
Combined Immune Defect in B-Cell Lymphoproliferative Disorders Is Associated with Severe Infection and Cancer Progression

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Methods
2.1. Patients and Study Design
2.2. Laboratory Assessment
2.2.1. Immunoglobulins’ and Complement Analysis
2.2.2. Measurement of B, T and NK Lymphocytes
2.2.3. Measurement of Differential White Blood Cell Count
2.3. Statistical Analysis
3. Results
Overall Overview
4. Immunological Assessment
4.1. Serum Immunoglobulins
4.2. Specific Antibody Responses to Immunisation
4.3. Lymphocyte Subpopulations
4.4. Neutrophils
4.5. Serum Complement Factors
5. Immunological Stratification of SID
6. Associated Conditions
6.1. Autoimmune Disease and Immune Dysregulation
6.2. Gastrointestinal Involvement
6.3. Second Primary Neoplasia
6.4. Suspicion of Underlying Primary Immunodeficiency Disease
7. Management and Therapeutic Strategies
7.1. Immunoglobulin Replacement Therapy
7.2. Trained Immunity-Based Vaccines
8. Prognostic
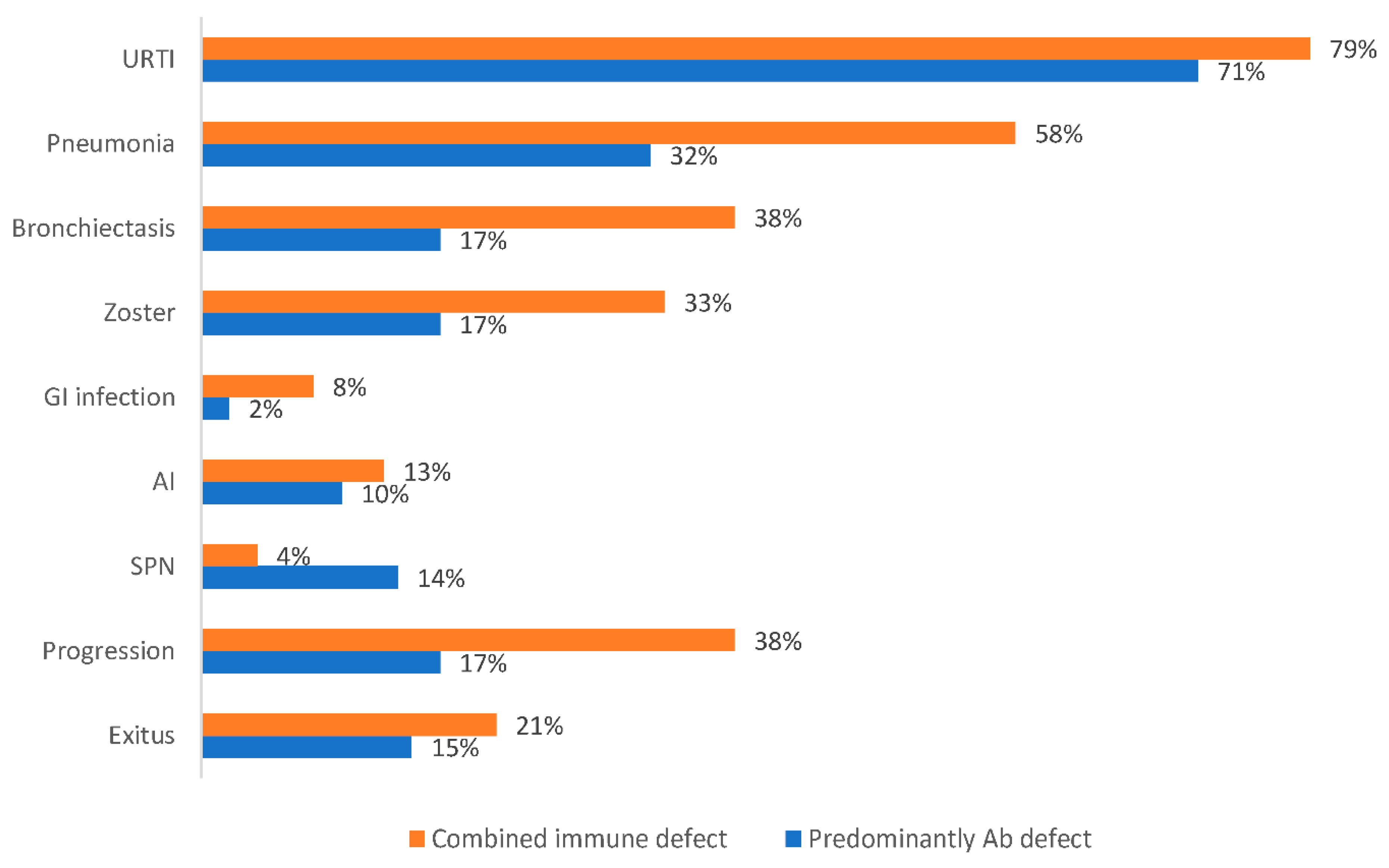
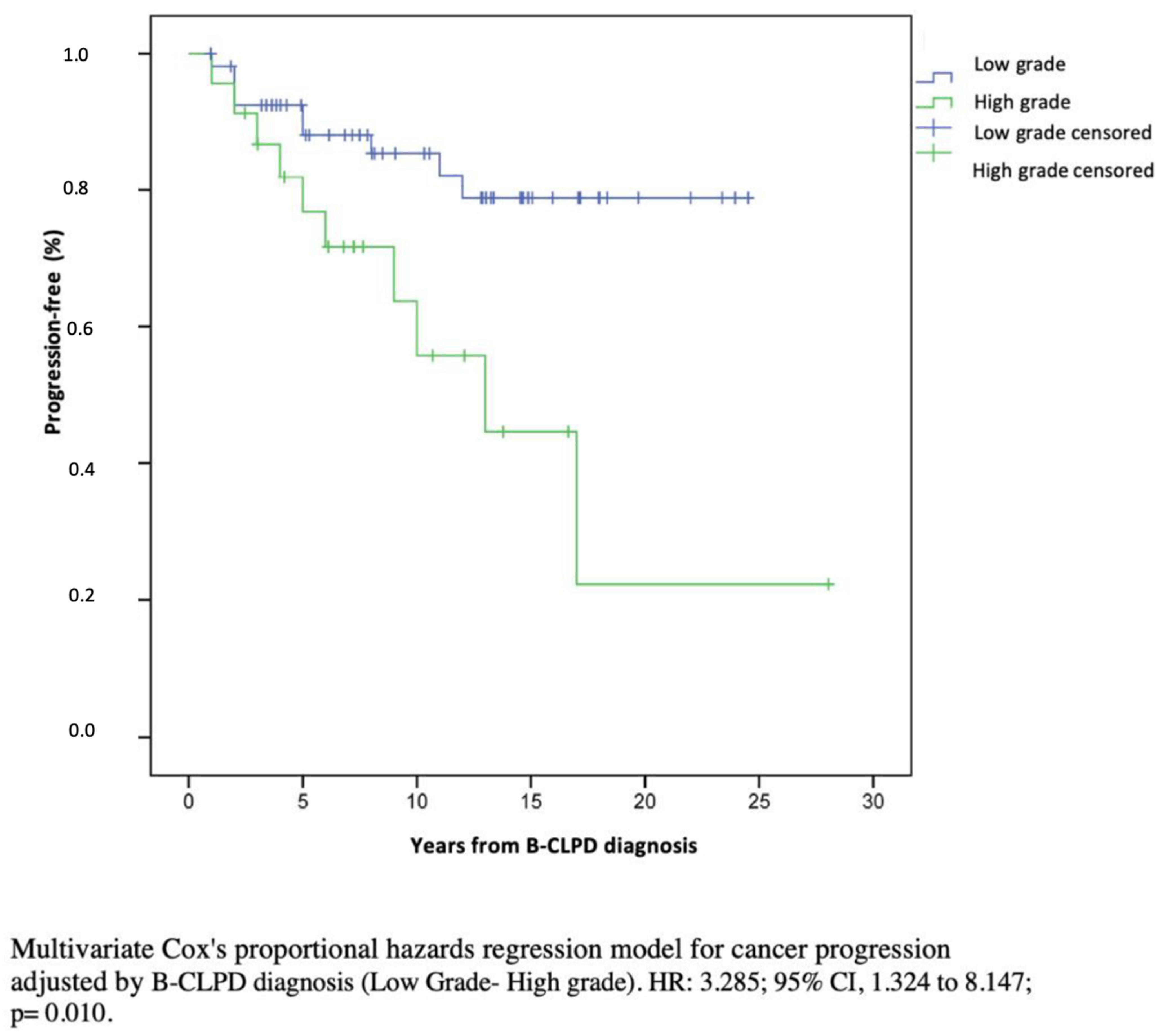
Survival, Progression, and Mortality
9. Discussion
10. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Kotlov, N.; Bagaev, A.; Revuelta, M.V.; Phillip, J.M.; Cacciapuoti, M.T.; Antysheva, Z.; Svekolkin, V.; Tikhonova, E.; Miheecheva, N.; Kuzkina, N.; et al. Clinical and Biological Subtypes of B-Cell Lymphoma Revealed by Microenvironmental Signatures. Cancer Discov. 2021, 11, 1468–1489. [Google Scholar] [CrossRef] [PubMed]
- Seifert, M.; Scholtysik, R.; Küppers, R. Origin and Pathogenesis of B Cell Lymphomas. In Lymphoma; Küppers, R., Ed.; Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2013; Volume 971, pp. 1–25. ISBN 978-1-62703-268-1. [Google Scholar]
- Griffiths, H.; Brennan, V.; Lea, J.; Bunch, C.; Lee, M.; Chapel, H. Crossover Study of Immunoglobulin Replacement Therapy in Patients with Low-Grade B-Cell Tumors. Blood 1989, 73, 366–368. [Google Scholar] [CrossRef] [PubMed]
- Chapel, H.M.; Bunch, C. Mechanisms of Infection in Chronic Lymphocytic Leukemia. Semin. Hematol. 1987, 24, 291–296. [Google Scholar] [PubMed]
- Boughton, B.J.; Jackson, N.; Lim, S.; Smith, N. Randomized Trial of Intravenous Immunoglobulin Prophylaxis for Patients with Chronic Lymphocytic Leukaemia and Secondary Hypogammaglobulinaemia. Clin. Lab. Haematol. 2008, 17, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Molica, S.; Musto, P.; Chiurazzi, F.; Specchia, G.; Brugiatelli, M.; Cicoira, L.; Levato, D.; Nobile, F.; Carotenuto, M.; Liso, V.; et al. Prophylaxis against Infections with Low-Dose Intravenous Immunoglobulins (IVIG) in Chronic Lymphocytic Leukemia. Results of a Crossover Study. Haematologica 1996, 81, 121–126. [Google Scholar]
- Sklenar, I.; Schiffman, G.; Jønsson, V.; Verhoef, G.; Birgens, H.; Boogaerts, M.; Ferrant, A.; Christensen, B.E.; Hasle, H.; Drivsholm, A.; et al. Effect of Various Doses of Intravenous Polyclonal IgG on in Vivo Levels of 12 Pneumococcal Antibodies in Patients with Chronic Lymphocytic Leukaemia and Multiple Myeloma. Oncology 1993, 50, 466–477. [Google Scholar] [CrossRef]
- A Randomized, Controlled Clinical Trial Cooperative Group for the Study of Immunoglobulin in Chronic Lymphocytic Leukemia* Intravenous Immunoglobulin for the Prevention of Infection in Chronic Lymphocytic Leukemia. N. Engl. J. Med. 1988, 319, 902–907. [CrossRef]
- Matanock, A.; Lee, G.; Gierke, R.; Kobayashi, M.; Leidner, A.; Pilishvili, T. Use of 13-Valent Pneumococcal Conjugate Vaccine and 23-Valent Pneumococcal Polysaccharide Vaccine Among Adults Aged ≥65 Years: Updated Recommendations of the Advisory Committee on Immunization Practices. MMWR Morb. Mortal. Wkly. Rep. 2019, 68, 1069–1075. [Google Scholar] [CrossRef]
- Griffiths, H.; Lea, J.; Bunch, C.; Lee, M.; Chapel, H. Predictors of Infection in Chronic Lymphocytic Leukaemia (CLL). Clin. Exp. Immunol. 1992, 89, 374–377. [Google Scholar] [CrossRef]
- European Medicines Agency Guideline on the Clinical Investigation of Human Normal Immunoglobulin for Intravenous Administration (IVIg). 2019. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-clinical-investigation-human-normal-immunoglobulin-intravenous-administration-ivig-rev-4_en.pdf (accessed on 11 August 2022).
- Parker, A.R.; Bradley, C.; Harding, S.; Sánchez-Ramón, S.; Jolles, S.; Kiani-Alikhan, S. Measurement and Interpretation of Salmonella Typhi Vi IgG Antibodies for the Assessment of Adaptive Immunity. J. Immunol. Methods 2018, 459, 1–10. [Google Scholar] [CrossRef]
- Ferry, B.L.; Misbah, S.A.; Stephens, P.; Sherrell, Z.; Lythgoe, H.; Bateman, E.; Banner, C.; Jones, J.; Groome, N.; Chapel, H.M. Development of an Anti-Salmonella Typhi Vi ELISA: Assessment of Immunocompetence in Healthy Donors. Clin. Exp. Immunol. 2004, 136, 297–303. [Google Scholar] [CrossRef] [PubMed]
- Ochoa-Grullón, J.; Benavente Cuesta, C.; Pérez López, C.; Peña Cortijo, A.; Rodríguez de la Peña, A.R.; Álvarez Carmona, A.; Mateo Morales, M.; LLano Hernández, K.; Williams, L.J.; Rodríguez de Frías, E.; et al. Evaluation of Polysaccharide Typhim vi Antibody Response as a Predictor of Humoral Immunodeficiency in Haematological Malignancies. Clin. Immunol. 2019, 210, 108307. [Google Scholar] [CrossRef] [PubMed]
- Watson, E.K.; Rose, P.W.; Neal, R.D.; Hulbert-Williams, N.; Donnelly, P.; Hubbard, G.; Elliott, J.; Campbell, C.; Weller, D.; Wilkinson, C. Personalised Cancer Follow-up: Risk Stratification, Needs Assessment or Both? Br. J. Cancer 2012, 106, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Condoluci, A.; Terzi di Bergamo, L.; Langerbeins, P.; Hoechstetter, M.A.; Herling, C.D.; De Paoli, L.; Delgado, J.; Rabe, K.G.; Gentile, M.; Doubek, M.; et al. International Prognostic Score for Asymptomatic Early-Stage Chronic Lymphocytic Leukemia. Blood 2020, 135, 1859–1869. [Google Scholar] [CrossRef] [PubMed]
- Hermans, J.; Krol, A.D.; van Groningen, K.; Kluin, P.M.; Kluin-Nelemans, J.C.; Kramer, M.H.; Noordijk, E.M.; Ong, F.; Wijermans, P.W. International Prognostic Index for Aggressive Non-Hodgkin’s Lymphoma Is Valid for All Malignancy Grades. Blood 1995, 86, 1460–1463. [Google Scholar] [CrossRef]
- Mailankody, S.; Mena, E.; Yuan, C.M.; Balakumaran, A.; Kuehl, W.M.; Landgren, O. Molecular and Biologic Markers of Progression in Monoclonal Gammopathy of Undetermined Significance to Multiple Myeloma. Leuk. Lymphoma 2010, 51, 2159–2170. [Google Scholar] [CrossRef]
- Gentile, M.; Shanafelt, T.D.; Cutrona, G.; Molica, S.; Tripepi, G.; Alvarez, I.; Mauro, F.R.; Di Renzo, N.; Di Raimondo, F.; Vincelli, I.; et al. A Progression-Risk Score to Predict Treatment-Free Survival for Early Stage Chronic Lymphocytic Leukemia Patients. Leukemia 2016, 30, 1440–1443. [Google Scholar] [CrossRef]
- Sánchez-Ramón, S.; Chapel, H.; Cunningham-Rundles, C. On the Relevance of Immunodeficiency Evaluation in Haematological Cancer. Hematol. Oncol. 2021, 39, 721–723. [Google Scholar] [CrossRef]
- Forconi, F.; Moss, P. Perturbation of the Normal Immune System in Patients with CLL. Blood 2015, 126, 573–581. [Google Scholar] [CrossRef]
- Ochoa-Grullón, J.; Peña Cortijo, A.; Guevara-Hoyer, K.; Jiménez García, C.; Fuente, E.; Peña, A.R.; Fernández-Arquero, M.; González Fernández, A.; Sánchez-Ramón, S. B-cell Haematological Malignancies and SARS-CoV-2 Infection: Could Immunological Interventions Influence the Outcome? eJHaem 2021, 2, 503–507. [Google Scholar] [CrossRef]
- Jolles, S.; Chapel, H.; Litzman, J. When to Initiate Immunoglobulin Replacement Therapy (IGRT) in Antibody Deficiency: A Practical Approach: When to Initiate IgG Therapy in Antibody Deficiency. Clin. Exp. Immunol. 2017, 188, 333–341. [Google Scholar] [CrossRef] [PubMed]
- Dhalla, F.; Lucas, M.; Schuh, A.; Bhole, M.; Jain, R.; Patel, S.Y.; Misbah, S.; Chapel, H. Antibody Deficiency Secondary to Chronic Lymphocytic Leukemia: Should Patients Be Treated with Prophylactic Replacement Immunoglobulin? J. Clin. Immunol. 2014, 34, 277–282. [Google Scholar] [CrossRef]
- Atta-ur-Rahman (Ed.) Frontiers in Clinical Drug Research: Anti-Infectives; Bentham Science Publishers: Sharjah, United Arab Emirates, 2016. [Google Scholar] [CrossRef]
- Mourits, V.P.; Wijkmans, J.C.; Joosten, L.A.; Netea, M.G. Trained Immunity as a Novel Therapeutic Strategy. Curr. Opin. Pharmacol. 2018, 41, 52–58. [Google Scholar] [CrossRef]
- Ochoa-Grullón, J.; Benavente Cuesta, C.; González Fernández, A.; Cordero Torres, G.; Pérez López, C.; Peña Cortijo, A.; Conejero Hall, L.; Mateo Morales, M.; Rodríguez de la Peña, A.; Díez-Rivero, C.M.; et al. Trained Immunity-Based Vaccine in B Cell Hematological Malignancies With Recurrent Infections: A New Therapeutic Approach. Front. Immunol. 2021, 11, 611566. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Ramón, S.; Fernández-Paredes, L.; Saz-Leal, P.; Diez-Rivero, C.M.; Ochoa-Grullón, J.; Morado, C.; Macarrón, P.; Martínez, C.; Villaverde, V.; de la Peña, A.R.; et al. Sublingual Bacterial Vaccination Reduces Recurrent Infections in Patients With Autoimmune Diseases Under Immunosuppressant Treatment. Front. Immunol. 2021, 12, 675735. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, S.H.; Campo, E.; Pileri, S.A.; Harris, N.L.; Stein, H.; Siebert, R.; Advani, R.; Ghielmini, M.; Salles, G.A.; Zelenetz, A.D.; et al. The 2016 Revision of the World Health Organization Classification of Lymphoid Neoplasms. Blood 2016, 127, 2375–2390. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.Y.; Carbone, J.; Jolles, S. The Expanding Field of Secondary Antibody Deficiency: Causes, Diagnosis, and Management. Front. Immunol. 2019, 10, 33. [Google Scholar] [CrossRef]
- Sánchez-Ramón, S.; Dhalla, F.; Chapel, H. Challenges in the Role of Gammaglobulin Replacement Therapy and Vaccination Strategies for Hematological Malignancy. Front. Immunol. 2016, 7, 317. [Google Scholar] [CrossRef]
- Jolles, S.; Michallet, M.; Agostini, C.; Albert, M.H.; Edgar, D.; Ria, R.; Trentin, L.; Lévy, V. Treating Secondary Antibody Deficiency in Patients with Haematological Malignancy: European Expert Consensus. Eur. J. Haematol. 2021, 106, 439–449. [Google Scholar] [CrossRef]
- Valdiglesias, V.; Sánchez-Flores, M.; Maseda, A.; Marcos-Pérez, D.; Millán-Calenti, J.C.; Pásaro, E.; Lorenzo-López, L.; Laffon, B. Lymphocyte Subsets in a Population of Nonfrail Elderly Individuals. J. Toxicol. Environ. Health Part A 2015, 78, 790–804. [Google Scholar] [CrossRef]
- Quinquenel, A.; Fornecker, L.-M.; Letestu, R.; Ysebaert, L.; Fleury, C.; Lazarian, G.; Dilhuydy, M.-S.; Nollet, D.; Guieze, R.; Feugier, P.; et al. Prevalence of BTK and PLCG2 Mutations in a Real-Life CLL Cohort Still on Ibrutinib after 3 Years: A FILO Group Study. Blood 2019, 134, 641–644. [Google Scholar] [CrossRef] [PubMed]
- Guevara-Hoyer, K.; Ochoa-Grullón, J.; Fernández-Arquero, M.; Cárdenas, M.; Pérez de Diego, R.; Sánchez-Ramón, S. Serum Free Immunoglobulins Light Chains: A Common Feature of Common Variable Immunodeficiency? Front. Immunol. 2020, 11, 2004. [Google Scholar] [CrossRef]
- Chatsirisupachai, K.; Lagger, C.; de Magalhães, J.P. Age-Associated Differences in the Cancer Molecular Landscape. Trends Cancer 2022, S2405803322001352. [Google Scholar] [CrossRef] [PubMed]
- Agostini, C.; Blau, I.-W.; Kimby, E.; Plesner, T. Prophylactic Immunoglobulin Therapy in Secondary Immune Deficiency—An Expert Opinion. Expert Rev. Clin. Immunol. 2016, 12, 921–926. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Ramón, S.; de Gracia, J.; García-Alonso, A.M.; Rodríguez Molina, J.J.; Melero, J.; de Andrés, A.; García Ruiz de Morales, J.M.; Ferreira, A.; Ocejo-Vinyals, J.G.; Cid, J.J.; et al. Multicenter Study for the Evaluation of the Antibody Response against Salmonella Typhi Vi Vaccination (EMPATHY) for the Diagnosis of Anti-Polysaccharide Antibody Production Deficiency in Patients with Primary Immunodeficiency. Clin. Immunol. 2016, 169, 80–84. [Google Scholar] [CrossRef]
- Bausch-Jurken, M.T.; Verbsky, J.W.; Gonzaga, K.A.; Elms, N.P.; Hintermeyer, M.K.; Gauld, S.B.; Routes, J.M. The Use of Salmonella Typhim Vaccine to Diagnose Antibody Deficiency. J. Clin. Immunol. 2017, 37, 427–433. [Google Scholar] [CrossRef]
- Arruga, F.; Gyau, B.B.; Iannello, A.; Vitale, N.; Vaisitti, T.; Deaglio, S. Immune Response Dysfunction in Chronic Lymphocytic Leukemia: Dissecting Molecular Mechanisms and Microenvironmental Conditions. Int. J. Mol. Sci. 2020, 21, 1825. [Google Scholar] [CrossRef]
- Nakamura, K.; Smyth, M.J.; Martinet, L. Cancer Immunoediting and Immune Dysregulation in Multiple Myeloma. Blood 2020, 136, 2731–2740. [Google Scholar] [CrossRef]
- Luo, S.; Wang, M.; Wang, H.; Hu, D.; Zipfel, P.F.; Hu, Y. How Does Complement Affect Hematological Malignancies: From Basic Mechanisms to Clinical Application. Front. Immunol. 2020, 11, 593610. [Google Scholar] [CrossRef]
- Hauck, F.; Voss, R.; Urban, C.; Seidel, M.G. Intrinsic and Extrinsic Causes of Malignancies in Patients with Primary Immunodeficiency Disorders. J. Allergy Clin. Immunol. 2018, 141, 59–68.e4. [Google Scholar] [CrossRef]
- Derpoorter, C.; Bordon, V.; Laureys, G.; Haerynck, F.; Lammens, T. Genes at the Crossroad of Primary Immunodeficiencies and Cancer. Front. Immunol. 2018, 9, 2544. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Ramón, S.; Bermúdez, A.; González-Granado, L.I.; Rodríguez-Gallego, C.; Sastre, A.; Soler-Palacín, P. the ID-Signal Onco-Haematology Group Primary and Secondary Immunodeficiency Diseases in Oncohaematology: Warning Signs, Diagnosis, and Management. Front. Immunol. 2019, 10, 586. [Google Scholar] [CrossRef] [PubMed]
- Bomken, S.; van der Werff Ten Bosch, J.; Attarbaschi, A.; Bacon, C.M.; Borkhardt, A.; Boztug, K.; Fischer, U.; Hauck, F.; Kuiper, R.P.; Lammens, T.; et al. Current Understanding and Future Research Priorities in Malignancy Associated With Inborn Errors of Immunity and DNA Repair Disorders: The Perspective of an Interdisciplinary Working Group. Front. Immunol. 2018, 9, 2912. [Google Scholar] [CrossRef] [PubMed]
- Claveau, J.-S.; Savary Bélanger, S.; Ahmad, I.; Delisle, J.-S.; De Guire, V.; Roy, J.; LeBlanc, R. Early Free Light Chain Reduction Following Treatment Initiation Predicts Favorable Outcome in Intact Immunoglobulin Myeloma. Blood Cancer J. 2022, 12, 3. [Google Scholar] [CrossRef]
- Solomon, B.M.; Chaffee, K.G.; Moreira, J.; Schwager, S.M.; Cerhan, J.R.; Call, T.G.; Kay, N.E.; Slager, S.L.; Shanafelt, T.D. Risk of Non-Hematologic Cancer in Individuals with High-Count Monoclonal B-Cell Lymphocytosis. Leukemia 2016, 30, 331–336. [Google Scholar] [CrossRef]
- Kumar, V.; Ailawadhi, S.; Bojanini, L.; Mehta, A.; Biswas, S.; Sher, T.; Roy, V.; Vishnu, P.; Marin-Acevedo, J.; Alegria, V.R.; et al. Trends in the Risk of Second Primary Malignancies among Survivors of Chronic Lymphocytic Leukemia. Blood Cancer J. 2019, 9, 75. [Google Scholar] [CrossRef]
- Hodgson, D.C.; Gilbert, E.S.; Dores, G.M.; Schonfeld, S.J.; Lynch, C.F.; Storm, H.; Hall, P.; Langmark, F.; Pukkala, E.; Andersson, M.; et al. Long-Term Solid Cancer Risk Among 5-Year Survivors of Hodgkin’s Lymphoma. J. Clin. Oncol. 2007, 25, 1489–1497. [Google Scholar] [CrossRef]
- Leung, W.; Sandlund, J.T.; Hudson, M.M.; Zhou, Y.; Hancock, M.L.; Zhu, Y.; Ribeiro, R.C.; Rubnitz, J.E.; Kun, L.E.; Razzouk, B.; et al. Second Malignancy after Treatment of Childhood Non-Hodgkin Lymphoma. Cancer 2001, 92, 1959–1966. [Google Scholar] [CrossRef]
- Castillo, J.J.; Gertz, M.A. Secondary Malignancies in Patients with Multiple Myeloma, Waldenström Macroglobulinemia and Monoclonal Gammopathy of Undetermined Significance. Leuk. Lymphoma 2017, 58, 773–780. [Google Scholar] [CrossRef]
- Itala, M.; Helenius, H.; Nikoskelainen, J.; Remes, K. Infections and Serum IgG Levels in Patients with Chronic Lymphocytic Leukemia. European Journal of Haematology 2009, 48, 266–270. [Google Scholar] [CrossRef]
- Young, H.J.-A. Epidemiology and Management of Infectious Complications in Contemporary Management of Chronic Leukemias. Infect. Disord.-Drug Targets 2011, 11, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Gafter-Gvili, A.; Fraser, A.; Paul, M.; Vidal, L.; Lawrie, T.A.; van de Wetering, M.D.; Kremer, L.C.; Leibovici, L. Antibiotic Prophylaxis for Bacterial Infections in Afebrile Neutropenic Patients Following Chemotherapy. Cochrane Database Syst. Rev. 2012, 1, CD004386. [Google Scholar] [CrossRef] [PubMed]
- Alecsandru, D.; Valor, L.; Sánchez-Ramón, S.; Gil, J.; Carbone, J.; Navarro, J.; Rodríguez, J.J.; Rodríguez-Sainz, C.; Fernández-Cruz, E. Sublingual Therapeutic Immunization with a Polyvalent Bacterial Preparation in Patients with Recurrent Respiratory Infections: Immunomodulatory Effect on Antigen-Specific Memory CD4+ T Cells and Impact on Clinical Outcome: Sublingual Immunization in Patients with Recurrent Respiratory Infections. Clin. Exp. Immunol. 2011, 164, 100–107. [Google Scholar] [CrossRef] [PubMed]
- Conejero Hall, L.; Nieto García, A.; Brandi, P.; Nieto Cid, M.; Mazón, Á.; Cueto, F.J.; Martínez-Cano, S.; Saz-Leal, P.; Enamorado, M.; Amores-Iniesta, J.; et al. Bacterial Immunotherapy in Children with Wheezing Attacks: Clinical Impact and Mechanism of Action. Eur. Respir. J. 2019, 54, PA4998. [Google Scholar]
- Sanchez-Ramon, S.; Diego, R.; Dieli-Crimi, R.; Subiza, J.-L. Extending the Clinical Horizons of Mucosal Bacterial Vaccines: Current Evidence and Future Prospects. Curr. Drug Targets 2014, 15, 1132–1143. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Variable | NHL (No. 41) | CLL (No. 18) | MGUS (No. 12) |
|---|---|---|---|
| IgG (mg/dL) | 516 ± 319 435 (471) | 480 ± 335 338 (481) | 1423 ± 797 1200 (1144) |
| IgA (mg/dL) | 59 ± 70 19 (87) | 62 ± 122 17 (33) | 133 ± 97 94 (207) |
| IgM (mg/dL) | 67 ± 124 26 (60) | 23 ± 25 14 (14) | 233 ± 378 91 (131) |
| IgG1 subclass (mg/dL) | 311 ± 198 285 (328) | 235 ± 142 196 (192) | 914 ± 588 905 (729) |
| IgG2 subclass (mg/dL) | 170 ± 128 120 (206) | 201 ± 178 118 (301) | 284 ± 184 264 (199) |
| C3 (mg/dL) | 127 ± 29 129 (38) | 129 ± 37 131(63) | 114 ± 28 118 (40) |
| C4 (mg/dL) | 29 ± 7 28 (9) | 25 ± 11 25 (14) | 20 ± 10 18 (11) |
| CD4+T-lymphocytes/mm3 | 521 ± 324 494 (419) | 1177 ± 931 1082 (927) | 852 ± 434 774 (739) |
| CD8+T-lymphocytes/mm3 | 612 ± 612 515 (362) | 1870 ± 3051 985 (1143) | 534 ± 257 521 (495) |
| CD19 B-lymphocytes/mm3 | 60 ± 121 103 (160) | 17,328 ± 3337 4722 (11,402) | 173 ± 53 143 (73) |
| NK cells/mm3 | 223 ± 193 139 (256) | 697 ± 651 591 (820) | 256 ± 132 229 (225) |
| Neutrophils ×103/uL) | 3572 ± 1886 3450 (2350) | 3508 ± 1425 3640 (2492) | 3650 ± 1884 3850 (1900) |
| No. of Patients n = 83 | % | |
|---|---|---|
| Recurrent bronchitis, sinusitis, otitis | 73 | 88 |
| Pneumonia | 33 | 40 |
| Sepsis (Pseudomona sp., pneumococcus, H. influenzae, S. agalactiae, CMV) | 22 | 27 |
| History of herpes zoster | 20 | 24 |
| Recurrent urinary tract infections | 12 | 14 |
| Recurrent oral herpes | 7 | 8 |
| Pulmonary TB | 5 | 6 |
| Oropharyngeal candidiasis | 4 | 5 |
| Viral hepatitis | 4 | 5 |
| Aspergillosis (pneumonia, brain abscess) | 4 | 5 |
| Campylobacter enteritis | 3 | 4 |
| Cellulitis | 3 | 4 |
| Cytomegalovirus pneumoniae | 2 | 3 |
| Human papillomavirus reactivation | 2 | 3 |
| Meningitis | 1 | 1 |
| Cryptogenic organizing pneumonia | 1 | 1 |
| Recurrent parotitis | 1 | 1 |
| Pneumocystis jirovecii infection | 1 | 1 |
| Osteomyelitis | 1 | 1 |
| Pyoderma gangrenosum | 1 | 1 |
| Patient | B-CLPD | Immune Defect | Time Interval (Years) | SPN |
|---|---|---|---|---|
| #4 | CLL | Predominantly Ab defect | 1 (2013–2014) | Lung adenocarcinoma |
| #12 | CLL | Combined immune defect | 4 (2005–2009) | Thyroid papillary carcinoma |
| #18 | NHL (FL) | Predominantly Ab defect | 3 (2003–2006–2008) | Peripheral nerve sheath tumor, Thyroid cancer and malignant nasal Ca |
| #28 | NHL (FL) | Predominantly Ab defect | 4 (2013–2017) | Colon adenocarcinoma |
| #48 | NHL | Predominantly Ab defect | 22 (1992–2014) | Prostate adenocarcinoma |
| #60 | MGUS | Predominantly Ab defect | 6 (2006–2012) | Breast cancer (infiltrating ductal carcinoma) |
| #61 | MM (IgA kappa) | Predominantly Ab defect | 7 (2013–2020) | Pancreatic intraductal papillary mucinous neoplasm |
| #62 | MGUS (IgA Lambda) | Predominantly Ab defect | 3 (2016–2019) | Breast cancer (infiltrating ductal carcinoma) |
| #68 | MGUS (IgA Lambda) | Predominantly Ab defect | 1 (2016–2017) | Endometrial cancer |
| Patient | Primary Neoplasia | Time Interval Years | Immune Defect | B-CLPD Diagnosis |
|---|---|---|---|---|
| #5 | Basal cell carcinoma | 5 (1998–2003) | Predominantly Ab defect | CLL |
| #24 | Breast cancer | 14 (1994–2008) | Combined immune defect | NHL (FL) |
| #25 | GIST | 2 (2009–2011) | Combined immune defect | NHL (FL) |
| #43 | Prostate adenocarcinoma | 12 (2002–2014) | Predominantly Ab defect | NHL (DLBCL) |
| #59 | Breast cancer (infiltrating ductal carcinoma) | 11 (2005–2016) | Combined immune defect | MM |
| Patient | B-CLPD Diagnosis | Age at Diagnosis (Years) | Date Last Chemotherapy | Serum Free Kappa (mg/L) | Serum Free Lambda (mg/L) | Past Medical History Previous B-CLPD Diagnosis | GI Involvement |
|---|---|---|---|---|---|---|---|
| #7 | CLL | 50 | 2012 | 2.0 | 1.5 | RRTI; Salmonella GI. | No Salmonella Typhi gastroenteritis |
| #10 | CLL | 38 | 2020 | 12.9 | 5.8 | RRTI | Genetic susceptibility to CD |
| #12 | CLL | 48 | 2017 | 12.3 | 12.3 | RRTI | Persistent H pylori infection |
| #15 | CLL | 54 | 2007 | 0.1 | 0.3 | RRTI | Campylobacter jejuni infection |
| #18 | NHL (FL) | 67 | 2008 | 0.0 | 0.0 | Severe infection | Celiac disease (MARSH III) |
| #23 | NHL (FL) | 50 | 1996 | 0.0 | 0.0 | Recurrent pneumonia | - |
| #32 | NHL (FL) | 45 | 2017 | 0.3 | 0.3 | RRTI | - |
| #34 | NHL (FL) | 34 | 2009 | 0.3 | 1.1 | RRTI, zoster infection. | - |
| #42 | NHL (DLBCL) | 59 | 2014 | 2.3 | 1.7 | Pneumonia | Persistent H. pylori infection |
| #51 | NHL (Burkit) | 7 | 2017 | 2.1 | 1.5 | Lymphoma recurrence | Persisten H. pylori infection |
| #54 | HL | 13 | - | 2.1 | 1.7 | RRTI, lymphoma recurrence | Genetic susceptibility to CD, elevated liver enzymes, H. pylori infection |
| #63 | MGUS | 41 | - | 19.0 | 25.0 | Lung lymphangiomatosis | Genetic susceptibility to CD |
| Patient | B-CLPD Diagnosis | Baseline | Post-Vaccination | Typhim Vi Booster |
|---|---|---|---|---|
| #3 | CLL | 7.4 | 7.4 | 12.4 |
| #4 | CLL | 7.4 | 7.4 | 8.8 |
| #7 | CLL | 7.4 | 7.4 | 15.4 |
| #8 | CLL | 7.4 | 10.3 | 32.1 |
| #10 | CLL+HL | 7.4 | 7.4 | 12.4 |
| #19 | NHL | 7.4 | 7.4 | 9.2 |
| #25 | NHL | 7.4 | 7.4 | 7.4 |
| #50 | NHL (MALT) | 7.4 | 7.4 | 7.4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ochoa-Grullón, J.; Guevara-Hoyer, K.; Pérez López, C.; Pérez de Diego, R.; Peña Cortijo, A.; Polo, M.; Mateo Morales, M.; Anguita Mandley, E.; Jiménez García, C.; Bolaños, E.; et al. Combined Immune Defect in B-Cell Lymphoproliferative Disorders Is Associated with Severe Infection and Cancer Progression. Biomedicines 2022, 10, 2020. https://doi.org/10.3390/biomedicines10082020
Ochoa-Grullón J, Guevara-Hoyer K, Pérez López C, Pérez de Diego R, Peña Cortijo A, Polo M, Mateo Morales M, Anguita Mandley E, Jiménez García C, Bolaños E, et al. Combined Immune Defect in B-Cell Lymphoproliferative Disorders Is Associated with Severe Infection and Cancer Progression. Biomedicines. 2022; 10(8):2020. https://doi.org/10.3390/biomedicines10082020
Chicago/Turabian StyleOchoa-Grullón, Juliana, Kissy Guevara-Hoyer, Cristina Pérez López, Rebeca Pérez de Diego, Ascensión Peña Cortijo, Marta Polo, Marta Mateo Morales, Eduardo Anguita Mandley, Carlos Jiménez García, Estefanía Bolaños, and et al. 2022. "Combined Immune Defect in B-Cell Lymphoproliferative Disorders Is Associated with Severe Infection and Cancer Progression" Biomedicines 10, no. 8: 2020. https://doi.org/10.3390/biomedicines10082020
APA StyleOchoa-Grullón, J., Guevara-Hoyer, K., Pérez López, C., Pérez de Diego, R., Peña Cortijo, A., Polo, M., Mateo Morales, M., Anguita Mandley, E., Jiménez García, C., Bolaños, E., Íñigo, B., Medina, F., Rodríguez de la Peña, A., Izquierdo Delgado, C., de la Fuente Muñoz, E., Mayol, E., Fernández-Arquero, M., González-Fernández, A., Benavente Cuesta, C., & Sánchez-Ramón, S. (2022). Combined Immune Defect in B-Cell Lymphoproliferative Disorders Is Associated with Severe Infection and Cancer Progression. Biomedicines, 10(8), 2020. https://doi.org/10.3390/biomedicines10082020