
Targeting Epigenetic Mechanisms: A Boon for Cancer Immunotherapy


Abstract
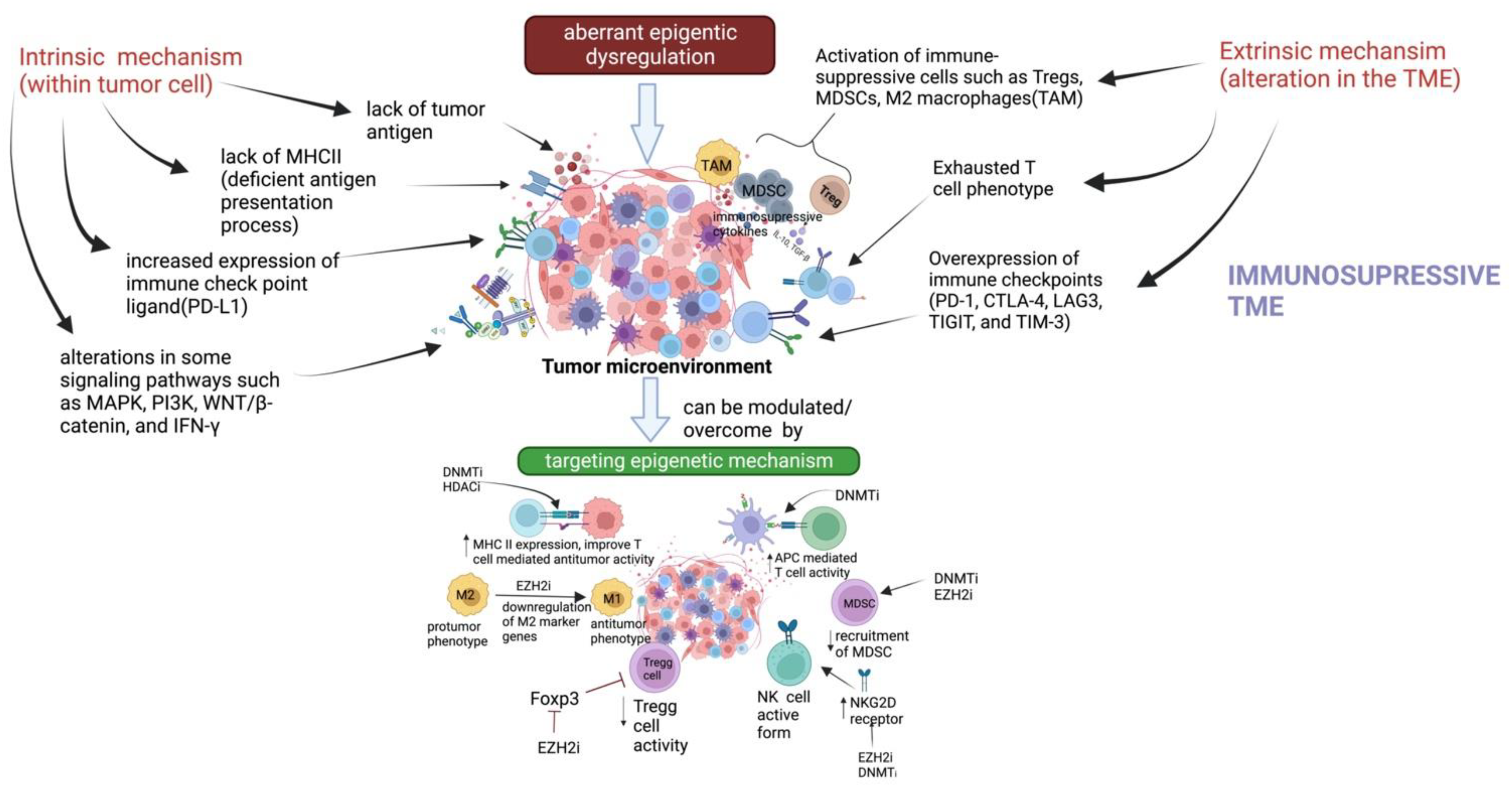
:1. Introduction
2. Epigenetic Regulation in Cancer
3. Epigenetic Regulation of Immune Cells
3.1. Epigenetic Regulation of Myeloid Cells (Differentiation, Activation)
3.1.1. Dendritic Cells
3.1.2. Macrophages
3.1.3. Myeloid-Derived Suppressor Cells (MDSC)
3.2. Epigenetic Regulation of Lymphoid Cells
3.2.1. Natural Killer Cells (NK Cells)
3.2.2. CD4+ T Cells
3.2.3. Treg Cell
3.2.4. CD8+ T Cell
3.2.5. B Cells
4. Targeting the Epigenetic Mechanism to Modulate Antitumor Immunity: A Strategy to Improve Cancer Immunotherapy
4.1. DNMT Inhibitors Targeting Antitumor Immunity
4.2. Tet-2 Role in Modulating Antitumor Immunity
4.3. HDAC Inhibitors Targeting Antitumor Immunity
4.4. SIRT-1 [Sirtuin (Silent Mating Type Information Regulation 2 Homolog) 1] Modulating Antitumor Immunity
4.5. HMT Inhibitors Targeting Antitumor Immunity
4.6. PRMT-5 Inhibitor Targeting Antitumor Immunity
4.7. Bromodomain Inhibitor Targeting Antitumor Immunity
5. Clinical Trial Combining Epigenetic Therapy with Immunotherapy
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Topper, M.J.; Vaz, M.; Marrone, K.A.; Brahmer, J.R.; Baylin, S.B. The emerging role of epigenetic therapeutics in immuno-oncology. Nat. Rev. Clin. Oncol. 2020, 17, 75–90. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Zhang, T.; Zheng, L.; Liu, H.; Song, W.; Liu, D.; Li, Z.; Pan, C.-X. Combination strategies to maximize the benefits of cancer immunotherapy. J. Hematol. Oncol. 2021, 14, 1–33. [Google Scholar] [CrossRef]
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer Immunoediting: Integrating Immunity’s Roles in Cancer Suppression and Promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef] [Green Version]
- O’Donnell, J.S.; Teng, M.W.L.; Smyth, M.J. Cancer immunoediting and resistance to T cell-based immunotherapy. Nat. Rev. Clin. Oncol. 2019, 16, 151–167. [Google Scholar] [CrossRef] [PubMed]
- Lodewijk, I.; Nunes, S.P.; Henrique, R.; Jerónimo, C.; Dueñas, M.; Paramio, J.M. Tackling tumor microenvironment through epigenetic tools to improve cancer immunotherapy. Clin. Epigenetics 2021, 13, 63. [Google Scholar] [CrossRef] [PubMed]
- Barbari, C.; Fontaine, T.; Parajuli, P.; Lamichhane, N.; Jakubski, S.; Lamichhane, P.; Deshmukh, R. Immunotherapies and Combination Strategies for Immuno-Oncology. Int. J. Mol. Sci. 2020, 21, 5009. [Google Scholar] [CrossRef]
- McDowell, S. Immunotherapy How Immunotherapy Is Used to Treat. Cancer 2019, 2019, 1–26. [Google Scholar]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [Green Version]
- Ohaegbulam, K.; Assal, A. EL-M-T in molecular, 2015 U. Human cancer immunotherapy with antibodies to the PD-1 and PD-L1 pathway. Trends Mol. Med. 2015, 21, 24–33. [Google Scholar] [CrossRef] [Green Version]
- Bashash, D.; Zandi, Z.; Kashani, B.; Pourbagheri-Sigaroodi, A.; Salari, S.; Ghaffari, S.H. Resistance to immunotherapy in human malignancies: Mechanisms, research progresses, challenges, and opportunities. J. Cell. Physiol. 2022, 237, 346–372. [Google Scholar] [CrossRef]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef] [PubMed]
- Bai, R.; Chen, N.; Li, L.; Du, N.; Bai, L.; Lv, Z.; Tian, H.; Cui, J. Mechanisms of Cancer Resistance to Immunotherapy. Front. Oncol. 2020, 10, 1290. [Google Scholar] [CrossRef]
- Bonaventura, P.; Shekarian, T.; Alcazer, V.; Valladeau-Guilemond, J.; Valsesia-Wittmann, S.; Amigorena, S.; Caux, C.; Depil, S. Cold Tumors: A Therapeutic Challenge for Immunotherapy. Front. Immunol. 2019, 10, 168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hogg, S.J.; Beavis, P.A.; Dawson, M.A.; Johnstone, R.W. Targeting the epigenetic regulation of antitumour immunity. Nat. Rev. Drug Discov. 2020, 19, 776–800. [Google Scholar] [CrossRef] [PubMed]
- Baylin, S.B.; Jones, P.A. Epigenetic Determinants of Cancer. Cold Spring Harb. Perspect. Biol. 2016, 8, a019505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quina, A.S.; Buschbeck, M.; di Croce, L. Chromatin structure and epigenetics. Biochem. Pharmacol. 2006, 72, 1563–1569. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.; Rao, C.M. Epigenetics in cancer: Fundamentals and Beyond. Pharmacol. Ther. 2017, 173, 118–134. [Google Scholar] [CrossRef]
- Skourti, E.; Dhillon, P. Cancer epigenetics: Promises and pitfalls for cancer therapy. FEBS J. 2022, 289, 1156–1159. [Google Scholar] [CrossRef]
- Baxter, E.; Windloch, K.; Gannon, F.; Lee, J.S. Epigenetic regulation in cancer progression. Cell Biosci. 2014, 4, 45. [Google Scholar] [CrossRef] [Green Version]
- Allis, C.; Januwein, T. The molecular hallmarks of epigenetic control. Nat. Rev. Genet. 2016, 17, 487–500. [Google Scholar] [CrossRef]
- Gibney, E.R.; Nolan, C.M. Epigenetics and gene expression. Heredity 2010, 105, 4–13. [Google Scholar] [CrossRef] [PubMed]
- Huether, R.; Dong, L.; Chen, X.; Wu, G.; Parker, M.; Wei, L.; Ma, J.; Edmonson, M.N.; Hedlund, E.K.; Rusch, M.C.; et al. The landscape of somatic mutations in epigenetic regulators across 1000 paediatric cancer genomes. Nat. Commun. 2014, 5, 3630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morrison, A.J. Cancer cell metabolism connects epigenetic modifications to transcriptional regulation. FEBS J. 2022, 289, 1302–1314. [Google Scholar] [CrossRef]
- Dos Santos, E.S.; Wagner, V.P.; Cabral Ramos, J.; Lambert, D.W.; Castilho, R.M.; Paes Leme, A.F. Epigenetic modulation of the tumor microenvironment in head and neck cancer: Challenges and opportunities. Crit. Rev. Oncol. Hematol. 2021, 164, 103397. [Google Scholar] [CrossRef]
- Labani-Motlagh, A.; Ashja-Mahdavi, M.; Loskog, A. The Tumor Microenvironment: A Milieu Hindering and Obstructing Antitumor Immune Responses. Front. Immunol. 2020, 11, 940. [Google Scholar] [CrossRef] [PubMed]
- Duan, Q.; Zhang, H.; Zheng, J.; Zhang, L. Turning Cold into Hot: Firing up the Tumor Microenvironment. Trends Cancer 2020, 6, 605–618. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Gomez, A.; Rodríguez-Ubreva, J.; Ballestar, E. Epigenetic interplay between immune, stromal and cancer cells in the tumor microenvironment. Clin. Immunol. 2018, 196, 64–71. [Google Scholar] [CrossRef]
- Balakrishnan, A.; Vig, M.; Dubey, S. Role of myeloid cells in the tumor microenvironment. J. Cancer Metastasis Treat. 2022, 8, 27. [Google Scholar] [CrossRef]
- Anderson, N.M.; Simon, M.C. The tumor microenvironment. Curr. Biol. 2020, 30, R921–R925. [Google Scholar] [CrossRef]
- Broz, M.L.; Binnewies, M.; Boldajipour, B.; Nelson, A.E.; Pollack, J.L.; Erle, D.J.; Barczak, A.; Rosenblum, M.D.; Daud, A.; Barber, D.L.; et al. Dissecting the tumor myeloid compartment reveals rare activating antigen-presenting cells critical for T cell immunity. Cancer Cell 2014, 26, 638–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tran Janco, J.M.; Lamichhane, P.; Karyampudi, L.; Knutson, K.L. Tumor-Infiltrating Dendritic Cells in Cancer Pathogenesis. J. Immunol. 2015, 194, 2985–2991. [Google Scholar] [CrossRef]
- Rodríguez-Ubreva, J.; Garcia-Gomez, A.; Ballestar, E. Epigenetic mechanisms of myeloid differentiation in the tumor microenvironment. Curr. Opin. Pharmacol. 2017, 35, 20–29. [Google Scholar] [CrossRef] [PubMed]
- Verneau, J.; Sautés-Fridman, C.; Sun, C.M. Dendritic cells in the tumor microenvironment: Prognostic and theranostic impact. Semin. Immunol. 2020, 48, 101410. [Google Scholar] [CrossRef] [PubMed]
- Stoitzner, P.; Green, L.K.; Jung, J.Y.; Price, K.; Atarea, H.; Kivell, B.; Ronchese, F. Inefficient presentation of tumor-derived antigen by tumor-infiltrating dendritic cells. Cancer Immunol. Immunother. 2008, 57, 1665–1673. [Google Scholar] [CrossRef]
- Nutt, S.L.; Chopin, M. Transcriptional Networks Driving Dendritic Cell Differentiation and Function. Immunity 2020, 52, 942–956. [Google Scholar] [CrossRef]
- Zhou, Z.; Chen, H.; Xie, R.; Wang, H.; Li, S.; Xu, Q.; Xu, N.; Cheng, Q.; Qian, Y.; Huang, R.; et al. Epigenetically modulated FOXM1 suppresses dendritic cell maturation in pancreatic cancer and colon cancer. Mol. Oncol. 2019, 13, 873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitamura, H.; Ohno, Y.; Toyoshima, Y.; Ohtake, J.; Homma, S.; Kawamura, H.; Takahashi, N.; Taketomi, A. Interleukin-6/STAT3 signaling as a promising target to improve the efficacy of cancer immunotherapy. Cancer Sci. 2017, 108, 1947. [Google Scholar] [CrossRef] [Green Version]
- Park, S.-J.; Nakagawa, T.; Kitamura, H.; Atsumi, T.; Kamon, H.; Sawa, S.-I.; Kamimura, D.; Ueda, N.; Iwakura, Y.; Ishihara, K.; et al. IL-6 regulates in vivo dendritic cell differentiation through STAT3 activation. J. Immunol. 2004, 173, 3844–3854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenzweig, J.M.; Glenn, J.D.; Calabresi, P.A.; Whartenby, K.A. KLF4 modulates expression of IL-6 in dendritic cells via both promoter activation and epigenetic modification. J. Biol. Chem. 2013, 288, 23868–23874. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.E.; Yu, H.N.; Yoon, C.H.; Bae, Y.S. Tumor-mediated down-regulation of MHC class II in DC development is attributable to the epigenetic control of the CIITA type I promoter. Eur. J. Immunol. 2009, 39, 858–868. [Google Scholar] [CrossRef]
- Guillot, A. Liver macrophages: Old dogmas and new insights. Hepatol. Commun. 2019, 3, 730–743. [Google Scholar] [CrossRef] [PubMed]
- Noy, R.; Pollard, J.W. Tumor-associated macrophages: From mechanisms to therapy. Immunity 2014, 41, 49–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiang, X.; Wang, J.; Lu, D.; Xu, X. Targeting tumor-associated macrophages to synergize tumor immunotherapy. Signal Transduct. Target. Ther. 2021, 6, 75. [Google Scholar] [CrossRef] [PubMed]
- Ivashkiv, L.B. Epigenetic regulation of macrophage polarization and function. Trends Immunol. 2013, 34, 216–223. [Google Scholar] [CrossRef] [Green Version]
- Ishii, M.; Wen, H.; Corsa, C.; Liu, T.; Coelho, A.L.; Allen, R.M.; Carson, W.F.; Cavassani, K.A.; Li, X.; Lukacs, N.W.; et al. Epigenetic regulation of the alternatively activated macrophage phenotype. Blood 2009, 114, 3244–3254. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Wang, X.; Liu, D.; Yu, L. Epigenetic regulation of macrophage polarization by DNA methyltransferase 3b. Mol. Endocrinol. 2014, 28, 565–574. [Google Scholar] [CrossRef] [Green Version]
- Tikhanovich, I.; Zhao, J.; Olson, J.; Adams, A.; Taylor, R.; Bridges, B.; Marshall, L.; Roberts, B.; Weinman, S.A. Protein arginine methyltransferase 1 modulates innate immune responses through regulation of peroxisome proliferator-activated receptor γ-dependent macrophage differentiation. J. Biol. Chem. 2017, 292, 6882. [Google Scholar] [CrossRef] [Green Version]
- Mullican, S.E.; Gaddis, C.A.; Alenghat, T.; Nair, M.G.; Giacomin, P.R.; Everett, L.J.; Feng, D.; Steger, D.J.; Schug, J.; Artis, D.; et al. Histone deacetylase 3 is an epigenomic brake in macrophage alternative activation. Genes Dev. 2011, 25, 2480–2488. [Google Scholar] [CrossRef] [Green Version]
- Yang, Q.; Wei, J.; Zhong, L.; Shi, M.; Zhou, P.; Zuo, S.; Wu, K.; Zhu, M.; Huang, X.; Yu, Y.; et al. Cross Talk between Histone Deacetylase 4 and STAT6 in the Transcriptional Regulation of Arginase 1 during Mouse Dendritic Cell Differentiation. Mol. Cell. Biol. 2015, 35, 63. [Google Scholar] [CrossRef] [Green Version]
- Veglia, F.; Sanseviero, E.; Gabrilovich, D.I. Myeloid-derived suppressor cells in the era of increasing myeloid cell diversity. Nat. Rev. Immunol. 2021, 21, 485–498. [Google Scholar] [CrossRef]
- Sido, J.M.; Yang, X.; Nagarkatti, P.S.; Nagarkatti, M. Δ 9-Tetrahydrocannabinol-mediated epigenetic modifications elicit myeloid-derived suppressor cell activation via STAT3/S100A8. J. Leukoc. Biol. 2015, 97, 677–688. [Google Scholar] [CrossRef] [PubMed]
- Vasquez-Dunddel, D.; Pan, F.; Zeng, Q.; Gorbounov, M.; Albesiano, E.; Fu, J.; Blosser, R.L.; Tam, A.J.; Bruno, T.; Zhang, H.; et al. STAT3 regulates arginase-I in myeloid-derived suppressor cells from cancer patients. J. Clin. Investig. 2013, 123, 1580–1589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, F.; Lienlaf, M.; Perez-Villarroel, P.; Wang, H.-W.; Lee, C.; Woan, K.; Woods, D.; Knox, T.; Bergman, J.; Pinilla-Ibarz, J.; et al. Divergent roles of histone deacetylase 6 (HDAC6) and histone deacetylase 11 (HDAC11) on the transcriptional regulation of IL10 in antigen presenting cells. Mol. Immunol. 2014, 60, 44–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sahakian, E.; Powers, J.; Chen, J.; Deng, S.L.; Cheng, F.; Distler, A.; Woods, D.M.; Rock-Klotz, J.; Sodre, A.L.; Youn, J.-I.; et al. Histone deacetylase 11: A novel epigenetic regulator of myeloid derived suppressor cell expansion and function. Mol. Immunol. 2015, 63, 579–585. [Google Scholar] [CrossRef] [Green Version]
- De Almeida Nagata, D.E.; Chiang, E.Y.; Jhunjhunwala, S.; Caplazi, P.; Arumugam, V.; Modrusan, Z.; Chan, E.; Merchant, M.; Jin, L.; Arnott, D.; et al. Regulation of Tumor-Associated Myeloid Cell Activity by CBP/EP300 Bromodomain Modulation of H3K27 Acetylation. Cell Rep. 2019, 27, 269–281. [Google Scholar] [CrossRef] [Green Version]
- Youn, J.-I.; Collazo, M.; Shalova, I.N.; Biswas, S.K.; Gabrilovich, D.I. Characterization of the nature of granulocytic myeloid-derived suppressor cells in tumor-bearing mice. J. Leukoc. Biol. 2012, 91, 167. [Google Scholar] [CrossRef] [Green Version]
- Youn, J.-I.; Kumar, V.; Collazo, M.; Nefedova, Y.; Condamine, T.; Cheng, P.; Villagra, A.; Antonia, S.; McCaffrey, J.C.; Fishman, M.; et al. Epigenetic silencing of retinoblastoma gene regulates pathologic differentiation of myeloid cells in cancer. Nat. Immunol. 2013, 14, 211–220. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Wang, S.; Liu, Y.; Yang, C. Epigenetics in myeloid derived suppressor cells: A sheathed sword towards cancer. Oncotarget 2016, 7, 57452. [Google Scholar] [CrossRef] [Green Version]
- Prager, I.; Watzl, C. Mechanisms of natural killer cell-mediated cellular cytotoxicity. J. Leukoc. Biol. 2019, 105, 1319–1329. [Google Scholar] [CrossRef]
- Schenk, A.; Bloch, W.; Zimmer, P. Natural Killer Cells—An Epigenetic Perspective of Development and Regulation. Int. J. Mol. Sci. 2016, 17, 326. [Google Scholar] [CrossRef] [Green Version]
- Bi, J.; Tian, Z. NK cell dysfunction and checkpoint immunotherapy. Front. Immunol. 2019, 10, 1999. [Google Scholar] [CrossRef] [PubMed]
- Xia, M.; Wang, B.; Wang, Z.; Zhang, X.; Wang, X. Epigenetic Regulation of NK Cell-Mediated Antitumor Immunity. Front Immunol. 2021, 12, 672328. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Sánchez, A.; Raneros, A.B.; Palao, R.C.; Sanz, A.B.; Ortiz, A.; Ortega, F.; Suárez-Álvarez, B.; López-Larrea, C. DNA demethylation and histone H3K9 acetylation determine the active transcription of the NKG2D gene in human CD8+ T and NK cells. Epigenetics 2013, 8, 66–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, N.-H.; Qian, Y.; Wu, C.-S.; Wang, J.-W.; Fang, Y.; Fan, X.-P.; Gao, S.; Fan, Y.-C.; Wang, K. Diagnostic value of NKG2D promoter methylation in hepatitis B virus-associated hepatocellular carcinoma. Biomark. Med. 2019, 13, 1093–1105. [Google Scholar] [CrossRef] [PubMed]
- Bugide, S.; Green, M.R.; Wajapeyee, N. Inhibition of Enhancer of zeste homolog 2 (EZH2) induces natural killer cell-mediated eradication of hepatocellular carcinoma cells. Proc. Natl. Acad. Sci. USA 2018, 115, E3509–E3518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, D.; Zhang, Q.; Liu, Y.; Li, X.; Zhao, K.; Ding, Y.; Li, Z.; Shen, Q.; Wang, C.; Li, N.; et al. H3K4me3 Demethylase Kdm5a Is Required for NK Cell Activation by Associating with p50 to Suppress SOCS1. Cell Rep. 2016, 15, 288–299. [Google Scholar] [CrossRef] [Green Version]
- Luckheeram, R.V.; Zhou, R.; Verma, A.D.; Xia, B. CD4 +T cells: Differentiation and functions. Clin. Dev. Immunol. 2012, 2012, 925135. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.J.; Cantor, H. CD4 T-cell Subsets and Tumor Immunity: The Helpful and the Not-so-Helpful. Cancer Immunol. Res. 2014, 2, 91–98. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Zou, J.; Wang, M.; Ding, X.; Chepelev, I.; Zhou, X.; Zhao, W.; Wei, G.; Cui, J.; Zhao, K.; et al. Critical role of histone demethylase Jmjd3 in the regulation of CD4+ T cell differentiation. Nat. Commun. 2014, 5, 5780. [Google Scholar] [CrossRef] [Green Version]
- Tumes, D.J.; Onodera, A.; Suzuki, A.; Shinoda, K.; Endo, Y.; Iwamura, C.; Hosokawa, H.; Koseki, H.; Tokoyoda, K.; Suzuki, Y.; et al. The polycomb protein Ezh2 regulates differentiation and plasticity of CD4(+) T helper type 1 and type 2 cells. Immunity 2013, 39, 819–832. [Google Scholar] [CrossRef] [Green Version]
- Miyatake, S.; Arai, N.; Arai, K.I. Chromatin remodeling and T helper subset differentiation. IUBMB Life 2000, 49, 473–478. [Google Scholar] [CrossRef] [PubMed]
- Morinobu, A.; Kanno, Y.; O’Shea, J.J. Discrete roles for histone acetylation in human T helper 1 cell-specific gene expression. J. Biol. Chem. 2004, 279, 40640–40646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bam, M.; Chintala, S.; Fetcko, K.; Williamsen, B.C.; Siraj, S.; Liu, S.; Wan, J.; Xuei, X.; Liu, Y.; Leibold, A.T.; et al. Genome wide DNA methylation landscape reveals glioblastoma’s influence on epigenetic changes in tumor infiltrating CD4+ T cells. Oncotarget 2021, 12, 967–981. [Google Scholar] [CrossRef] [PubMed]
- Saito, T.; Nishikawa, H.; Wada, H.; Nagano, Y.; Sugiyama, D.; Atarashi, K.; Maeda, Y.; Hamaguchi, M.; Ohkura, N.; Sato, E.; et al. Two FOXP3+CD4+ T cell subpopulations distinctly control the prognosis of colorectal cancers. Nat. Med. 2016, 22, 679–684. [Google Scholar] [CrossRef]
- Deng, G. Tumor-infiltrating regulatory T cells: Origins and features. Am. J. Clin. Exp. Immunol. 2018, 7, 81. [Google Scholar]
- Klarquist, J.; Tobin, K.; Oskuei, P.F.; Henning, S.W.; Fernandez, M.F.; Dellacecca, E.R.; Navarro, F.C.; Eby, J.M.; Chatterjee, S.; Mehrotra, S.; et al. Ccl22 Diverts T Regulatory Cells and Controls the Growth of Melanoma. Cancer Res. 2016, 76, 6230–6240. [Google Scholar] [CrossRef] [Green Version]
- Halvorsen, E.C.; Hamilton, M.J.; Young, A.; Wadsworth, B.J.; LePard, N.E.; Lee, H.N.; Firmino, N.; Collier, J.L.; Bennewith, K.L. Maraviroc decreases CCL8-mediated migration of CCR5(+) regulatory T cells and reduces metastatic tumor growth in the lungs. Oncoimmunology 2016, 5, e1150398. [Google Scholar] [CrossRef] [Green Version]
- Waight, J.D.; Takai, S.; Marelli, B.; Qin, G.; Hance, K.W.; Zhang, D.; Tighe, R.; Lan, Y.; Lo, K.-M.; Sabzevari, H.; et al. Cutting edge: Epigenetic regulation of Foxp3 defines a stable population of CD4+ regulatory T cells in tumors from mice and humans. J. Immunol. 2015, 194, 878–882. [Google Scholar] [CrossRef] [Green Version]
- DuPage, M.; Chopra, G.; Quiros, J.; Rosenthal, W.L.; Morar, M.M.; Holohan, D.; Zhang, R.; Turka, L.; Marson, A.; Bluestone, J.A. The chromatin-modifying enzyme Ezh2 is critical for the maintenance of regulatory T cell identity after activation. Immunity 2015, 42, 227–238. [Google Scholar] [CrossRef] [Green Version]
- Marson, A.; Kretschmer, K.; Frampton, G.M.; Jacobsen, E.S.; Polansky, J.K.; MacIsaac, K.D.; Levine, S.S.; Fraenkel, E.; Von Boehmer, H.; Young, R.A. Foxp3 occupancy and regulation of key target genes during T-cell stimulation. Nature 2007, 445, 931–935. [Google Scholar] [CrossRef] [Green Version]
- Morikawa, H.; Ohkura, N.; Vandenbon, A.; Itoh, M.; Nagao-Sato, S.; Kawaji, H. Differential roles of epigenetic changes and Foxp3 expression in regulatory T cell-specific transcriptional regulation. Proc. Natl. Acad. Sci. USA 2014, 111, 5289–5294. [Google Scholar] [CrossRef] [Green Version]
- Arvey, A.; van der Veeken, J.; Samstein, R.M.; Feng, Y.; Stamatoyannopoulos, J.A.; Rudensky, A.Y. Inflammation-induced repression of chromatin bound by the transcription factor Foxp3 in regulatory T cells. Nat. Immunol. 2014, 15, 580–587. [Google Scholar] [CrossRef]
- Wang, D.; Quiros, J.; Mahuron, K.; Pai, C.-C.; Ranzani, V.; Young, A.; Silveria, S.; Harwin, T.; Abnousian, A.; Pagani, M.; et al. Targeting EZH2 Reprograms Intratumoral Regulatory T Cells to Enhance Cancer Immunity. Cell Rep. 2018, 23, 3262–3274. [Google Scholar] [CrossRef] [PubMed]
- Henning, A.; Roychoudhuri, R.; Restifo, N.P. Epigenetic control of CD8+ T cell differentiation. Nat. Rev. Immunol. 2018, 18, 340–356. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Li, F.; Ping, Y.; Wang, L.; Chen, X.; Wang, D.; Cao, L.; Zhao, S.; Li, B.; Kalinski, P.; et al. Local production of the chemokines CCL5 and CXCL10 attracts CD8+ T lymphocytes into esophageal squamous cell carcinoma. Oncotarget 2015, 6, 24978–24989. [Google Scholar] [CrossRef] [Green Version]
- Slaney, C.Y.; Kershaw, M.H.; Darcy, P.K. Trafficking of T Cells into Tumors. Cancer Res 2014, 74, 7168–7174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maimela, N.R.; Liu, S.; Zhang, Y. Fates of CD8+ T cells in Tumor Microenvironment. Comput. Struct. Biotechnol. J. 2019, 17, 1–13. [Google Scholar] [CrossRef]
- Sanmamed, M.F.; Nie, X.; Desai, S.S.; Villaroel-Espindola, F.; Badri, T.; Zhao, D. A burned-out cd8+ t-cell subset ex-pands in the tumor microenvironment and curbs cancer immunotherapy. Cancer Discov. 2021, 11, 1700–1715. [Google Scholar] [CrossRef] [PubMed]
- Peng, D.; Kryczek, I.; Nagarsheth, N.; Zhao, L.; Wei, S.; Wang, W.; Sun, Y.; Zhao, E.; Vatan, L.; Szeliga, W.; et al. Epigenetic silencing of TH1-type chemokines shapes tumour immunity and immunotherapy. Nature 2015, 527, 249–253. [Google Scholar] [CrossRef] [Green Version]
- Yang, R.; Cheng, S.-J.; Luo, N.; Gao, R.; Yu, K.; Kang, B.; Wang, L.; Zhang, Q.; Fang, Q.; Zhang, L.; et al. Distinct epigenetic features of tumor-reactive CD8+ T cells in colorectal cancer patients revealed by genome-wide DNA methylation analysis. Genome Biol. 2019, 21, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Crompton, J.G.; Narayanan, M.; Cuddapah, S.; Roychoudhuri, R.; Ji, Y.; Yang, W.; Patel, S.J.; Sukumar, M.; Palmer, D.; Peng, W.; et al. Lineage relationship of CD8+ T cell subsets is revealed by progressive changes in the epigenetic landscape. Cell. Mol. Immunol. 2015, 13, 502–513. [Google Scholar] [CrossRef] [PubMed]
- Bian, Y.; Li, W.; Kremer, D.M.; Sajjakulnukit, P.; Li, S.; Crespo, J.; Nwosu, Z.C.; Zhang, L.; Czerwonka, A.; Pawłowska, A.; et al. Cancer SLC43A2 alters T cell methionine metabolism and histone methylation. Nature 2020, 585, 277–282. [Google Scholar] [CrossRef] [PubMed]
- Downs-Canner, S.M.; Meier, J.; Vincent, B.G.; Serody, J.S. B Cell Function in the Tumor Microenvironment. Annu. Rev. Immunol. 2022, 40, 169–193. [Google Scholar] [CrossRef]
- Mondello, P.; Ansell, S.M.; Nowakowski, G.S. Immune Epigenetic Crosstalk Between Malignant B Cells and the Tumor Microenvironment in B Cell Lymphoma. Front. Genet. 2022, 13, 826594. [Google Scholar] [CrossRef] [PubMed]
- Chiappinelli, K.B.; Strissel, P.L.; Desrichard, A.; Li, H.; Henke, C.; Akman, B.; Hein, A.; Rote, N.S.; Cope, L.M.; Snyder, A.; et al. Inhibiting DNA Methylation Causes an Interferon Response in Cancer via dsRNA Including Endogenous Retroviruses. Cell 2015, 162, 974–986. [Google Scholar] [CrossRef] [Green Version]
- Costantini, B.; Kordasti, S.Y.; Kulasekararaj, A.G.; Jiang, J.; Seidl, T.; Abellan, P.P.; Mohamedali, A.; Thomas, N.S.B.; Farzaneh, F.; Mufti, G.J. The effects of 5-azacytidine on the function and number of regulatory T cells and T-effectors in myelodysplastic syndrome. Haematologica 2012, 98, 1196–1205. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Yao, Y.; Shen, Q.; Li, G.; Hu, L.; Zhang, X. Demethylating agent decitabine disrupts tumor-induced immune tolerance by depleting myeloid-derived suppressor cells. J. Cancer Res. Clin. Oncol. 2017, 143, 1371–1380. [Google Scholar] [CrossRef]
- Cany, J.; Roeven, M.W.H.; Evert, J.S.H.-V.; Hobo, W.; Maas, F.; Fernandez, R.F.; Blijlevens, N.M.A.; Van Der Velden, W.J.; Huls, G.; Jansen, J.H.; et al. Decitabine enhances targeting of AML cells by CD34+ progenitor-derived NK cells in NOD/SCID/IL2Rgnull mice. Blood 2018, 131, 202–214. [Google Scholar] [CrossRef]
- Ghoneim, H.E.; Fan, Y.; Moustaki, A.; Abdelsamed, H.A.; Dash, P.; Dogra, P.; Carter, R.; Awad, W.; Neale, G.; Thomas, P.G.; et al. De Novo Epigenetic Programs Inhibit PD-1 Blockade-Mediated T Cell Rejuvenation. Cell 2017, 170, 142–157.e19. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Amoozgar, Z.; Huang, J.; Saleh, M.H.; Xing, D.; Orsulic, S.; Goldberg, M.S. Decitabine Enhances Lymphocyte Migration and Function and Synergizes with CTLA-4 Blockade in a Murine Ovarian Cancer Model. Cancer Immunol. Res. 2015, 3, 1030–1041. [Google Scholar] [CrossRef] [Green Version]
- Shen, L.; Ciesielski, M.; Ramakrishnan, S.; Miles, K.M.; Ellis, L.; Sotomayor, P.; Shrikant, P.; Fenstermaker, R.; Pili, R. Class I Histone Deacetylase Inhibitor Entinostat Suppresses Regulatory T Cells and Enhances Immunotherapies in Renal and Prostate Cancer Models. PLoS ONE 2012, 7, e30815. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Cherukuri, P.; Luo, J. Activation of Stat3 Sequence-specific DNA Binding and Transcription by p300/CREB-binding Protein-mediated Acetylation. J. Biol. Chem. 2005, 280, 11528–11534. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.; Son, W.-C.; Lee, Y.-S.; Youn, E.; Kang, C.-D.; Park, Y.-S.; Bae, J. Differential Effects of Histone Deacetylases on the Expression of NKG2D Ligands and NK Cell-Mediated Anticancer Immunity in Lung Cancer Cells. Molecules 2021, 26, 3952. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Su, X.; Liu, R.; Pan, Y.; Fang, J.; Cao, L.; Feng, C.; Shang, Q.; Chen, Y.; Shao, C.; et al. HDAC inhibition potentiates anti-tumor activity of macrophages and enhances anti-PD-L1-mediated tumor suppression. Oncogene 2021, 40, 1836–1850. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.-D.; Park, S.-M.; Ha, H.C.; Lee, A.R.; Won, H.; Cha, H.; Cho, S.; Cho, J.M. HDAC Inhibitor, CG-745, Enhances the Anti-Cancer Effect of Anti-PD-1 Immune Checkpoint Inhibitor by Modulation of the Immune Microenvironment. J. Cancer 2020, 11, 4059–4072. [Google Scholar] [CrossRef] [Green Version]
- Goswami, S.; Apostolou, I.; Zhang, J.; Skepner, J.; Anandhan, S.; Zhang, X.; Xiong, L.; Trojer, P.; Aparicio, A.; Subudhi, S.K.; et al. Modulation of EZH2 expression in T cells improves efficacy of anti–CTLA-4 therapy. J. Clin. Investig. 2018, 128, 3813–3818. [Google Scholar] [CrossRef]
- Zhou, L.; Mudianto, T.; Ma, X.; Riley, R.; Uppaluri, R. Targeting EZH2 enhances antigen presentation, antitumor immunity, and circumvents anti–PD-1 resistance in head and neck cancer. Clin. Cancer Res. 2020, 26, 290–300. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Y.; Yuan, Y.; Chen, M.; Li, S.; Bai, J.; Zhang, Y.; Sun, Y.; Wang, G.; Xu, H.; Wang, Z.; et al. PRMT5 disruption drives antitumor immunity in cervical cancer by reprogramming T cell-mediated response and regulating PD-L1 expression. Theranostics 2021, 11, 9162–9176. [Google Scholar] [CrossRef]
- Hu, R.; Zhou, B.; Chen, Z.; Chen, S.; Chen, N.; Shen, L. PRMT5 Inhibition Promotes PD-L1 Expression and Immuno-Resistance in Lung Cancer. Front. Immunol. 2021, 12, 722188. [Google Scholar] [CrossRef]
- Hogg, S.J.; Wellinger, L.; Rohle, D.; Johnstone, R.W. Abstract 4485: Enhancing antitumor immune responses with clinical BET bromodomain inhibitor RG6146. Cancer Res. 2019, 79, 4485. [Google Scholar] [CrossRef]
- Wrangle, J.; Wang, W.; Koch, A.; Easwaran, H.; Mohammad, H.P.; Pan, X.; Vendetti, F.; VanCriekinge, W.; DeMeyer, T.; Du, Z.; et al. Alterations of immune response of non-small cell lung cancer with Azacytidine. Oncotarget 2013, 4, 2067–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juergens, R.A.; Wrangle, J.; Vendetti, F.P.; Murphy, S.C.; Zhao, M.; Coleman, B.; Sebree, R.; Rodgers, K.; Hooker, C.M.; Franco, N.; et al. Combination Epigenetic Therapy Has Efficacy in Patients with Refractory Advanced Non–Small Cell Lung Cancer. Cancer Discov. 2011, 1, 598–607. [Google Scholar] [CrossRef] [PubMed]
- Goswami, M.; Gui, G.; Dillon, L.W.; E Lindblad, K.; Thompson, J.; Valdez, J.; Kim, D.-Y.; Ghannam, J.Y.; A Oetjen, K.; Destefano, C.B.; et al. Pembrolizumab and decitabine for refractory or relapsed acute myeloid leukemia. J. Immunother. Cancer 2022, 10, e003392. [Google Scholar] [CrossRef] [PubMed]
- Gros, C.; Fahy, J.; Halby, L.; Dufau, I.; Erdmann, A.; Gregoire, J.-M.; Ausseil, F.; Vispé, S.; Arimondo, P.B. DNA methylation inhibitors in cancer: Recent and future approaches. Biochimie 2012, 94, 2280–2296. [Google Scholar] [CrossRef]
- Brueckner, B.; Boy, R.G.; Siedlecki, P.; Musch, T.; Kliem, H.C.; Zielenkiewicz, P.; Suhai, S.; Wiessler, M.; Lyko, F. Epigenetic Reactivation of Tumor Suppressor Genes by a Novel Small-Molecule Inhibitor of Human DNA Methyltransferases. Cancer Res. 2005, 65, 6305–6311. [Google Scholar] [CrossRef] [Green Version]
- Ghoshal, K.; Datta, J.; Majumder, S.; Bai, S.; Kutay, H.; Motiwala, T. 5-Aza-Deoxycytidine Induces Selective Degrada-tion of DNA Methyltransferase 1 by a Proteasomal Pathway That Requires the KEN Box, Bromo-Adjacent Homology Do-main, and Nuclear Localization Signal. Mol. Cell. Biol. 2005, 25, 4727–4741. [Google Scholar] [CrossRef] [Green Version]
- Kondo, Y. Epigenetic Cross-Talk between DNA Methylation and Histone Modifications in Human Cancers. Yonsei Med. J. 2009, 50, 455–463. [Google Scholar] [CrossRef] [Green Version]
- Panda, A.; De Cubas, A.A.; Stein, M.; Riedlinger, G.; Kra, J.; Mayer, T.; Smith, C.C.; Vincent, B.G.; Serody, J.S.; Beckermann, K.E.; et al. Endogenous retrovirus expression is associated with response to immune checkpoint blockade in clear cell renal cell carcinoma. JCI Insight 2018, 3, e121522. [Google Scholar] [CrossRef]
- Krishnadas, D.K.; Bao, L.; Bai, F.; Chencheri, S.C.; Lucas, K. Decitabine facilitates immune recognition of sarcoma cells by upregulating CT antigens, MHC molecules, and ICAM-1. Tumor Biol. 2014, 35, 5753–5762. [Google Scholar] [CrossRef]
- Siebenkäs, C.; Chiappinelli, K.B.; Guzzetta, A.A.; Sharma, A.; Jeschke, J.; Vatapalli, R.; Baylin, S.B.; Ahuja, N. Inhibiting DNA methylation activates cancer testis antigens and expression of the antigen processing and presentation machinery in colon and ovarian cancer cells. PLoS ONE 2017, 12, e0179501. [Google Scholar] [CrossRef]
- Dan, H.; Zhang, S.; Zhou, Y.; Guan, Q. DNA Methyltransferase Inhibitors: Catalysts For Antitumour Immune Responses. Onco Targets Ther. 2019, ume 12, 10903–10916. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Zhang, X.W.Y. TET-mediated active DNA demethylation: Mechanism, function and beyond. Nat. Rev. Genet. 2017, 18, 517–534. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S. Tet2 at the interface between cancer and immunity. Commun. Biol. 2020, 3, 667. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.-P.; Lv, L.; Liu, Y.; Smith, M.D.; Li, W.-C.; Tan, X.-M. Tumor suppressor TET2 promotes cancer immunity and immunotherapy efficacy. J. Clin. Investig. 2019, 129, 4316–4331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Feng, J.; Wu, F.; Cai, J.; Zhang, X.; Wang, H. TET2 promotes anti-tumor immunity by governing G-MDSCs and CD8(+) T-cell numbers. EMBO Rep. 2020, 21, e49425. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.; Li, J.; Li, J.; Fang, S.; Zhang, J.; Vo, A.T.T.; Han, W.; Zeng, H.; Isgandarova, S.; Martinez-Moczygemba, M.; et al. Tet2 Inactivation Enhances the Antitumor Activity of Tumor-Infiltrating Lymphocytes. Cancer Res. 2021, 81, 1965–1976. [Google Scholar] [CrossRef] [PubMed]
- West, A.C.; Johnstone, R.W. New and emerging HDAC inhibitors for cancer treatment. J. Clin. Investig. 2014, 124, 30–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Özdağ, H.; E Teschendorff, A.; Ahmed, A.A.; Hyland, S.J.; Blenkiron, C.; Bobrow, L.; Veerakumarasivam, A.; Burtt, G.; Subkhankulova, T.; Arends, M.J.; et al. Differential expression of selected histone modifier genes in human solid cancers. BMC Genom. 2006, 7, 90. [Google Scholar] [CrossRef] [Green Version]
- Tzogani, K.; van Hennik, P.; Walsh, I.; De Graeff, P.; Folin, A.; Sjöberg, J.; Salmonson, T.; Bergh, J.; Laane, E.; Ludwig, H.; et al. EMA Review of Panobinostat (Farydak) for the Treatment of Adult Patients with Relapsed and/or Refractory Multiple Myeloma. Oncologist 2019, 23, 631–636. [Google Scholar] [CrossRef] [Green Version]
- Licciardi, P.V.; Ververis, K.; Hiong, A.; Karagiannis, T.C. Histone deacetylase inhibitors (HDACIs): Multitargeted anticancer agents. Biol. Targets Ther. 2013, 7, 47–60. [Google Scholar] [CrossRef] [Green Version]
- Setiadi, A.F.; Omilusik, K.; David, M.D.; Seipp, R.P.; Hartikainen, J.; Gopaul, R.; Choi, K.B.; Jefferies, W.A. Epigenetic Enhancement of Antigen Processing and Presentation Promotes Immune Recognition of Tumors. Cancer Res. 2008, 68, 9601–9607. [Google Scholar] [CrossRef] [Green Version]
- McCaw, T.R.; Randall, T.D.; Forero, A.; Buchsbaum, D.J. Modulation of antitumor immunity with histone deacetylase inhibitors. Immunotherapy 2017, 9, 1359–1372. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Peterson, L.M.; Li, X. Trending topics of SIRT1 in tumorigenicity. Biochim. Biophys. Acta BBA Gen. Subj. 2021, 1865, 129952. [Google Scholar] [CrossRef]
- Limagne, E.; Thibaudin, M.; Euvrard, R.; Berger, H.; Chalons, P.; Végan, F.; Humblin, E.; Boidot, R.; Rébé, C.; Derangère, V.; et al. Sirtuin-1 Activation Controls Tumor Growth by Impeding Th17 Differentiation via STAT3 Deacetylation. Cell Rep. 2017, 19, 746–759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, F.; Jiang, J.; Zong, C.; Yang, X.; Gao, L.; Meng, Y.; Li, R.; Zhao, Q.; Han, Z.; Wei, L. Sirt1-Overexpressing Mesenchymal Stem Cells Drive the Anti-tumor Effect through Their Pro-inflammatory Capacity. Mol. Ther. 2020, 28, 874–888. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Yang, Y.; Li, C. SIRT1 inhibits hepatocellular carcinoma metastasis by promoting M1 macrophage polarization via NF-κB pathway. Onco Targets Ther. 2019, ume 12, 2519–2529. [Google Scholar] [CrossRef] [Green Version]
- Rugo, H.S.; Jacobs, I.; Sharma, S.; Scappaticci, F.; Paul, T.A.; Jensen-Pergakes, K.; Malouf, G.G. The Promise for Histone Methyltransferase Inhibitors for Epigenetic Therapy in Clinical Oncology: A Narrative Review. Adv. Ther. 2020, 37, 3059–3082. [Google Scholar] [CrossRef]
- Christofides, A.; Karantanos, T.; Bardhan, K.; Boussiotis, V.A. Epigenetic regulation of cancer biology and anti-tumor immunity by EZH2. Oncotarget 2016, 7, 85624–85640. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.H.; Roberts, K.H.K.C.W.M. Targeting EZH2 in cancer. Nat. Med. 2016, 22, 128–134. [Google Scholar] [CrossRef] [Green Version]
- Kang, N.; Eccleston, M.; Clermont, P.-L.; Latarani, M.; Male, D.K.; Wang, Y.; Crea, F. EZH2 inhibition: A promising strategy to prevent cancer immune editing. Epigenomics 2020, 12, 1457–1476. [Google Scholar] [CrossRef]
- Kim, H.; Ronai, Z.A. PRMT5 function and targeting in cancer. Cell Stress. 2020, 4, 199–215. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.W.; Cho, Y.; Bae, G.-U.; Kim, S.-N.; Kim, Y.K. Protein arginine methyltransferases: Promising targets for cancer therapy. Exp. Mol. Med. 2021, 53, 788–808. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Salvia, M.; Esteller, M. Bromodomain inhibitors and cancer therapy: From structures to applications. Epigenetics 2017, 12, 323–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shorstova, T.; Foulkes, W.D.; Witcher, M. Achieving clinical success with BET inhibitors as anti-cancer agents. Br. J. Cancer 2021, 124, 1478–1490. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Bengsch, F.; Svoronos, N.; Rutkowski, M.R.; Bitler, B.G.; Allegrezza, M.J.; Yokoyama, Y.; Kossenkov, A.V.; Bradner, J.E.; Conejo-Garcia, J.R.; et al. BET Bromodomain Inhibition Promotes Anti-tumor Immunity by Suppressing PD-L1 Expression. Cell Rep. 2016, 16, 2829–2837. [Google Scholar] [CrossRef] [Green Version]
- Mao, W.; Ghasemzadeh, A.; Freeman, Z.; Obradovic, A.; Chaimowitz, M.G.; Nirschl, T.R.; McKiernan, E.; Yegnasubramanian, S.; Drake, C.G. Immunogenicity of prostate cancer is augmented by BET bromodomain inhibition. J. Immunother. Cancer 2019, 7, 277. [Google Scholar] [CrossRef]
- Adeegbe, D.O.; Liu, Y.; Lizotte, P.H.; Kamihara, Y.; Aref, A.R.; Almonte, C.; Dries, R.; Li, Y.; Liu, S.; Wang, X.; et al. Synergistic Immunostimulatory Effects and Therapeutic Benefit of Combined Histone Deacetylase and Bromodomain Inhibition in Non–Small Cell Lung Cancer. Cancer Discov. 2017, 7, 852–867. [Google Scholar] [CrossRef] [Green Version]
- Sarnik, J.; Popławski, T.; Tokarz, P. BET Proteins as Attractive Targets for Cancer Therapeutics. Int. J. Mol. Sci. 2021, 22, 11102. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Epigenetic Modulator | Clinical Status | Studies Targeting Antitumor Immunity | Reference | ||
|---|---|---|---|---|---|
| Cancer Model | Type of Immune Cell Modulated | Effects | |||
| DNMTi | |||||
| Ovarian cancer cell line | Activate ERVs expression Upregulate immune genes (IFNβ, IRF7, STAT3) | ||||
| Treatment of acute myeloid leukemia and myelodysplastic syndrome | B16-F10 mouse melanoma model | Type 1 Interferon signaling pathway | Enhance immune checkpoint inhibitors (anti-CTLA4) antitumor effects | [95] | |
| Azacytidine | Patient with myelodysplastic syndrome | Treg cell | Increased Foxp3 expression but reduced Treg cell function, producing a large amount of IL-17 | [96] | |
| Leukemia mice model | MDSC | Depletion of MDSC | [97] | ||
| Decitabine | Acute myeloid leukemia model | NK cells | Modulation of NK cells phenotype, activity, and trafficking through upregulation of activating receptors (NKG2D, DNAM-1), inflammatory cytokines (IFN-γ, TNF-α), and perforin | [98] | |
| Treatment of Myelodysplastic syndrome | Ovarian cancer model | CD8+ and helper CD4+ T cells | Upregulation of chemokines (CXCL9 and CXCL10) expression and increase T cell infiltration in TME | [89] | |
| Tramp-C2 tumor model | CD8+ T cell | Inhibit exhaustion-specific gene expression responsible for CD8+ cell exhaustion. In combination with ICB, improve ICB efficacy that restricts ICB-responsiveness | [99] | ||
| Syngeneic murine ovarian cancer model | Nk cells, CD8+ T cell | Enhance T cell recruitment and function, and improve the efficacy of anti-CTLA4) | [100] | ||
| HDACi | |||||
| Entinostat | Phase 2 studies in breast and NSCLC cancer showed promising results | Renal and prostate cancer model | Treg cell | Reduced Treg cell function, downregulation of Foxp3 expression due to STAT3 acetylation | [101,102] |
| Romidepsin | Treatment of cutaneous T-cell lymphoma | Lung cancer cell lines: NCI-H23 and A549 | NK cell | Increase expression of NKG2DL, improve NK cell-mediated antitumor immunity | [103] |
| Trichostatin | Phase 1 study in subjects with relapsed or refractory hematologic malignancies | Syngeneic mouse model | Macrophage (M2) & MDSC | Modulation of M2 phenotype (due to decreased mRNA expression levels of the M2 markers) Increased number of M1 phenotypes within TME. Reduced infiltration of MDSC within TME | [104] |
| CG-745 | Phase II clinical trial pancreatic cancer | Syngeneic mouse model | Macrophage (M2), T cell & MDSC | Suppress M2 macrophage polarization. Effective T cell activity (due to an increase in expression of IL-2 and IFN-γ. Depletion of immunosuppressive cells (MDSC, Treg) | [105] |
| HMTi | |||||
| CPI-1205 | Phase Ib/II in combination with enzalutamide (E) or abiraterone/prednisone (A/P) in patients with metastatic castration-resistant prostate cancer (mCRPC) | Murine colorectal (M38) tumor model | Treg cell | Reduced Foxp3 expression, Treg cell depletion. Enhanced CD8+ activity. Improve the efficacy of anti-CTLA-4 therapy | [83,106] |
| GSK126 & EPZ6438 (Tazemetostat) | Phase 1 clinical trial in subject with B cell lymphoma, treatment of metastatic or locally advanced epithelioid sarcoma | Human and mouse HNSCC cell lines CAL-33, CAL-27, SCC-9, and SCC-25 | Antigen presentation | Upregulation of MHC class I expression | [107] |
| PRMT-5i | |||||
| EPZ015666 | Potent, selective and orally bioavailable PRMT-5 inhibitor | Cervical cancer mice model | CD4+ and CD8+ T cell | Increased the cytokine secretion from CD4+ and CD8+ T cells, such as IFN-γ, TNF-α and granzyme B | [108] |
| GSK591 | Chemical probe of PRMT-5 inhibitor | Human and mouse lung cancer cell lines | Tumor-infiltrating T cells | Combination with ani-PD-L1 therapy increases the number and function of tumor-infiltrating T cells | [109] |
| BETi | |||||
| RG6146 | Early phase clinical trials for the treatment of hematological and solid malignancies | Syngeneic colon and breast tumor model | CD8+ T-cell | Enhance anti-tumor CD8+ T-cell responses | [110] |
| JQ1 | Tested in clinical trials for a variety of cancers such as NUT midline carcinoma | Syngeneic mouse model | Expression of immune checkpoint | Decreased expression of PD-L1 on tumor cells, tumor-associated dendritic cells, and macrophages | [111] |
| JQ1 + anti- CTLA4 | Prostate cancer model | Antigen processing and immune checkpoint molecules | Increased MHC I expression, decreased PD-L1 expression, increased CD8 infiltration | [112] | |
| JQ1 + HDACi | Murine lung cancer model | Tregg cell | Suppressing Tregg cell function | [113] | |
| Epigenetic Modulator | Immunotherapy Agent | Condition | Clinical Trial ID | Phase | Status |
|---|---|---|---|---|---|
| DNMT inhibitors | |||||
| Azacytidine | Visilizumab (Anti-CD3 mAb) | Relapsed and refractory acute myeloid leukemia | NCT04722952 | III | Recruiting |
| Pembrolizumab (Anti-PD1 mAb) | Myelodysplastic Syndrome | NCT03094637 | II | Active | |
| Nivolumab (Anti-PD1 mAb) | Squamous Cell Carcinoma of Head and Neck | NCT05317000 | I | Not yet recruiting | |
| Pembrolizumab (Anti-PD1 mAb)Epacadostat (IDO1 inhibitor) | Solid Tumors Advanced Malignancies Metastatic Cancer | NCT02959437 | I/II | Terminated | |
| Decitabine | Nivolumab (Anti-PD1 mAb) | Lung Cancer, Non-small Cell Lung Cancer | NCT02664181 | II | Active |
| Ipilimumab (Anti-CTLA4 mAb) | Relapsed or Refractory Myelodysplastic Syndrome Acute Myeloid Leukemia | NCT02890329 | I | Active | |
| Pembrolizumab (Anti-PD1 mAb) | Refractory or Relapsed Acute Myeloid Leukemia | NCT02996474 | I/II | Completed | |
| Guadecitabine | Atezolizumab (Anti-PD1 mAb), CDX-1401 (cancer vaccine) | Recurrent ovarian, Fallopian tube, or primary peritoneal cancer | NCT03206047 | I/II | Active |
| Ipilimumab (Anti-CTLA4 mAb) | Metastatic Melanoma | NCT02608437 | I | Unknown | |
| HDAC inhibitors | |||||
| Entinostat | Pembrolizumab (Anti-PD1 mAb) | Myelodysplastic Syndrome | NCT02936752 | I | Active |
| Ipilimumab (Anti-CTLA4 mAb), Nivolumab (Anti-PD1 mAb) | Breast cancer | NCT02453620 | I | Active | |
| vorinostat | Pembrolizumab (Anti-PD1 mAb) | Squamous Cell Lung Cancer, Vulvar Cancer, Penile Cancer, Cervix Cancer, Head, and Neck Squamous Cell Carcinoma, Anal Cancer | NCT04357873 | II | Active |
| Mocetinostat | Durvalumab (IgG1κ mAb) | Advanced or Metastatic Solid Tumors and Non-Small Cell Lung Cancer | NCT02805660 | I/II | Terminated |
| Romidepsin | Nivolumab (Anti-PD1 mAb) | Triple-Negative Breast Cancer | NCT02393794 | I/II | Active |
| DNMTi + HDACi | |||||
| Guadecitabine, Mocetinostat | Pembrolizumab (Anti-PD1 mAb) | Lung Cancer | NCT03220477 | I | Active |
| EZH2 inhibitors | |||||
| Tazemetostat | Durvalumab (IgG1κ mAb) | Advanced Solid Tumor Advanced Colorectal Carcinoma Advanced Soft-tissue Sarcoma Advanced Pancreatic Adenocarcinoma Adult Solid Tumor | NCT04705818 | II | Recruiting |
| Pembrolizumab (Anti-PD1 mAb) | Advanced Urothelial Carcinoma | NCT03854474 | I/II | Recruiting | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Parab, A.; Kumar Bhatt, L.; Omri, A. Targeting Epigenetic Mechanisms: A Boon for Cancer Immunotherapy. Biomedicines 2023, 11, 169. https://doi.org/10.3390/biomedicines11010169
Parab A, Kumar Bhatt L, Omri A. Targeting Epigenetic Mechanisms: A Boon for Cancer Immunotherapy. Biomedicines. 2023; 11(1):169. https://doi.org/10.3390/biomedicines11010169
Chicago/Turabian StyleParab, Asmita, Lokesh Kumar Bhatt, and Abdelwahab Omri. 2023. "Targeting Epigenetic Mechanisms: A Boon for Cancer Immunotherapy" Biomedicines 11, no. 1: 169. https://doi.org/10.3390/biomedicines11010169
APA StyleParab, A., Kumar Bhatt, L., & Omri, A. (2023). Targeting Epigenetic Mechanisms: A Boon for Cancer Immunotherapy. Biomedicines, 11(1), 169. https://doi.org/10.3390/biomedicines11010169