Haan, G.; Lazare, S.S. Aging of hematopoietic stem cells. Blood J. Am. Soc. Hematol. 2018, 131, 479–487. [Google Scholar] [CrossRef] [PubMed]
- Laurenti, E.; Göttgens, B. From haematopoietic stem cells to complex differentiation landscapes. Nature 2018, 553, 418–426. [Google Scholar] [CrossRef]
- Zhang, X.; Yan, C.; Zhan, X.; Li, L.; Lei, J.; Shi, Y. Structure of the human activated spliceosome in three conformational states. Cell Res. 2018, 28, 307–322. [Google Scholar] [CrossRef]
- Visconte, V.; O Nakashima, M.; J Rogers, H. Mutations in splicing factor genes in myeloid malignancies: Significance and impact on clinical features. Cancers 2019, 11, 1844. [Google Scholar] [CrossRef]
- Chen, H.-C.; Cheng, S.-C. Functional roles of protein splicing factors. Biosci. Rep. 2012, 32, 345–359. [Google Scholar] [CrossRef] [PubMed]
- Larsson, C.A.; Cote, G.; Quintás-Cardama, A. The changing mutational landscape of acute myeloid leukemia and myelodysplastic syndrome. Mol. Cancer Res. 2013, 11, 815–827. [Google Scholar] [CrossRef]
- Boultwood, J.; Dolatshad, H.; Varanasi, S.S.; Yip, B.H.; Pellagatti, A. The role of splicing factor mutations in the pathogenesis of the myelodysplastic syndromes. Adv. Biol. Regul. 2014, 54, 153–161. [Google Scholar] [CrossRef]
- Brunner, A.M.; Steensma, D.P. Targeting aberrant splicing in myelodysplastic syndromes: Biologic rationale and clinical opportunity. Hematol. Oncol. Clin. 2020, 34, 379–391. [Google Scholar] [CrossRef]
- Ilagan, J.O.; Ramakrishnan, A.; Hayes, B.; Murphy, M.E.; Zebari, A.S.; Bradley, P.; Bradley, R.K. U2AF1 mutations alter splice site recognition in hematological malignancies. Genome Res. 2015, 25, 14–26. [Google Scholar] [CrossRef] [PubMed]
- Malcovati, L.; Stevenson, K.; Papaemmanuil, E.; Neuberg, D.; Bejar, R.; Boultwood, J.; Bowen, D.T.; Campbell, P.J.; Ebert, B.L.; Fenaux, P. SF3B1-mutant MDS as a distinct disease subtype: A proposal from the International Working Group for the Prognosis of MDS. Blood 2020, 136, 157–170. [Google Scholar] [CrossRef] [PubMed]
- Inoue, D.; Polaski, J.T.; Taylor, J.; Castel, P.; Chen, S.; Kobayashi, S.; Hogg, S.J.; Hayashi, Y.; Pineda, J.M.B.; El Marabti, E. Minor intron retention drives clonal hematopoietic disorders and diverse cancer predisposition. Nat. Genet. 2021, 53, 707–718. [Google Scholar] [CrossRef] [PubMed]
- Togami, K.; Chung, S.S.; Madan, V.; Booth, C.A.; Kenyon, C.M.; Cabal-Hierro, L.; Taylor, J.; Kim, S.S.; Griffin, G.K.; Ghandi, M. Sex-biased ZRSR2 mutations in myeloid malignancies impair plasmacytoid dendritic cell activation and apoptosis. Cancer Discov. 2022, 12, 522–541. [Google Scholar] [CrossRef]
- Madan, V.; Kanojia, D.; Li, J.; Okamoto, R.; Sato-Otsubo, A.; Kohlmann, A.; Sanada, M.; Grossmann, V.; Sundaresan, J.; Shiraishi, Y. Aberrant splicing of U12-type introns is the hallmark of ZRSR2 mutant myelodysplastic syndrome. Nat. Commun. 2015, 6, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Madan, V.; Cao, Z.; Teoh, W.W.; Dakle, P.; Han, L.; Shyamsunder, P.; Jeitany, M.; Zhou, S.; Li, J.; Nordin, H.B.M. ZRSR1 co-operates with ZRSR2 in regulating splicing of U12-type introns in murine hematopoietic cells. Haematologica 2022, 107, 680. [Google Scholar] [PubMed]
- Shiozawa, Y.; Malcovati, L.; Gallì, A.; Sato-Otsubo, A.; Kataoka, K.; Sato, Y.; Watatani, Y.; Suzuki, H.; Yoshizato, T.; Yoshida, K. Aberrant splicing and defective mRNA production induced by somatic spliceosome mutations in myelodysplasia. Nat. Commun. 2018, 9, 1–16. [Google Scholar] [CrossRef]
- Choudhary, G.S.; Smith, M.A.; Pellagatti, A.; Bhagat, T.D.; Gordon, S.; Pandey, S.; Shah, N.; Aluri, S.; Booher, R.N.; Ramachandra, M. SF3B1 mutations induce oncogenic IRAK4 isoforms and activate targetable innate immune pathways in MDS and AML. Blood 2019, 134, 4224. [Google Scholar] [CrossRef]
- Akgul, C.; Moulding, D.; Edwards, S. Alternative splicing of Bcl-2-related genes: Functional consequences and potential therapeutic applications. Cell. Mol. Life Sci. CMLS 2004, 61, 2189–2199. [Google Scholar] [CrossRef]
- Wahl, M.C.; Lührmann, R. SnapShot: Spliceosome dynamics I. Cell 2015, 161, 1474. [Google Scholar] [CrossRef]
- Merdzhanova, G.; Edmond, V.; De Seranno, S.; Van den Broeck, A.; Corcos, L.; Brambilla, C.; Brambilla, E.; Gazzeri, S.; Eymin, B. E2F1 controls alternative splicing pattern of genes involved in apoptosis through upregulation of the splicing factor SC35. Cell Death Differ. 2008, 15, 1815–1823. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.C.-W.; North, K.; Kim, E.; Jang, E.; Obeng, E.; Lu, S.X.; Liu, B.; Inoue, D.; Yoshimi, A.; Ki, M. Synthetic lethal and convergent biological effects of cancer-associated spliceosomal gene mutations. Cancer Cell 2018, 34, 225–241.e8. [Google Scholar] [CrossRef] [PubMed]
- Quesada, V.; Conde, L.; Villamor, N.; Ordóñez, G.R.; Jares, P.; Bassaganyas, L.; Ramsay, A.J.; Beà, S.; Pinyol, M.; Martínez-Trillos, A. Exome sequencing identifies recurrent mutations of the splicing factor SF3B1 gene in chronic lymphocytic leukemia. Nat. Genet. 2012, 44, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Gallenga, C.E.; Franco, E.; Adamo, G.G.; Violanti, S.S.; Tassinari, P.; Tognon, M.; Perri, P. Genetic Basis and Molecular Mechanisms of Uveal Melanoma Metastasis: A Focus on Prognosis. Front. Oncol. 2022, 12, 828112. [Google Scholar] [CrossRef]
- Pagliuca, S.; Gurnari, C.; Visconte, V. Molecular targeted therapy in myelodysplastic syndromes: New options for tailored treatments. Cancers 2021, 13, 784. [Google Scholar] [CrossRef]
- Teng, T.; Tsai, J.H.; Puyang, X.; Seiler, M.; Peng, S.; Prajapati, S.; Aird, D.; Buonamici, S.; Caleb, B.; Chan, B. Splicing modulators act at the branch point adenosine binding pocket defined by the PHF5A–SF3b complex. Nat. Commun. 2017, 8, 1–16. [Google Scholar] [CrossRef]
- Inoue, D.; Chew, G.-L.; Liu, B.; Michel, B.C.; Pangallo, J.; D’Avino, A.R.; Hitchman, T.; North, K.; Lee, S.C.-W.; Bitner, L. Spliceosomal disruption of the non-canonical BAF complex in cancer. Nature 2019, 574, 432–436. [Google Scholar] [CrossRef]
- Cazzola, M.; Rossi, M.; Malcovati, L. Biologic and clinical significance of somatic mutations of SF3B1 in myeloid and lymphoid neoplasms. Blood J. Am. Soc. Hematol. 2013, 121, 260–269. [Google Scholar] [CrossRef]
- Makishima, H.; Visconte, V.; Sakaguchi, H.; Jankowska, A.M.; Abu Kar, S.; Jerez, A.; Przychodzen, B.; Bupathi, M.; Guinta, K.; Afable, M.G. Mutations in the spliceosome machinery, a novel and ubiquitous pathway in leukemogenesis. Blood J. Am. Soc. Hematol. 2012, 119, 3203–3210. [Google Scholar] [CrossRef]
- Seo, J.Y.; Lee, K.-O.; Kim, S.-H.; Kim, K.; Jung, C.W.; Jang, J.H.; Kim, H.-J. Clinical significance of SF3B1 mutations in Korean patients with myelodysplastic syndromes and myelodysplasia/myeloproliferative neoplasms with ring sideroblasts. Ann. Hematol. 2014, 93, 603–608. [Google Scholar] [CrossRef]
- Patnaik, M.M.; Lasho, T.L.; Hodnefield, J.M.; Knudson, R.A.; Ketterling, R.P.; Garcia-Manero, G.; Steensma, D.P.; Pardanani, A.; Hanson, C.A.; Tefferi, A. SF3B1 mutations are prevalent in myelodysplastic syndromes with ring sideroblasts but do not hold independent prognostic value. Blood J. Am. Soc. Hematol. 2012, 119, 569–572. [Google Scholar] [CrossRef] [PubMed]
- Hosono, N. Genetic abnormalities and pathophysiology of MDS. Int. J. Clin. Oncol. 2019, 24, 885–892. [Google Scholar] [CrossRef] [PubMed]
- Harbour, J.W.; Roberson, E.; Anbunathan, H.; Onken, M.D.; Worley, L.A.; Bowcock, A.M. Recurrent mutations at codon 625 of the splicing factor SF3B1 in uveal melanoma. Nat. Genet. 2013, 45, 133–135. [Google Scholar] [CrossRef] [PubMed]
- Quek, C.; Rawson, R.V.; Ferguson, P.M.; Shang, P.; Silva, I.; Saw, R.P.; Shannon, K.; Thompson, J.F.; Hayward, N.K.; Long, G.V. Recurrent hotspot SF3B1 mutations at codon 625 in vulvovaginal mucosal melanoma identified in a study of 27 Australian mucosal melanomas. Oncotarget 2019, 10, 930. [Google Scholar] [CrossRef] [PubMed]
- Damm, F.; Kosmider, O.; Gelsi-Boyer, V.; Renneville, A.; Carbuccia, N.; Hidalgo-Curtis, C.; Della Valle, V.; Couronné, L.; Scourzic, L.; Chesnais, V. Mutations affecting mRNA splicing define distinct clinical phenotypes and correlate with patient outcome in myelodysplastic syndromes. Blood J. Am. Soc. Hematol. 2012, 119, 3211–3218. [Google Scholar] [CrossRef] [PubMed]
- Thol, F.; Kade, S.; Schlarmann, C.; Löffeld, P.; Morgan, M.; Krauter, J.; Wlodarski, M.W.; Kölking, B.; Wichmann, M.; Görlich, K. Frequency and prognostic impact of mutations in SRSF2, U2AF1, and ZRSR2 in patients with myelodysplastic syndromes. Blood J. Am. Soc. Hematol. 2012, 119, 3578–3584. [Google Scholar] [CrossRef]
- Yavuzyigitoglu, S.; Koopmans, A.E.; Verdijk, R.M.; Vaarwater, J.; Eussen, B.; Van Bodegom, A.; Paridaens, D.; Kiliç, E.; de Klein, A.; Group, R.O.M.S. Uveal melanomas with SF3B1 mutations: A distinct subclass associated with late-onset metastases. Ophthalmology 2016, 123, 1118–1128. [Google Scholar] [CrossRef]
- Liu, Z.; Yoshimi, A.; Wang, J.; Cho, H.; Lee, S.C.-W.; Ki, M.; Bitner, L.; Chu, T.; Shah, H.; Liu, B. Mutations in the RNA splicing factor SF3B1 promote tumorigenesis through MYC stabilization. Cancer Discov. 2020, 10, 806–821. [Google Scholar] [CrossRef]
- Yoshida, H.; Park, S.-Y.; Sakashita, G.; Nariai, Y.; Kuwasako, K.; Muto, Y.; Urano, T.; Obayashi, E. Elucidation of the aberrant 3′ splice site selection by cancer-associated mutations on the U2AF1. Nat. Commun. 2020, 11, 1–9. [Google Scholar] [CrossRef]
- Meggendorfer, M.; Roller, A.; Haferlach, T.; Eder, C.; Dicker, F.; Grossmann, V.; Kohlmann, A.; Alpermann, T.; Yoshida, K.; Ogawa, S. SRSF2 mutations in 275 cases with chronic myelomonocytic leukemia (CMML). Blood J. Am. Soc. Hematol. 2012, 120, 3080–3088. [Google Scholar] [CrossRef]
- Pellagatti, A.; Armstrong, R.N.; Steeples, V.; Sharma, E.; Repapi, E.; Singh, S.; Sanchi, A.; Radujkovic, A.; Horn, P.; Dolatshad, H. Impact of spliceosome mutations on RNA splicing in myelodysplasia: Dysregulated genes/pathways and clinical associations. Blood J. Am. Soc. Hematol. 2018, 132, 1225–1240. [Google Scholar] [CrossRef]
- Suzuki, H.; Kumar, S.A.; Shuai, S.; Diaz-Navarro, A.; Gutierrez-Fernandez, A.; De Antonellis, P.; Cavalli, F.M.; Juraschka, K.; Farooq, H.; Shibahara, I. Recurrent noncoding U1 snRNA mutations drive cryptic splicing in SHH medulloblastoma. Nature 2019, 574, 707–711. [Google Scholar] [CrossRef] [PubMed]
- Niño, C.A.; Scotto di Perrotolo, R.; Polo, S. Recurrent Spliceosome Mutations in Cancer: Mechanisms and Consequences of Aberrant Splice Site Selection. Cancers 2022, 14, 281. [Google Scholar] [CrossRef] [PubMed]
- Van Nostrand, E.L.; Freese, P.; Pratt, G.A.; Wang, X.; Wei, X.; Xiao, R.; Blue, S.M.; Chen, J.-Y.; Cody, N.A.; Dominguez, D. A large-scale binding and functional map of human RNA-binding proteins. Nature 2021, 589, 711–719. [Google Scholar] [CrossRef] [PubMed]
- Kurtovic-Kozaric, A.; Przychodzen, B.; Singh, J.; Konarska, M.M.; Clemente, M.J.; Otrock, Z.K.; Nakashima, M.; Hsi, E.D.; Yoshida, K.; Shiraishi, Y. PRPF8 defects cause missplicing in myeloid malignancies. Leukemia 2015, 29, 126–136. [Google Scholar] [CrossRef]
- Polprasert, C.; Schulze, I.; Sekeres, M.A.; Makishima, H.; Przychodzen, B.; Hosono, N.; Singh, J.; Padgett, R.A.; Gu, X.; Phillips, J.G. Inherited and somatic defects in DDX41 in myeloid neoplasms. Cancer Cell 2015, 27, 658–670. [Google Scholar] [CrossRef]
- Hershberger, C.; Hiznay, J.; Dietrich, R.; Gu, X.; Hirsch, C.M.; Graham, A.; Przychodzen, B.P.; Visconte, V.; Adema, V.; Parker, Y. LUC7L2 is a novel RNA-splicing regulatory factor mutated in myelodysplastic syndromes. Blood 2018, 132, 3073. [Google Scholar] [CrossRef]
- Kadono, M.; Kanai, A.; Nagamachi, A.; Shinriki, S.; Kawata, J.; Iwato, K.; Kyo, T.; Oshima, K.; Yokoyama, A.; Kawamura, T. Biological implications of somatic DDX41 p. R525H mutation in acute myeloid leukemia. Exp. Hematol. 2016, 44, 745–754.e4. [Google Scholar] [CrossRef]
- Makishima, H.; Nannya, Y.; Takeda, J.; Momozawa, Y.; Saiki, R.; Yoshizato, T.; Atsuta, Y.; Iijima-Yamashita, Y.; Yoshida, K.; Shiraishi, Y. Clinical impacts of germline DDX41 mutations on myeloid neoplasms. Blood 2020, 136, 38–40. [Google Scholar] [CrossRef]
- Abou Dalle, I.; Kantarjian, H.; Bannon, S.A.; Kanagal-Shamanna, R.; Routbort, M.; Patel, K.P.; Hu, S.; Bhalla, K.; Garcia-Manero, G.; DiNardo, C.D. Successful lenalidomide treatment in high risk myelodysplastic syndrome with germline DDX41 mutation. Am. J. Hematol. 2020, 95, 227–229. [Google Scholar] [CrossRef]
- Negoro, E.; Radivoyevitch, T.; Polprasert, C.; Adema, V.; Hosono, N.; Makishima, H.; Przychodzen, B.; Hirsch, C.; Clemente, M.J.; Nazha, A. Molecular predictors of response in patients with myeloid neoplasms treated with lenalidomide. Leukemia 2016, 30, 2405–2409. [Google Scholar] [CrossRef] [PubMed]
- Biechonski, S.; Yassin, M.; Milyavsky, M. DNA-damage response in hematopoietic stem cells: An evolutionary trade-off between blood regeneration and leukemia suppression. Carcinogenesis 2017, 38, 367–377. [Google Scholar] [CrossRef] [PubMed]
- Flach, J.; Jann, J.-C.; Knaflic, A.; Riabov, V.; Streuer, A.; Altrock, E.; Xu, Q.; Schmitt, N.; Obländer, J.; Nowak, V. Replication stress signaling is a therapeutic target in myelodysplastic syndromes with splicing factor mutations. Haematologica 2021, 106, 2906. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.D.; Zou, L.; Graubert, T.A. Targeting R-loop-associated ATR response in myelodysplastic syndrome. Oncotarget 2019, 10, 2581. [Google Scholar] [CrossRef]
- Welch, J.S.; Ley, T.J.; Link, D.C.; Miller, C.A.; Larson, D.E.; Koboldt, D.C.; Wartman, L.D.; Lamprecht, T.L.; Liu, F.; Xia, J. The origin and evolution of mutations in acute myeloid leukemia. Cell 2012, 150, 264–278. [Google Scholar] [CrossRef] [PubMed]
- Shlush, L.I.; Mitchell, A.; Heisler, L.; Abelson, S.; Ng, S.W.; Trotman-Grant, A.; Medeiros, J.J.; Rao-Bhatia, A.; Jaciw-Zurakowsky, I.; Marke, R. Tracing the origins of relapse in acute myeloid leukaemia to stem cells. Nature 2017, 547, 104–108. [Google Scholar] [CrossRef]
- Osorio, F.G.; Huber, A.R.; Oka, R.; Verheul, M.; Patel, S.H.; Hasaart, K.; de la Fonteijne, L.; Varela, I.; Camargo, F.D.; van Boxtel, R. Somatic mutations reveal lineage relationships and age-related mutagenesis in human hematopoiesis. Cell Rep. 2018, 25, 2308–2316.e4. [Google Scholar] [CrossRef]
- Minchom, A.; Aversa, C.; Lopez, J. Dancing with the DNA damage response: Next-generation anti-cancer therapeutic strategies. Ther. Adv. Med. Oncol. 2018, 10, 1–18. [Google Scholar] [CrossRef]
- Gorthi, A.; Romero, J.C.; Loranc, E.; Cao, L.; Lawrence, L.A.; Goodale, E.; Iniguez, A.B.; Bernard, X.; Masamsetti, V.P.; Roston, S. EWS–FLI1 increases transcription to cause R-loops and block BRCA1 repair in Ewing sarcoma. Nature 2018, 555, 387–391. [Google Scholar] [CrossRef]
- Pilié, P.G.; Tang, C.; Mills, G.B.; Yap, T.A. State-of-the-art strategies for targeting the DNA damage response in cancer. Nat. Rev. Clin. Oncol. 2019, 16, 81–104. [Google Scholar] [CrossRef]
- Qiu, J.; Zhou, B.; Thol, F.; Zhou, Y.; Chen, L.; Shao, C.; DeBoever, C.; Hou, J.; Li, H.; Chaturvedi, A. Distinct splicing signatures affect converged pathways in myelodysplastic syndrome patients carrying mutations in different splicing regulators. RNA 2016, 22, 1535–1549. [Google Scholar] [CrossRef]
- Wang, C.; Yang, Y.; Gao, S.; Chen, J.; Yu, J.; Zhang, H.; Li, M.; Zhan, X.; Li, W. Immune dysregulation in myelodysplastic syndrome: Clinical features, pathogenesis and therapeutic strategies. Crit. Rev. Oncol. /Hematol. 2018, 122, 123–132. [Google Scholar] [CrossRef]
- Matos, A.; Magalhães, S.M.; Rauh, M.J. Immune Dysregulation and Recurring Mutations in Myelodysplastic Syndromes Pathogenesis. Cell Biol. Transl. Med. 2020, 12, 1–10. [Google Scholar]
- Glenthøj, A.; Ørskov, A.D.; Hansen, J.W.; Hadrup, S.R.; O’Connell, C.; Grønbæk, K. Immune mechanisms in myelodysplastic syndrome. Int. J. Mol. Sci. 2016, 17, 944. [Google Scholar] [CrossRef]
- Banerjee, T.; Calvi, L.M.; Becker, M.W.; Liesveld, J.L. Flaming and fanning: The spectrum of inflammatory influences in myelodysplastic syndromes. Blood Rev. 2019, 36, 57–69. [Google Scholar] [CrossRef]
- Ramos, N.R.; Mo, C.C.; Karp, J.E.; Hourigan, C.S. Current approaches in the treatment of relapsed and refractory acute myeloid leukemia. J. Clin. Med. 2015, 4, 665–695. [Google Scholar] [CrossRef]
- Kotake, Y.; Sagane, K.; Owa, T.; Mimori-Kiyosue, Y.; Shimizu, H.; Uesugi, M.; Ishihama, Y.; Iwata, M.; Mizui, Y. Splicing factor SF3b as a target of the antitumor natural product pladienolide. Nat. Chem. Biol. 2007, 3, 570–575. [Google Scholar] [CrossRef]
- Kaida, D.; Motoyoshi, H.; Tashiro, E.; Nojima, T.; Hagiwara, M.; Ishigami, K.; Watanabe, H.; Kitahara, T.; Yoshida, T.; Nakajima, H. Spliceostatin A targets SF3b and inhibits both splicing and nuclear retention of pre-mRNA. Nat. Chem. Biol. 2007, 3, 576–583. [Google Scholar] [CrossRef]
- Sridhar, P.; Chan, S.; Lock, Y.-J.; Petrocca, F. Preclinical evaluation of the SF3B1 inhibitor E7107 in triple negative breast cancer. Cancer Res. 2017, 77, 420. [Google Scholar] [CrossRef]
- Hasegawa, M.; Miura, T.; Kuzuya, K.; Inoue, A.; Won Ki, S.; Horinouchi, S.; Yoshida, T.; Kunoh, T.; Koseki, K.; Mino, K. Identification of SAP155 as the target of GEX1A (Herboxidiene), an antitumor natural product. ACS Chem. Biol. 2011, 6, 229–233. [Google Scholar] [CrossRef]
- Fan, L.; Lagisetti, C.; Edwards, C.C.; Webb, T.R.; Potter, P.M. Sudemycins, novel small molecule analogues of FR901464, induce alternative gene splicing. ACS Chem. Biol. 2011, 6, 582–589. [Google Scholar] [CrossRef] [PubMed]
- Motoyoshi, H.; Horigome, M.; Ishigami, K.; Yoshida, T.; Horinouchi, S.; Yoshida, M.; Watanabe, H.; Kitahara, T. Structure-activity relationship for FR901464: A versatile method for the conversion and preparation of biologically active biotinylated probes. Biosci. Biotechnol. Biochem. 2004, 68, 2178–2182. [Google Scholar] [CrossRef] [PubMed]
- Seiler, M.; Yoshimi, A.; Darman, R.; Chan, B.; Keaney, G.; Thomas, M.; Agrawal, A.A.; Caleb, B.; Csibi, A.; Sean, E. H3B-8800, an orally available small-molecule splicing modulator, induces lethality in spliceosome-mutant cancers. Nat. Med. 2018, 24, 497–504. [Google Scholar] [CrossRef] [PubMed]
- Steensma, D.P.; Wermke, M.; Klimek, V.M.; Greenberg, P.L.; Font, P.; Komrokji, R.S.; Yang, J.; Brunner, A.M.; Carraway, H.E.; Ades, L. Results of a clinical trial of H3B-8800, a splicing modulator, in patients with myelodysplastic syndromes (MDS), acute myeloid leukemia (AML) or chronic myelomonocytic leukemia (CMML). Blood 2019, 134, 673. [Google Scholar] [CrossRef]
- Steensma, D.P.; Wermke, M.; Klimek, V.M.; Greenberg, P.L.; Font, P.; Komrokji, R.S.; Yang, J.; Brunner, A.M.; Carraway, H.E.; Ades, L. Phase I First-in-Human Dose Escalation Study of the oral SF3B1 modulator H3B-8800 in myeloid neoplasms. Leukemia 2021, 35, 3542–3550. [Google Scholar] [CrossRef]
- Schneider-Poetsch, T.; Chhipi-Shrestha, J.K.; Yoshida, M. Splicing modulators: On the way from nature to clinic. J. Antibiot. 2021, 74, 603–616. [Google Scholar] [CrossRef]
- Assi, R.; Kantarjian, H.M.; Kadia, T.M.; Pemmaraju, N.; Jabbour, E.; Jain, N.; Daver, N.; Estrov, Z.; Uehara, T.; Owa, T. Final results of a phase 2, open-label study of indisulam, idarubicin, and cytarabine in patients with relapsed or refractory acute myeloid leukemia and high-risk myelodysplastic syndrome. Cancer 2018, 124, 2758–2765. [Google Scholar] [CrossRef]
- Rugo, H.S.; Jacobs, I.; Sharma, S.; Scappaticci, F.; Paul, T.A.; Jensen-Pergakes, K.; Malouf, G.G. The promise for histone methyltransferase inhibitors for epigenetic therapy in clinical oncology: A narrative review. Adv. Ther. 2020, 37, 3059–3082. [Google Scholar] [CrossRef]
- Gerhart, S.V.; Kellner, W.A.; Thompson, C.; Pappalardi, M.B.; Zhang, X.-P.; Montes de Oca, R.; Penebre, E.; Duncan, K.; Boriack-Sjodin, A.; Le, B. Activation of the p53-MDM4 regulatory axis defines the anti-tumour response to PRMT5 inhibition through its role in regulating cellular splicing. Sci. Rep. 2018, 8, 1–15. [Google Scholar] [CrossRef]
- Foote, K.M.; Nissink, J.W.M.; McGuire, T.; Turner, P.; Guichard, S.; Yates, J.W.; Lau, A.; Blades, K.; Heathcote, D.; Odedra, R. Discovery and characterization of AZD6738, a potent inhibitor of ataxia telangiectasia mutated and Rad3 related (ATR) kinase with application as an anticancer agent. J. Med. Chem. 2018, 61, 9889–9907. [Google Scholar] [CrossRef]
- Fordham, S.E.; Blair, H.J.; Elstob, C.J.; Plummer, R.; Drew, Y.; Curtin, N.J.; Heidenreich, O.; Pal, D.; Jamieson, D.; Park, C. Inhibition of ATR acutely sensitizes acute myeloid leukemia cells to nucleoside analogs that target ribonucleotide reductase. Blood Adv. 2018, 2, 1157–1169. [Google Scholar] [CrossRef] [PubMed]
- Green, A.M.; Budagyan, K.; Hayer, K.E.; Reed, M.A.; Savani, M.R.; Wertheim, G.B.; Weitzman, M.D. Cytosine deaminase APOBEC3A sensitizes leukemia cells to inhibition of the DNA replication checkpoint. Cancer Res. 2017, 77, 4579–4588. [Google Scholar] [CrossRef]
- Chaudhuri, L.; Vincelette, N.D.; Koh, B.D.; Naylor, R.M.; Flatten, K.S.; Peterson, K.L.; McNally, A.; Gojo, I.; Karp, J.E.; Mesa, R.A. CHK1 and WEE1 inhibition combine synergistically to enhance therapeutic efficacy in acute myeloid leukemia ex vivo. Haematologica 2014, 99, 688. [Google Scholar] [CrossRef] [PubMed]
- Schenk, E.L.; Koh, B.D.; Flatten, K.S.; Peterson, K.L.; Parry, D.; Hess, A.D.; Smith, B.D.; Karp, J.E.; Karnitz, L.M.; Kaufmann, S.H. Effects of selective checkpoint kinase 1 inhibition on cytarabine cytotoxicity in acute myelogenous leukemia cells in vitro. Clin. Cancer Res. 2012, 18, 5364–5373. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Niu, X.; Li, X.; Edwards, H.; Wang, G.; Wang, Y.; Taub, J.W.; Lin, H.; Ge, Y. Inhibition of CHK1 enhances cell death induced by the Bcl-2-selective inhibitor ABT-199 in acute myeloid leukemia cells. Oncotarget 2016, 7, 34785. [Google Scholar] [CrossRef]
- Garcia-Manero, G.; Platzbecker, U.; Tarantolo, S.R.; Gropper, S.; Talati, C.; Götze, K.S.; Dugan, J.; Winer, E.S.; Martinez, E.; Lieberman, C. A Phase 1, Open Label Dose Escalation Trial Evaluating the Safety, Pharmacokinetics, Pharmacodynamics, and Clinical Activity of Orally Administered CA-4948 in Patients with Acute Myelogenous Leukemia or Myelodysplastic Syndrome. Blood 2020, 136, 16. [Google Scholar] [CrossRef]
- Fenaux, P.; Platzbecker, U.; Mufti, G.J.; Garcia-Manero, G.; Buckstein, R.; Santini, V.; Díez-Campelo, M.; Finelli, C.; Cazzola, M.; Ilhan, O. Luspatercept in patients with lower-risk myelodysplastic syndromes. N. Engl. J. Med. 2020, 382, 140–151. [Google Scholar] [CrossRef]
- Platzbecker, U.; Germing, U.; Götze, K.S.; Kiewe, P.; Mayer, K.; Chromik, J.; Radsak, M.; Wolff, T.; Zhang, X.; Laadem, A. Luspatercept for the treatment of anaemia in patients with lower-risk myelodysplastic syndromes (PACE-MDS): A multicentre, open-label phase 2 dose-finding study with long-term extension study. Lancet Oncol. 2017, 18, 1338–1347. [Google Scholar] [CrossRef]
- Eskens, F.A.; Ramos, F.J.; Burger, H.; O’Brien, J.P.; Piera, A.; De Jonge, M.J.; Mizui, Y.; Wiemer, E.A.; Carreras, M.J.; Baselga, J. Phase I pharmacokinetic and pharmacodynamic study of the first-in-class spliceosome inhibitor E7107 in patients with advanced solid tumors. Clin. Cancer Res. 2013, 19, 6296–6304. [Google Scholar] [CrossRef]
- Hong, D.; Kurzrock, R.; Naing, A.; Wheler, J.; Falchook, G.; Schiffman, J.; Faulkner, N.; Pilat, M.; O’Brien, J.; LoRusso, P. A phase I, open-label, single-arm, dose-escalation study of E7107, a precursor messenger ribonucleic acid (pre-mRNA) splicesome inhibitor administered intravenously on days 1 and 8 every 21 days to patients with solid tumors. Investig. New Drugs 2014, 32, 436–444. [Google Scholar] [CrossRef]
- Roybal, G.A.; Jurica, M.S. Spliceostatin A inhibits spliceosome assembly subsequent to prespliceosome formation. Nucleic Acids Res. 2010, 38, 6664–6672. [Google Scholar] [CrossRef] [PubMed]
- Larrayoz, M.; Blakemore, S.J.; Dobson, R.C.; Blunt, M.D.; Rose-Zerilli, M.J.; Walewska, R.; Duncombe, A.; Oscier, D.; Koide, K.; Forconi, F. The SF3B1 inhibitor spliceostatin A (SSA) elicits apoptosis in chronic lymphocytic leukaemia cells through downregulation of Mcl-1. Leukemia 2016, 30, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Shirai, C.L.; White, B.S.; Tripathi, M.; Tapia, R.; Ley, J.N.; Ndonwi, M.; Kim, S.; Shao, J.; Carver, A.; Saez, B. Mutant U2AF1-expressing cells are sensitive to pharmacological modulation of the spliceosome. Nat. Commun. 2017, 8, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Shirai, C.L.; Tripathi, M.; Ley, J.N.; Ndonwi, M.; White, B.S.; Tapia, R.; Saez, B.; Bertino, A.; Shao, J.; Kim, S. Preclinical activity of splicing modulators in U2AF1 mutant MDS/AML. Blood 2015, 126, 1653. [Google Scholar] [CrossRef]
- Bakker, S.T.; Passegué, E. Resilient and resourceful: Genome maintenance strategies in hematopoietic stem cells. Exp. Hematol. 2013, 41, 915–923. [Google Scholar] [CrossRef]
- Cavelier, C.; Didier, C.; Prade, N.; Mansat-De Mas, V.; Manenti, S.; Recher, C.; Demur, C.; Ducommun, B. Constitutive activation of the DNA damage signaling pathway in acute myeloid leukemia with complex karyotype: Potential importance for checkpoint targeting therapy. Cancer Res. 2009, 69, 8652–8661. [Google Scholar] [CrossRef]
- Schoppy, D.W.; Ragland, R.L.; Gilad, O.; Shastri, N.; Peters, A.A.; Murga, M.; Fernandez-Capetillo, O.; Diehl, J.A.; Brown, E.J. Oncogenic stress sensitizes murine cancers to hypomorphic suppression of ATR. J. Clin. Investig. 2012, 122, 241–252. [Google Scholar] [CrossRef]
- Ma, J.; Li, X.; Su, Y.; Zhao, J.; Luedtke, D.A.; Epshteyn, V.; Edwards, H.; Wang, G.; Wang, Z.; Chu, R. Mechanisms responsible for the synergistic antileukemic interactions between ATR inhibition and cytarabine in acute myeloid leukemia cells. Sci. Rep. 2017, 7, 1–14. [Google Scholar] [CrossRef]
- Yip, B.H.; Steeples, V.; Repapi, E.; Armstrong, R.N.; Llorian, M.; Roy, S.; Shaw, J.; Dolatshad, H.; Taylor, S.; Verma, A. The U2AF1 S34F mutation induces lineage-specific splicing alterations in myelodysplastic syndromes. J. Clin. Investig. 2017, 127, 2206–2221. [Google Scholar] [CrossRef]
- Kwok, M.; Davies, N.; Agathanggelou, A.; Smith, E.; Oldreive, C.; Petermann, E.; Stewart, G.; Brown, J.; Lau, A.; Pratt, G. ATR inhibition induces synthetic lethality and overcomes chemoresistance in TP53-or ATM-defective chronic lymphocytic leukemia cells. Blood J. Am. Soc. Hematol. 2016, 127, 582–595. [Google Scholar] [CrossRef]
- Sanchez, Y.; Wong, C.; Thoma, R.S.; Richman, R.; Wu, Z.; Piwnica-Worms, H.; Elledge, S.J. Conservation of the Chk1 checkpoint pathway in mammals: Linkage of DNA damage to Cdk regulation through Cdc25. Science 1997, 277, 1497–1501. [Google Scholar] [CrossRef] [PubMed]
- Short, N.J.; Konopleva, M.; Kadia, T.M.; Borthakur, G.; Ravandi, F.; DiNardo, C.D.; Daver, N. Advances in the treatment of acute myeloid leukemia: New drugs and new challenges. Cancer Discov. 2020, 10, 506–525. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.-I.; Su, C.-C.; Yang, C.-Y.; Hung, D.-Z.; Lin, C.-T.; Lu, T.-H.; Liu, S.-H.; Huang, C.-F. Etoposide induces pancreatic β-cells cytotoxicity via the JNK/ERK/GSK-3 signaling-mediated mitochondria-dependent apoptosis pathway. Toxicol. Vitr. 2016, 36, 142–152. [Google Scholar] [CrossRef]
- Vincelette, N.D.; Ding, H.; Huehls, A.M.; Flatten, K.S.; Kelly, R.L.; Kohorst, M.A.; Webster, J.; Hess, A.D.; Pratz, K.W.; Karnitz, L.M. Effect of CHK1 Inhibition on CPX-351 Cytotoxicity in vitro and ex vivo. Sci. Rep. 2019, 9, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Qi, W.; Xie, C.; Li, C.; Caldwell, J.T.; Edwards, H.; Taub, J.W.; Wang, Y.; Lin, H.; Ge, Y. CHK1 plays a critical role in the anti-leukemic activity of the wee1 inhibitor MK-1775 in acute myeloid leukemia cells. J. Hematol. Oncol. 2014, 7, 1–12. [Google Scholar] [CrossRef]
- Boudny, M.; Trbusek, M. ATR-CHK1 pathway as a therapeutic target for acute and chronic leukemias. Cancer Treat. Rev. 2020, 88, 102026. [Google Scholar] [CrossRef]
- Eldfors, S.; Rai, S.; Sharma, V.; Gilbert, A.N.; Porkka, K.; Graubert, T.A. ATR/CHK1/WEE1 Dependency in SRSF2-Mutated MDS/AML. Blood 2021, 138, 3661. [Google Scholar] [CrossRef]
- Garcia-Manero, G.; Chien, K.S.; Montalban-Bravo, G. Myelodysplastic syndromes: 2021 update on diagnosis, risk stratification and management. Am. J. Hematol. 2020, 95, 1399–1420. [Google Scholar] [CrossRef]
- Kubasch, A.S.; Fenaux, P.; Platzbecker, U. Development of luspatercept to treat ineffective erythropoiesis. Blood Adv. 2021, 5, 1565–1575. [Google Scholar] [CrossRef]
- Garcia-Manero, G.; Winer, E.S.; DeAngelo, D.J.; Tarantolo, S.R.; Sallman, D.A.; Dugan, J.; Groepper, S.; Giagounidis, A.; Gotze, K.S.; Metzeler, K. Phase 1/2a study of the IRAK4 inhibitor CA-4948 as monotherapy or in combination with azacitidine or venetoclax in patients with relapsed/refractory (R/R) acute myeloid leukemia or lyelodysplastic syndrome. J. Clin. Oncol. 2022, 40, 7016. [Google Scholar] [CrossRef]
- Garcia-Manero, G.; Tarantolo, S.; Verma, A.; Dugan, J.; Winer, E.; Giagounidis, A. A Phase 1, dose escalation trial with novel oral irak4 inhibitor ca-4948 in patients with acute myelogenous leukemia or myelodysplastic syndrome–interim report. Proc. EHA Annu. Meet. 2021, 324573, S165. [Google Scholar]
- Restelli, V.; Lupi, M.; Chilà, R.; Vagni, M.; Tarantelli, C.; Spriano, F.; Gaudio, E.; Bertoni, F.; Damia, G.; Carrassa, L. DNA damage response inhibitor combinations exert synergistic antitumor activity in aggressive B-cell lymphomas. Mol. Cancer Ther. 2019, 18, 1255–1264. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, Y.; Hejna, J.; Takata, M. Regulation of R-loops and genome instability in Fanconi anemia. J. Biochem. 2019, 165, 465–470. [Google Scholar] [CrossRef] [PubMed]
- Dvinge, H. Regulation of alternative mRNA splicing: Old players and new perspectives. FEBS Lett. 2018, 592, 2987–3006. [Google Scholar] [CrossRef] [PubMed]
- Bejar, R. Splicing factor mutations in cancer. In RNA processing; Springer: Berlin/Heidelberg, Germany, 2016; pp. 215–228. [Google Scholar]


{kind=link}
| Mutations | Pathway/Functions | Target Genes | Key References |
|---|---|---|---|
| ZRSR2 | Transcription | LZTR1, IRF7, E2F1, E2F2, E2F3, E2F4, E2F6 | [33,34,35] |
| MAPK pathway | MAPK1, MAPK3, MAPK9 | [35,36] | |
| Other | RASGRP1, RASGRP2, RASGRP4, ARAF, BRAF, RAF1, PTEN | [35] | |
| SF3B1 | MAPK pathway | MAP3K7 | [10,37] |
| Immune response or inflammation | IRAK4 | [38] | |
| DNA damage | ATR/CHK1 | [14] | |
| Other | NF1, DICER1, PML, PDS5A, PPP2R5A, BRD9 | [37,39] | |
| U2AF1 | Epigenetic regulation | H2AFY, ASXL1, BCOR, DNMT3B | [11,31] |
| Apoptosis | CASP8 | [11,31] | |
| DNA damage | ATR/CHK1, FANCA | [11,31] | |
| Immune response or inflammation | IRAK4 | [17] | |
| Other | STRAP, CEP164, EHMT1, WAC, PABPC4, PPWD1, PTBP1, UPF3B | [40] | |
| SRSF2 | Transcription | EZH2, E2F1 | [9,41] |
| Apoptosis | CASP8 | [42] |
| Category | Target | Agent | Pre-Clinical/Clinical Evaluation | Key References |
|---|---|---|---|---|
| Splicing factor modulators | SF3B complex | Pladienolides | Anti-tumor activities in various mouse xenograft models | [87] |
| FR901464 | Anti-tumor activities in mouse xenograft models | [87,88] | ||
| Spliceostatin A | [87,88,92] | |||
| Herboxidiene | Inhibit tumor growth in mouse xenograft models | [90] | ||
| H3B-8800 | NCT02841540/I/Recruiting | [3,93,94,95] | ||
| E7107 | Anti-tumor activities in mouse xenograft models | [12,96] | ||
| Sudemycin D6 | [91] | |||
| Other splicing modulators | RBM39 | E7070 | NCT01692197/II/Completed | [97] |
| PRMT5 | GSK3326595 | NCT03614728/ I/Terminated | [98,99] | |
| JNJ-64619178 | NCT03573310/I/Active | [98] | ||
| PRT543 | NCT03886831/I/Active | |||
| DDR inhibitors | ATR | AZD6738 | NCT03770429/I/Recruiting | [74,100] |
| VE-822 | Anti-tumor activities in various mouse xenograft models | [101,102] | ||
| VE-821 | Cellular/xenograft | [103] | ||
| CHK1 | UCN-01 | NCT00301938/I/Completed | [14] | |
| NCT00004263/I/Completed | ||||
| MK-8776 (SCH900776) | NCT00907517/I/Terminated | [104] | ||
| NCT01870596/II/Completed | ||||
| LY2603618 | NCT02649764/I/Active | [105] | ||
| WEE1 | MK1775 | Cellular | [103] | |
| Inflammasome | IRAK4 | CA-4948 | NCT04278768/I/II/Active | [106] |
| Cytokines | TGF-β | Luspatercept | NCT02604433/III/Completed NCT02631070/III/Completed | [107] [107,108] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, F.; Chen, L. Molecular Threat of Splicing Factor Mutations to Myeloid Malignancies and Potential Therapeutic Modulations. Biomedicines 2022, 10, 1972. https://doi.org/10.3390/biomedicines10081972
Zhang F, Chen L. Molecular Threat of Splicing Factor Mutations to Myeloid Malignancies and Potential Therapeutic Modulations. Biomedicines. 2022; 10(8):1972. https://doi.org/10.3390/biomedicines10081972
Chicago/Turabian StyleZhang, Fangliang, and Liang Chen. 2022. "Molecular Threat of Splicing Factor Mutations to Myeloid Malignancies and Potential Therapeutic Modulations" Biomedicines 10, no. 8: 1972. https://doi.org/10.3390/biomedicines10081972
APA StyleZhang, F., & Chen, L. (2022). Molecular Threat of Splicing Factor Mutations to Myeloid Malignancies and Potential Therapeutic Modulations. Biomedicines, 10(8), 1972. https://doi.org/10.3390/biomedicines10081972