
The Biology of Lysosomes: From Order to Disorder


 and
and
Abstract
:1. Introduction
2. The Ordered Lysosome
2.1. Lysosome Biogenesis
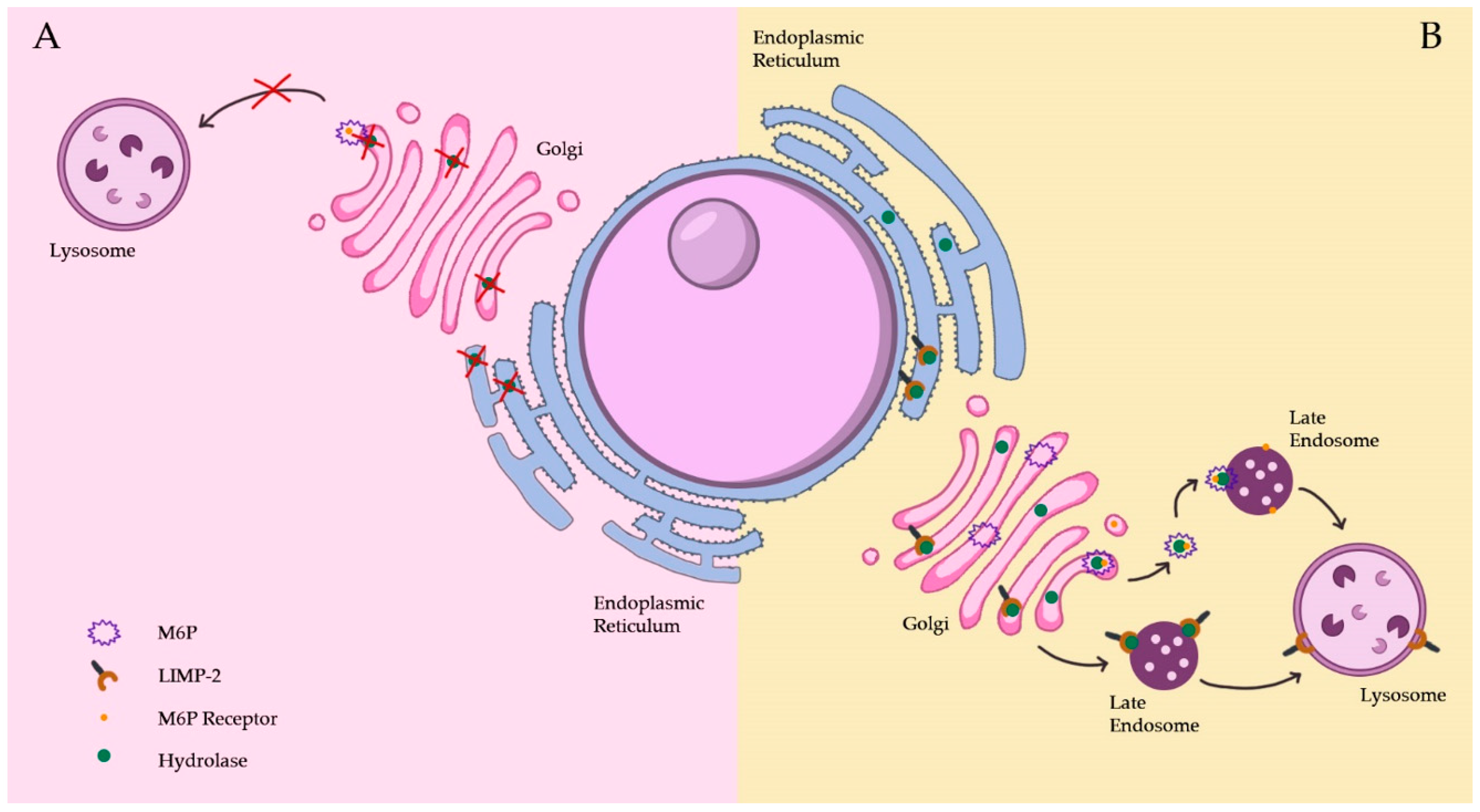
2.1.1. The Biosynthetic Pathway
2.1.2. The Endocytic Pathway
2.1.3. Lysosomal Membrane Protein Pathways
2.2. Lysosomal Functions
2.3. Pathogenesis
2.3.1. The Lysosome as a Signalling Hub
2.3.2. Lysosomal Nutrient Sensing and mTORC1 Signalling
2.3.3. Lysosomal Calcium Signalling
2.3.4. Lysosomal Adaptation
2.3.5. TFEB and the CLEAR Gene Network
2.3.6. Regulation of TFEB by Environmental Signals
3. From Order to Disorder
3.1. Lysosome Dysfunction in Disease
Lysosomal Storage Disorders
3.2. Lysosomal Hydrolase Deficiency
3.3. Integral Membrane Protein Deficiency
3.4. Lipid and Ion Transporters Deficiency
3.5. Enzyme Modifiers and Activator Deficiency

{kind=link}
{kind=link}
| Lysosomal Disorder | Genetic Mutations | Primary Defects | Cellular Consequences |
|---|---|---|---|
| Gaucher Disease (Autosomal recessive) | GBA1 | GCase impaired hydrolytic activity or premature degradation [134] | GlcCer accumulation in macrophages, which become Gaucher cells [135] and infiltrate several organs; GlcCer burden is associated with tissue inflammation processes [107]. |
| LIMP-2 | Impaired GCase transport [43,103] | ||
| PSAP | Saposin C deficiency and impaired GCase function [9,104,105] | ||
| Danon Disease (Dominant X-linked) | LAMP-2 | LAMP-2A dysfunction [119] | Accumulation of immature autophagic vacuoles [136]. Glycogen accumulation in autophagic vacuoles [137]. Block in autophagy leads to impaired autophagosome–lysosome fusion and/or inefficient lysosome biogenesis and maturation [138]. |
| Mucolipidosis IV (Autosomal recessive) | MCOLN1 | Mucolipin-1 absence [65] | Altered endocytic pathway
|
Altered mTORC1/TFEB signalling axis
| |||
| GM2 Gangliosidosis AB Variant (Autosomal recessive) | GM2A | GM2 activator deficiency, lack of formation of beta-hexosaminidase A/ GM2 complex [9,130] | Deficient GM2 removal from membrane, deficient degradation leads to the intralysosomal accumulation of GM2 and related glycolipids. |
4. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AP-# | Adaptor Protein # |
| ARF | ADP ribosylation factor |
| β-HEXA | β-hexosaminidase A |
| Ca2+ | Calcium |
| CLEAR | Coordinated lysosomal expression and regulation element |
| DD | Danon Disease |
| EE | Early endosomes |
| EL | Endolysosomes |
| ER | Endoplasmic reticulum |
| FGE | Formylglycine-generating enzyme |
| FLCN | Folliculin |
| FNIP | Folliculin-interacting proteins |
| GATOR1 | GTPase-activating protein (GAP) for RAG A |
| GCase | Glucocerebrosidase |
| GD | Gaucher Disease |
| GGA | Golgi-localized, γ ear–containing, ARF–binding protein |
| GlcCer | Glucosylceramide |
| GM2 | Ganglioside monosialic 2 |
| GM2A | GM2 ganglioside activator protein |
| GTPase | GTP (guanosine triphosphate)-binding proteins |
| HOPS | Homotypic fusion and vacuole protein sorting |
| ILVs | Intraluminal vesicles |
| LAMP# | Lysosomal-associated membrane protein # |
| LD | Lysosomal Disease |
| LIMP-# | Lysosomal integral membrane protein- # |
| LMP | Lysosomal membrane proteins |
| LRO | Lysosome-related organelles |
| LRP | Lipoprotein receptor-related protein |
| LYCHOS | Lysosomal cholesterol sensing protein (G protein–coupled receptor 155) |
| M6P | Mannose 6-phosphate |
| M6PR | Mannose-6-phosphate receptors |
| MCOLN1 | Mucolipin TRP Cation Channel 1; protein coding gene |
| MITF | Microphthalmia-associated transcription factor |
| MiT-TEF | Family of transcriptions factors |
| ML1 | Mucolipin-1 |
| ML IV | Mucolipidosis type IV |
| MSD | Multiple sulphatase deficiency |
| mTOR | Mammalian target of rapamycin |
| mTORC # | Mammalian target of rapamycin complex # |
| NPC1 | Niemann–Pick type C1 protein |
| NSF | N-ethyl-maleimide-sensitive factor |
| PSAP | Gene that encodes prosaposin |
| RER | Rough Endoplasmic Reticulum |
| Sap C | Saposin C |
| SNARE | Soluble N-ethylmaleimide-sensitive factor-attachment protein receptor |
| SUMF1 | Sulphatase Modifying Factor 1 |
| TFE3 | Transcription factor E3 |
| TFEB | Transcription factor EB |
| TFEC | Transcription Factor EC |
| TGN | Trans-Golgi network |
| TPC | Two-pore channel |
| TRP | Transient receptor potential |
| TRPML | Transient receptor potential of the mucolipin family |
| TRPML1 | Transient receptor potential of mucolipin 1 |
| TSC | Tuberous sclerosis complex |
| TSE | Tubular sorting endosome |
| UCE | Uncovering Enzyme |
| UDP-GlcNac | Uridine diphosphate N-acetylglucosamine |
| ULK1 | Unc-51- like kinase 1 |
| VSE | Vacuolar sorting endosome |
References
- de Duve, C. The lysosome turns fifty. Nat. Cell Biol. 2005, 7, 847–849. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Ren, D. Lysosomal Physiology. Annu. Rev. Physiol. 2015, 77, 57–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Platt, F.M.; d’Azzo, A.; Davidson, B.L.; Neufeld, E.F.; Tifft, C.J. Lysosomal storage diseases. Nat. Rev. Dis. Primer 2018, 4, 27. [Google Scholar] [CrossRef]
- de O.Poswar, F.; Vairo, F.; Burin, M.; Michelin-Tirelli, K.; Brusius-Facchin, A.C.; Kubaski, F.; de Souza, C.F.M.; Baldo, G.; Giugliani, R. Lysosomal diseases: Overview on current diagnosis and treatment. Genet. Mol. Biol. 2019, 42, 165–177. [Google Scholar] [CrossRef] [PubMed]
- Alroy, J.; Lyons, J.A. Lysosomal Storage Diseases. J. Inborn Errors Metab. Screen. 2014, 2, 232640981351766. [Google Scholar] [CrossRef]
- Boustany, R.-M.N. Lysosomal storage diseases—the horizon expands. Nat. Rev. Neurol. 2013, 9, 583–598. [Google Scholar] [CrossRef]
- Ballabio, A.; Gieselmann, V. Lysosomal disorders: From storage to cellular damage. Biochim. Biophys. Acta BBA-Mol. Cell Res. 2009, 1793, 684–696. [Google Scholar] [CrossRef] [Green Version]
- Vitner, E.B.; Platt, F.M.; Futerman, A.H. Common and Uncommon Pathogenic Cascades in Lysosomal Storage Diseases. J. Biol. Chem. 2010, 285, 20423–20427. [Google Scholar] [CrossRef] [Green Version]
- Kolter, T.; Sandhoff, K. Principles of lysosomal membrane digestion: Stimulation of Sphingolipid Degradation by Sphingolipid Activator Proteins and Anionic Lysosomal Lipids. Annu. Rev. Cell Dev. Biol. 2005, 21, 81–103. [Google Scholar] [CrossRef] [Green Version]
- Settembre, C.; Fraldi, A.; Medina, D.L.; Ballabio, A. Signals from the lysosome: A control centre for cellular clearance and energy metabolism. Nat. Rev. Mol. Cell Biol. 2013, 14, 283–296. [Google Scholar] [CrossRef]
- Palmieri, M.; Impey, S.; Kang, H.; di Ronza, A.; Pelz, C.; Sardiello, M.; Ballabio, A. Characterization of the CLEAR network reveals an integrated control of cellular clearance pathways. Hum. Mol. Genet. 2011, 20, 3852–3866. [Google Scholar] [CrossRef] [Green Version]
- Settembre, C.; Di Malta, C.; Polito, V.A.; Arencibia, M.G.; Vetrini, F.; Erdin, S.; Erdin, S.U.; Huynh, T.; Medina, D.; Colella, P.; et al. TFEB Links Autophagy to Lysosomal Biogenesis. Science 2011, 332, 1429–1433. [Google Scholar] [CrossRef] [Green Version]
- Martina, J.A.; Diab, H.I.; Lishu, L.; Jeong-A, L.; Patange, S.; Raben, N.; Puertollano, R. The Nutrient-Responsive Transcription Factor TFE3 Promotes Autophagy, Lysosomal Biogenesis, and Clearance of Cellular Debris. Sci. Signal. 2014, 7. [Google Scholar] [CrossRef] [Green Version]
- Braulke, T.; Bonifacino, J.S. Sorting of lysosomal proteins. Biochim. Biophys. Acta BBA Mol. Cell Res. 2009, 1793, 605–614. [Google Scholar] [CrossRef] [Green Version]
- Saftig, P.; Klumperman, J. Lysosome biogenesis and lysosomal membrane proteins: Trafficking meets function. Nat. Rev. Mol. Cell Biol. 2009, 10, 623–635. [Google Scholar] [CrossRef] [PubMed]
- Klumperman, J.; Kuliawat, R.; Griffith, J.M.; Geuze, H.J.; Arvan, P. Mannose 6–Phosphate Receptors Are Sorted from Immature Secretory Granules via Adaptor Protein AP-1, Clathrin, and Syntaxin 6–positive Vesicles. J. Cell Biol. 1998, 141, 359–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowman, G.D.; Nodelman, I.M.; Hong, Y.; Chua, N.-H.; Lindberg, U.; Schutt, C.E. A comparative structural analysis of the ADF/Cofilin family. Proteins Struct. Funct. Genet. 2000, 41, 374–384. [Google Scholar] [CrossRef]
- Ghosh, P.; Griffith, J.; Geuze, H.J.; Kornfeld, S. Mammalian GGAs act together to sort mannose 6-phosphate receptors. J. Cell Biol. 2003, 163, 755–766. [Google Scholar] [CrossRef] [Green Version]
- Meel, E.; Klumperman, J. Imaging and imagination: Understanding the endo-lysosomal system. Histochem. Cell Biol. 2008, 129, 253–266. [Google Scholar] [CrossRef] [Green Version]
- Braulke, T.; Gartung, C.; Hasilik, A.; von Figura, K. Is movement of mannose 6-phosphate-specific receptor triggered by binding of lysosomal enzymes? J. Cell Biol. 1987, 104, 1735–1742. [Google Scholar] [CrossRef] [PubMed]
- Pohlmann, R.; Boeker, M.W.C.; von Figura, K. The Two Mannose 6-Phosphate Receptors Transport Distinct Complements of Lysosomal Proteins. J. Biol. Chem. 1995, 270, 27311–27318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stoorvogel, W.; Strous, G.J.; Geuze, H.J.; Oorschot, V.; Schwartzt, A.L. Late endosomes derive from early endosomes by maturation. Cell 1991, 65, 417–427. [Google Scholar] [CrossRef] [PubMed]
- Luzio, J.P.; Pryor, P.R.; Bright, N.A. Lysosomes: Fusion and function. Nat. Rev. Mol. Cell Biol. 2007, 8, 622–632. [Google Scholar] [CrossRef] [PubMed]
- Bonifacino, J.S.; Rojas, R. Retrograde transport from endosomes to the trans-Golgi network. Nat. Rev. Mol. Cell Biol. 2006, 7, 568–579. [Google Scholar] [CrossRef] [PubMed]
- Colombo, M.; Raposo, G.; Théry, C. Biogenesis, Secretion, and Intercellular Interactions of Exosomes and Other Extracellular Vesicles. Annu. Rev. Cell Dev. Biol. 2014, 30, 255–289. [Google Scholar] [CrossRef]
- Gurung, S.; Perocheau, D.; Touramanidou, L.; Baruteau, J. The exosome journey: From biogenesis to uptake and intracellular signalling. Cell Commun. Signal. 2021, 19, 47. [Google Scholar] [CrossRef] [PubMed]
- Fader, C.M.; Colombo, M.I. Autophagy and multivesicular bodies: Two closely related partners. Cell Death Differ. 2009, 16, 70–78. [Google Scholar] [CrossRef] [Green Version]
- Peng, X.; Yang, L.; Ma, Y.; Li, Y.; Li, H. Focus on the morphogenesis, fate and the role in tumor progression of multivesicular bodies. Cell Commun. Signal. 2020, 18, 122. [Google Scholar] [CrossRef]
- Doyle, L.; Wang, M. Overview of Extracellular Vesicles, Their Origin, Composition, Purpose, and Methods for Exosome Isolation and Analysis. Cells 2019, 8, 727. [Google Scholar] [CrossRef] [Green Version]
- Madhyastha, R.; Madhyastha, H.; Nurrahmah, Q.I.; Purbasari, B.; Maruyama, M.; Nakajima, Y. MicroRNA 21 Elicits a Pro-inflammatory Response in Macrophages, with Exosomes Functioning as Delivery Vehicles. Inflammation 2021, 44, 1274–1287. [Google Scholar] [CrossRef]
- Luzio, J.P.; Hackmann, Y.; Dieckmann, N.M.G.; Griffiths, G.M. The Biogenesis of Lysosomes and Lysosome-Related Organelles. Cold Spring Harb. Perspect. Biol. 2014, 6, a016840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pryor, P.R.; Mullock, B.M.; Bright, N.A.; Gray, S.R.; Luzio, J.P. The Role of Intraorganellar Ca2+ in Late Endosome–Lysosome Heterotypic Fusion and in the Reformation of Lysosomes from Hybrid Organelles. J. Cell Biol. 2000, 149, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirota, Y.; Masuyama, N.; Kuronita, T.; Fujita, H.; Himeno, M.; Tanaka, Y. Analysis of post-lysosomal compartments. Biochem. Biophys. Res. Commun. 2004, 314, 306–312. [Google Scholar] [CrossRef] [PubMed]
- Bright, N.A.; Gratian, M.J.; Luzio, J.P. Endocytic Delivery to Lysosomes Mediated by Concurrent Fusion and Kissing Events in Living Cells. Curr. Biol. 2005, 15, 360–365. [Google Scholar] [CrossRef] [Green Version]
- Jahreiss, L.; Menzies, F.M.; Rubinsztein, D.C. The Itinerary of Autophagosomes: From Peripheral Formation to Kiss-and-Run Fusion with Lysosomes. Traffic 2008, 9, 574–587. [Google Scholar] [CrossRef] [Green Version]
- Yu, L.; McPhee, C.K.; Zheng, L.; Mardones, G.A.; Rong, Y.; Peng, J.; Mi, N.; Zhao, Y.; Liu, Z.; Wan, F.; et al. Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature 2010, 465, 942–946. [Google Scholar] [CrossRef] [Green Version]
- Bonifacino, J.S.; Traub, L.M. Signals for Sorting of Transmembrane Proteins to Endosomes and Lysosomes. Annu. Rev. Biochem. 2003, 72, 395–447. [Google Scholar] [CrossRef] [Green Version]
- Janvier, K.; Bonifacino, J.S. Role of the Endocytic Machinery in the Sorting of Lysosome-associated Membrane Proteins. Mol. Biol. Cell 2005, 16, 4231–4242. [Google Scholar] [CrossRef]
- Traub, L.M.; Bonifacino, J.S. Cargo Recognition in Clathrin-Mediated Endocytosis. Cold Spring Harb. Perspect. Biol. 2013, 5, a016790. [Google Scholar] [CrossRef] [Green Version]
- Schwake, M.; Schröder, B.; Saftig, P. Lysosomal Membrane Proteins and Their Central Role in Physiology: Lysosomal Membrane Proteins. Traffic 2013, 14, 739–748. [Google Scholar] [CrossRef]
- Winchester, B.G. Lysosomal membrane proteins. Eur. J. Paediatr. Neurol. 2001, 5, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Eskelinen, E.-L.; Tanaka, Y.; Saftig, P. At the acidic edge: Emerging functions for lysosomal membrane proteins. Trends Cell Biol. 2003, 13, 137–145. [Google Scholar] [CrossRef]
- Reczek, D.; Schwake, M.; Schröder, J.; Hughes, H.; Blanz, J.; Jin, X.; Brondyk, W.; Van Patten, S.; Edmunds, T.; Saftig, P. LIMP-2 Is a Receptor for Lysosomal Mannose-6-Phosphate-Independent Targeting of β-Glucocerebrosidase. Cell 2007, 131, 770–783. [Google Scholar] [CrossRef] [Green Version]
- Bassi, M.T.; Manzoni, M.; Monti, E.; Pizzo, M.T.; Ballabio, A.; Borsani, G. Cloning of the Gene Encoding a Novel Integral Membrane Protein, Mucolipidin—and Identification of the Two Major Founder Mutations Causing Mucolipidosis Type IV. Am. J. Hum. Genet. 2000, 67, 1110–1120. [Google Scholar] [CrossRef] [PubMed]
- LaPlante, J.M.; Falardeau, J.; Sun, M.; Kanazirska, M.; Brown, E.M.; Slaugenhaupt, S.A.; Vassilev, P.M. Identification and characterization of the single channel function of human mucolipin-1 implicated in mucolipidosis type IV, a disorder affecting the lysosomal pathway. FEBS Lett. 2002, 532, 183–187. [Google Scholar] [CrossRef] [Green Version]
- Huizing, M.; Helip-Wooley, A.; Westbroek, W.; Gunay-Aygun, M.; Gahl, W.A. Disorders of Lysosome-Related Organelle Biogenesis: Clinical and Molecular Genetics. Annu. Rev. Genomics Hum. Genet. 2008, 9, 359–386. [Google Scholar] [CrossRef] [Green Version]
- Marques, A.R.A.; Saftig, P. Lysosomal storage disorders—challenges, concepts and avenues for therapy: Beyond rare diseases. J. Cell Sci. 2019, 132, jcs221739. [Google Scholar] [CrossRef]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 169, 361–371. [Google Scholar] [CrossRef]
- Hosokawa, N.; Hara, T.; Kaizuka, T.; Kishi, C.; Takamura, A.; Miura, Y.; Iemura, S.; Natsume, T.; Takehana, K.; Yamada, N.; et al. Nutrient-dependent mTORC1 Association with the ULK1–Atg13–FIP200 Complex Required for Autophagy. Mol. Biol. Cell 2009, 20, 1981–1991. [Google Scholar] [CrossRef] [Green Version]
- Kim, E.; Goraksha-Hicks, P.; Li, L.; Neufeld, T.P.; Guan, K.-L. Regulation of TORC1 by Rag GTPases in nutrient response. Nat. Cell Biol. 2008, 10, 935–945. [Google Scholar] [CrossRef] [PubMed]
- Castellano, B.M.; Thelen, A.M.; Moldavski, O.; Feltes, M.; van der Welle, R.E.N.; Mydock-McGrane, L.; Jiang, X.; van Eijkeren, R.J.; Davis, O.B.; Louie, S.M.; et al. Lysosomal cholesterol activates mTORC1 via an SLC38A9–Niemann-Pick C1 signaling complex. Science 2017, 355, 1306–1311. [Google Scholar] [CrossRef] [Green Version]
- Shin, H.R.; Citron, Y.R.; Wang, L.; Tribouillard, L.; Goul, C.S.; Stipp, R.; Sugasawa, Y.; Jain, A.; Samson, N.; Lim, C.-Y.; et al. Lysosomal GPCR-like protein LYCHOS signals cholesterol sufficiency to mTORC1. Science 2022, 377, 1290–1298. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Jones, E.; Inoki, K. Lysosomal Regulation of mTORC1 by Amino Acids in Mammalian Cells. Biomolecules 2017, 7, 51. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Lin, X.; Hou, Q.; Hu, Z.; Wang, Y.; Wang, Z. Regulation of mTORC1 by amino acids in mammalian cells: A general picture of recent advances. Anim. Nutr. 2021, 7, 1009–1023. [Google Scholar] [CrossRef]
- Brailoiu, G.C.; Brailoiu, E. Modulation of Calcium Entry by the Endo-lysosomal System. In Calcium Entry Pathways in Non-Excitable Cells; Rosado, J.A., Ed.; Advances in Experimental Medicine and Biology; Springer International Publishing: Cham, Germany, 2016; Volume 898, pp. 423–447. ISBN 978-3-319-26972-6. [Google Scholar]
- Hesketh, G.G.; Wartosch, L.; Davis, L.J.; Bright, N.A.; Luzio, J.P. The Lysosome and Intracellular Signalling. In Endocytosis and Signaling; Lamaze, C., Prior, I., Eds.; Progress in Molecular and Subcellular Biology; Springer International Publishing: Cham, Germany, 2018; Volume 57, pp. 151–180. ISBN 978-3-319-96703-5. [Google Scholar]
- Morgan, A.J.; Platt, F.M.; Lloyd-Evans, E.; Galione, A. Molecular mechanisms of endolysosomal Ca2+ signalling in health and disease. Biochem. J. 2011, 439, 349–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mindell, J.A. Lysosomal Acidification Mechanisms. Annu. Rev. Physiol. 2012, 74, 69–86. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Gao, Q.; Yang, M.; Zhang, X.; Yu, L.; Lawas, M.; Li, X.; Bryant-Genevier, M.; Southall, N.T.; Marugan, J.; et al. Up-regulation of lysosomal TRPML1 channels is essential for lysosomal adaptation to nutrient starvation. Proc. Natl. Acad. Sci. USA 2015, 112, E1373–E1381. [Google Scholar] [CrossRef] [Green Version]
- Medina, D.L. Lysosomal calcium and autophagy. In International Review of Cell and Molecular Biology; Elsevier: Amsterdam, The Netherlands, 2021; Volume 362, pp. 141–170. ISBN 978-0-12-824034-2. [Google Scholar]
- Zhang, X.; Cheng, X.; Yu, L.; Yang, J.; Calvo, R.; Patnaik, S.; Hu, X.; Gao, Q.; Yang, M.; Lawas, M.; et al. MCOLN1 is a ROS sensor in lysosomes that regulates autophagy. Nat. Commun. 2016, 7, 12109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, X.; Shen, D.; Wang, X.; Dawson, T.; Li, X.; Zhang, Q.; Cheng, X.; Zhang, Y.; Weisman, L.S.; Delling, M.; et al. PI(3,5)P2 controls membrane trafficking by direct activation of mucolipin Ca2+ release channels in the endolysosome. Nat. Commun. 2010, 1, 38. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; She, J.; Zeng, W.; Guo, J.; Xu, H.; Bai, X.; Jiang, Y. Structure of mammalian endolysosomal TRPML1 channel in nanodiscs. Nature 2017, 550, 415–418. [Google Scholar] [CrossRef]
- Bargal, R.; Avidan, N.; Ben-Asher, E.; Olender, Z.; Zeigler, M.; Frumkin, A.; Raas-Rothschild, A.; Glusman, G.; Lancet, D.; Bach, G. Identification of the gene causing mucolipidosis type IV. Nat. Genet. 2000, 26, 118–122. [Google Scholar] [CrossRef] [PubMed]
- Boudewyn, L.C.; Walkley, S.U. Current concepts in the neuropathogenesis of mucolipidosis type IV. J. Neurochem. 2019, 148, 669–689. [Google Scholar] [CrossRef] [Green Version]
- Samie, M.; Wang, X.; Zhang, X.; Goschka, A.; Li, X.; Cheng, X.; Gregg, E.; Azar, M.; Zhuo, Y.; Garrity, A.G.; et al. A TRP Channel in the Lysosome Regulates Large Particle Phagocytosis via Focal Exocytosis. Dev. Cell 2013, 26, 511–524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bretou, M.; Sáez, P.J.; Sanséau, D.; Maurin, M.; Lankar, D.; Chabaud, M.; Spampanato, C.; Malbec, O.; Barbier, L.; Muallem, S.; et al. Lysosome signaling controls the migration of dendritic cells. Sci. Immunol. 2017, 2, eaak9573. [Google Scholar] [CrossRef] [Green Version]
- Medina, D.L.; Di Paola, S.; Peluso, I.; Armani, A.; De Stefani, D.; Venditti, R.; Montefusco, S.; Scotto-Rosato, A.; Prezioso, C.; Forrester, A.; et al. Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat. Cell Biol. 2015, 17, 288–299. [Google Scholar] [CrossRef] [Green Version]
- Di Paola, S.; Medina, D.L. TRPML1-/TFEB-Dependent Regulation of Lysosomal Exocytosis. In Calcium Signalling; Raffaello, A., Vecellio Reane, D., Eds.; Methods in Molecular Biology; Springer: New York, NY, USA, 2019; Volume 1925, pp. 143–144. ISBN 978-1-4939-9017-7. [Google Scholar]
- Medina, D.L.; Fraldi, A.; Bouche, V.; Annunziata, F.; Mansueto, G.; Spampanato, C.; Puri, C.; Pignata, A.; Martina, J.A.; Sardiello, M.; et al. Transcriptional Activation of Lysosomal Exocytosis Promotes Cellular Clearance. Dev. Cell 2011, 21, 421–430. [Google Scholar] [CrossRef]
- Franco-Juárez, B.; Coronel-Cruz, C.; Hernández-Ochoa, B.; Gómez-Manzo, S.; Cárdenas-Rodríguez, N.; Arreguin-Espinosa, R.; Bandala, C.; Canseco-Ávila, L.M.; Ortega-Cuellar, D. TFEB; Beyond Its Role as an Autophagy and Lysosomes Regulator. Cells 2022, 11, 3153. [Google Scholar] [CrossRef] [PubMed]
- Rega, L.R.; Polishchuk, E.; Montefusco, S.; Napolitano, G.; Tozzi, G.; Zhang, J.; Bellomo, F.; Taranta, A.; Pastore, A.; Polishchuk, R.; et al. Activation of the transcription factor EB rescues lysosomal abnormalities in cystinotic kidney cells. Kidney Int. 2016, 89, 862–873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- La Spina, M.; Contreras, P.S.; Rissone, A.; Meena, N.K.; Jeong, E.; Martina, J.A. MiT/TFE Family of Transcription Factors: An Evolutionary Perspective. Front. Cell Dev. Biol. 2021, 8, 609683. [Google Scholar] [CrossRef] [PubMed]
- Song, T.-T.; Cai, R.-S.; Hu, R.; Xu, Y.-S.; Qi, B.-N.; Xiong, Y.-A. The important role of TFEB in autophagy-lysosomal pathway and autophagy-related diseases: A systematic review. Eur. Rev. Med. Pharmacol. Sci. 2021, 25, 1641–1649. [Google Scholar] [CrossRef]
- Sardiello, M.; Palmieri, M.; di Ronza, A.; Medina, D.L.; Valenza, M.; Gennarino, V.A.; Di Malta, C.; Donaudy, F.; Embrione, V.; Polishchuk, R.S.; et al. A Gene Network Regulating Lysosomal Biogenesis and Function. Science 2009, 325, 473–477. [Google Scholar] [CrossRef] [Green Version]
- Song, W.; Wang, F.; Savini, M.; Ake, A.; di Ronza, A.; Sardiello, M.; Segatori, L. TFEB regulates lysosomal proteostasis. Hum. Mol. Genet. 2013, 22, 1994–2009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willett, R.; Martina, J.A.; Zewe, J.P.; Wills, R.; Hammond, G.R.V.; Puertollano, R. TFEB regulates lysosomal positioning by modulating TMEM55B expression and JIP4 recruitment to lysosomes. Nat. Commun. 2017, 8, 1580. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.; Dai, Y.; Liu, S.; Fan, Y.; Ding, Z.; Li, D. TFEB Biology and Agonists at a Glance. Cells 2021, 10, 333. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Liu, H.; Guan, Y.; Wang, Q.; Zhou, F.; Jie, L.; Ju, J.; Pu, L.; Du, H.; Wang, X. The altered autophagy mediated by TFEB in animal and cell models of amyotrophic lateral sclerosis. Am. J. Transl. Res. 2015, 7, 1574–1587. [Google Scholar]
- Bonam, S.R.; Wang, F.; Muller, S. Lysosomes as a therapeutic target. Nat. Rev. Drug Discov. 2019, 18, 923–948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lipton, J.O.; Sahin, M. The Neurology of mTOR. Neuron 2014, 84, 275–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lie, P.P.Y.; Nixon, R.A. Lysosome trafficking and signaling in health and neurodegenerative diseases. Neurobiol. Dis. 2019, 122, 94–105. [Google Scholar] [CrossRef]
- Tang, T.; Yang, Z.; Wang, D.; Yang, X.; Wang, J.; Li, L.; Wen, Q.; Gao, L.; Bian, X.; Yu, S. The role of lysosomes in cancer development and progression. Cell Biosci. 2020, 10, 131. [Google Scholar] [CrossRef]
- Almeida, M.F.; Bahr, B.A.; Kinsey, S.T. Endosomal-lysosomal dysfunction in metabolic diseases and Alzheimer’s disease. In International Review of Neurobiology; Elsevier: Amsterdam, The Netherlands, 2020; Volume 154, pp. 303–324. ISBN 978-0-12-820076-6. [Google Scholar]
- Peng, W.; Minakaki, G.; Nguyen, M.; Krainc, D. Preserving Lysosomal Function in the Aging Brain: Insights from Neurodegeneration. Neurotherapeutics 2019, 16, 611–634. [Google Scholar] [CrossRef]
- Farizatto, K.L.G.; Ikonne, U.S.; Almeida, M.F.; Ferrari, M.F.R.; Bahr, B.A. Aβ42-mediated proteasome inhibition and associated tau pathology in hippocampus are governed by a lysosomal response involving cathepsin B: Evidence for protective crosstalk between protein clearance pathways. PloS ONE 2017, 12, e0182895. [Google Scholar] [CrossRef]
- Koh, J.-Y.; Kim, H.N.; Hwang, J.J.; Kim, Y.-H.; Park, S.E. Lysosomal dysfunction in proteinopathic neurodegenerative disorders: Possible therapeutic roles of cAMP and zinc. Mol. Brain 2019, 12, 18. [Google Scholar] [CrossRef] [Green Version]
- Sullivan, P.M.; Zhou, X.; Robins, A.M.; Paushter, D.H.; Kim, D.; Smolka, M.B.; Hu, F. The ALS/FTLD associated protein C9orf72 associates with SMCR8 and WDR41 to regulate the autophagy-lysosome pathway. Acta Neuropathol. Commun. 2016, 4, 51. [Google Scholar] [CrossRef] [Green Version]
- Danon, M.J.; Oh, S.J.; DiMauro, S.; Manaligod, J.R.; Eastwood, A.; Naidu, S.; Schliselfeld, L.H. Lysosomal glycogen storage disease with normal acid maltase. Neurology 1981, 31, 51. [Google Scholar] [CrossRef] [PubMed]
- Montell, C. The TRP Superfamily of Cation Channels. Sci. STKE 2005, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Parenti, G.; Andria, G.; Ballabio, A. Lysosomal Storage Diseases: From Pathophysiology to Therapy. Annu. Rev. Med. 2015, 66, 471–486. [Google Scholar] [CrossRef] [PubMed]
- Platt, F.M. Emptying the stores: Lysosomal diseases and therapeutic strategies. Nat. Rev. Drug Discov. 2018, 17, 133–150. [Google Scholar] [CrossRef] [PubMed]
- Platt, F.M.; Boland, B.; van der Spoel, A.C. Lysosomal storage disorders: The cellular impact of lysosomal dysfunction. J. Cell Biol. 2012, 199, 723–734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Myerowitz, R.; Puertollano, R.; Raben, N. Impaired autophagy: The collateral damage of lysosomal storage disorders. EBioMedicine 2021, 63, 103166. [Google Scholar] [CrossRef]
- Schlotawa, L.; Adang, L.A.; Radhakrishnan, K.; Ahrens-Nicklas, R.C. Multiple Sulfatase Deficiency: A Disease Comprising Mucopolysaccharidosis, Sphingolipidosis, and More Caused by a Defect in Posttranslational Modification. Int. J. Mol. Sci. 2020, 21, 3448. [Google Scholar] [CrossRef]
- Fraldi, A.; Annunziata, F.; Lombardi, A.; Kaiser, H.-J.; Medina, D.L.; Spampanato, C.; Fedele, A.O.; Polishchuk, R.; Sorrentino, N.C.; Simons, K.; et al. Lysosomal fusion and SNARE function are impaired by cholesterol accumulation in lysosomal storage disorders. EMBO J. 2010, 29, 3607–3620. [Google Scholar] [CrossRef] [PubMed]
- Settembre, C.; Fraldi, A.; Rubinsztein, D.C.; Ballabio, A. Lysosomal storage diseases as disorders of autophagy. Autophagy 2008, 4, 113–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fürst, W.; Machleidt, W.; Sandhoff, K. The Precursor of Sulfatide Activator Protein is Processed to Three Different Proteins. Biol. Chem. Hoppe. Seyler 1988, 369, 317–328. [Google Scholar] [CrossRef]
- O’Brien, J.S.; Kretz, K.A.; Dewji, N.; Wenger, D.A.; Esch, F.; Fluharty, A.L. Coding of Two Sphingolipid Activator Proteins (SAP-1 and SAP-2) by Same Genetic Locus. Science 1988, 241, 1098–1101. [Google Scholar] [CrossRef]
- Alaei, M.R.; Tabrizi, A.; Jafari, N.; Mozafari, H. Gaucher Disease: New Expanded Classification Emphasizing Neurological Features. Iran J Child Neurol 2019, 13, 7–24. [Google Scholar]
- Yang, C.; Wang, H.; Zhu, D.; Hong, C.S.; Dmitriev, P.; Zhang, C.; Li, Y.; Ikejiri, B.; Brady, R.O.; Zhuang, Z. Mutant glucocerebrosidase in Gaucher disease recruits Hsp27 to the Hsp90 chaperone complex for proteasomal degradation. Proc. Natl. Acad. Sci. USA 2015, 112, 1137–1142. [Google Scholar] [CrossRef] [Green Version]
- Velayati, A.; DePaolo, J.; Gupta, N.; Choi, J.H.; Moaven, N.; Westbroek, W.; Goker-Alpan, O.; Goldin, E.; Stubblefield, B.K.; Kolodny, E.; et al. A mutation in SCARB2 is a modifier in gaucher disease. Hum. Mutat. 2011, 32, 1232–1238. [Google Scholar] [CrossRef] [Green Version]
- Balreira, A.; Gaspar, P.; Caiola, D.; Chaves, J.; Beirao, I.; Lima, J.L.; Azevedo, J.E.; Miranda, M.C.S. A nonsense mutation in the LIMP-2 gene associated with progressive myoclonic epilepsy and nephrotic syndrome. Hum. Mol. Genet. 2008, 17, 2238–2243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaccaro, A.M.; Motta, M.; Tatti, M.; Scarpa, S.; Masuelli, L.; Bhat, M.; Vanier, M.T.; Tylki-Szymanska, A.; Salvioli, R. Saposin C mutations in Gaucher disease patients resulting in lysosomal lipid accumulation, saposin C deficiency, but normal prosaposin processing and sorting. Hum. Mol. Genet. 2010, 19, 2987–2997. [Google Scholar] [CrossRef] [Green Version]
- Tylki-Szymańska, A.; Czartoryska, B.; Vanier, M.-T.; Poorthuis, B.; Groener, J.; Ługowska, A.; Millat, G.; Vaccaro, A.; Jurkiewicz, E. Non-neuronopathic Gaucher disease due to saposin C deficiency. Clin. Genet. 2007, 72, 538–542. [Google Scholar] [CrossRef]
- Lee, R.E. The fine structure of the cerebroside occurring in Gaucher’s disease. Proc. Natl. Acad. Sci. USA 1968, 61, 484–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pandey, M.K.; Burrow, T.A.; Rani, R.; Martin, L.J.; Witte, D.; Setchell, K.D.; Mckay, M.A.; Magnusen, A.F.; Zhang, W.; Liou, B.; et al. Complement drives glucosylceramide accumulation and tissue inflammation in Gaucher disease. Nature 2017, 543, 108–112. [Google Scholar] [CrossRef]
- Sidransky, E. Gaucher Disease: Insights from a Rare Mendelian Disorder. Discov. Med. 2012, 14, 273–281. [Google Scholar] [PubMed]
- Gaucher, E. De L’epithélioma Primitif de la Rate: Hypertrophie Idiopathique de la Rate Sans Leucémie. Ph.D Thesis, University of Paris, Paris, France, 1882. [Google Scholar]
- Wong, K.; Sidransky, E.; Verma, A.; Mixon, T.; Sandberg, G.D.; Wakefield, L.K.; Morrison, A.; Lwin, A.; Colegial, C.; Allman, J.M.; et al. Neuropathology provides clues to the pathophysiology of Gaucher disease. Mol. Genet. Metab. 2004, 82, 192–207. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.; Fang, Q. Danon disease: Focusing on heart. J. Hum. Genet. 2012, 57, 407–410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eskelinen, E.-L. Roles of LAMP-1 and LAMP-2 in lysosome biogenesis and autophagy. Mol. Aspects Med. 2006, 27, 495–502. [Google Scholar] [CrossRef]
- Gough, N.R.; Fambrough, D.M. Different Steady State Subcellular Distributions of the Three Splice Variants of Lysosome-associated Membrane Protein LAMP-2 Are Determined Largely by the COOH-terminal Amino Acid Residue. J. Cell Biol. 1997, 137, 1161–1169. [Google Scholar] [CrossRef]
- Terasawa, K.; Tomabechi, Y.; Ikeda, M.; Ehara, H.; Kukimoto-Niino, M.; Wakiyama, M.; Podyma-Inoue, K.A.; Rajapakshe, A.R.; Watabe, T.; Shirouzu, M.; et al. Lysosome-associated membrane proteins-1 and -2 (LAMP-1 and LAMP-2) assemble via distinct modes. Biochem. Biophys. Res. Commun. 2016, 479, 489–495. [Google Scholar] [CrossRef]
- Eskelinen, E.-L.; Illert, A.L.; Tanaka, Y.; Schwarzmann, G.; Blanz, J.; von Figura, K.; Saftig, P. Role of LAMP-2 in Lysosome Biogenesis and Autophagy. Mol. Biol. Cell 2002, 13, 3355–3368. [Google Scholar] [CrossRef] [Green Version]
- Schneede, A.; Schmidt, C.K.; Hölttä-Vuori, M.; Heeren, J.; Willenborg, M.; Blanz, J.; Domanskyy, M.; Breiden, B.; Brodesser, S.; Landgrebe, J.; et al. Role for LAMP-2 in endosomal cholesterol transport. J. Cell. Mol. Med. 2011, 15, 280–295. [Google Scholar] [CrossRef] [Green Version]
- Guo, S.; Zhou, L.; Wang, R.; Lv, Z.; Xu, H.; Han, B.; Korantzopoulos, P.; Hu, F.; Liu, T. Danon disease: Two patients with atrial fibrillation in a single family and review of the literature. Exp. Ther. Med. 2019, 18, 1527–1532. [Google Scholar] [CrossRef] [PubMed]
- Eskelinen, E.-L.; Cuervo, A.M.; Taylor, M.R.G.; Nishino, I.; Blum, J.S.; Dice, J.F.; Sandoval, I.V.; Lippincott-Schwartz, J.; August, J.T.; Saftig, P. Unifying Nomenclature for the Isoforms of the Lysosomal Membrane Protein LAMP-2: LAMP-2 Isoform Nomenclature. Traffic 2005, 6, 1058–1061. [Google Scholar] [CrossRef]
- Nishino, I.; Fu, J.; Tanji, K.; Yamada, T.; Shimojo, S.; Koori, T.; Mora, M.; Riggs, J.E.; Oh, S.J.; Koga, Y.; et al. Primary LAMP-2 deficiency causes X-linked vacuolar cardiomyopathy and myopathy (Danon disease). Nature 2000, 406, 906–910. [Google Scholar] [CrossRef] [PubMed]
- Endo, Y.; Furuta, A.; Nishino, I. Danon disease: A phenotypic expression of LAMP-2 deficiency. Acta Neuropathol. 2015, 129, 391–398. [Google Scholar] [CrossRef] [PubMed]
- Konecki, D.S.; Foetisch, K.; Zimmer, K.P.; Schlotter, M.; Konecki, U.L. An Alternatively Spliced Form of the Human Lysosome-Associated Membrane Protein-2 Gene Is Expressed in a Tissue-Specific Manner. Biochem. Biophys. Res. Commun. 1995, 215, 757–767. [Google Scholar] [CrossRef] [PubMed]
- Peters, C.; Mayer, A. Ca2+/calmodulin signals the completion of docking and triggers a late step of vacuole fusion. Nature 1998, 396, 575–580. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Shen, D.; Samie, M.; Xu, H. Mucolipins: Intracellular TRPML1-3 channels. FEBS Lett. 2010, 584, 2013–2021. [Google Scholar] [CrossRef] [Green Version]
- Kochumon, S.; Yesodharan, D.; Vinayan, K.; Radhakrishnan, N.; Sheth, J.; Nampoothiri, S. GM2 activator protein deficiency, mimic of Tay-Sachs disease. Int. J. Epilepsy 2017, 04, 184–187. [Google Scholar] [CrossRef]
- Kinoshita, T.; Schnaar, R.L. Glycosphingolipids. In Essentials of Glycobiology; Varki, A., Cummings, R.D., Esko, J.D., Stanley, P., Hart, G.W., Aebi, M., Darvill, A.G., Packer, N.H., Prestegard, J.H., Seeberger, P.H., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2017; Volume 11. Available online: http://www.ncbi.nlm.nih.gov/books/NBK310274/ (accessed on 21 November 2022).
- Conzelmann, E. The Role of Activator Proteins in Glycolipid Degradation and Storage Diseases. In Molecular Basis of Membrane-Associated Diseases; Azzi, A., Drahota, Z., Papa, S., Eds.; Springer: Berlin/Heidelberg, Germany, 1989; pp. 379–394. ISBN 978-3-642-74417-4. [Google Scholar]
- Komura, N.; Suzuki, K.G.N.; Ando, H.; Konishi, M.; Imamura, A.; Ishida, H.; Kusumi, A.; Kiso, M. Syntheses of Fluorescent Gangliosides for the Studies of Raft Domains. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 2017; Volume 597, pp. 239–263. ISBN 978-0-12-811469-8. [Google Scholar]
- Schröder, M.; Schnabel, D.; Suzuki, K.; Sandhoff, K. A mutation in the gene of a glycolipid-binding protein (GM2 activator) that causes GM2-gangliosidosis variant AB. FEBS Lett. 1991, 290, 1–3. [Google Scholar] [CrossRef] [Green Version]
- Renaud, D.; Brodsky, M. GM2-Gangliosidosis, AB Variant: Clinical, Ophthalmological, MRI, and Molecular Findings. In JIMD Reports, Volume 25; Morava, E., Baumgartner, M., Patterson, M., Rahman, S., Zschocke, J., Peters, V., Eds.; JIMD Reports; Springer: Berlin/Heidelberg, Germany, 2015; Volume 25, pp. 83–86. ISBN 978-3-662-49667-1. [Google Scholar]
- Gravel, R.A.; Kaback, M.M.; Proia, R.L.; Sandhoff, K.; Suzuki, K.; Suzuki, K. The GM2 Gangliosidoses. In The Online Metabolic and Molecular Bases of Inherited Disease; Valle, D.L., Antonarakis, S., Ballabio, A., Beaudet, A.L., Mitchell, G.A., Eds.; McGraw-Hill Education: New York, NY, USA, 2019; Available online: http://www.ommbid.mhmedical.com/content.aspx?aid=1181467110 (accessed on 21 November 2022).
- Dierks, T.; Schlotawa, L.; Frese, M.-A.; Radhakrishnan, K.; von Figura, K.; Schmidt, B. Molecular basis of multiple sulfatase deficiency, mucolipidosis II/III and Niemann–Pick C1 disease—Lysosomal storage disorders caused by defects of non-lysosomal proteins. Biochim. Biophys. Acta BBA Mol. Cell Res. 2009, 1793, 710–725. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, B.; Selmer, T.; Ingendoh, A.; Figurat, K. von A novel amino acid modification in sulfatases that is defective in multiple sulfatase deficiency. Cell 1995, 82, 271–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dierks, T.; Schmidt, B.; Borissenko, L.V.; Peng, J.; Preusser, A.; Mariappan, M.; von Figura, K. Multiple Sulfatase Deficiency Is Caused by Mutations in the Gene Encoding the Human Cα-Formylglycine Generating Enzyme. Cell 2003, 113, 435–444. [Google Scholar] [CrossRef] [PubMed]
- Brady, R.O.; Kanfer, J.N.; Shapiro, D. Metabolism of glucocerebrosides II. Evidence of an enzymatic deficiency in Gaucher’s disease. Biochem. Biophys. Res. Commun. 1965, 18, 221–225. [Google Scholar] [CrossRef]
- Jmoudiak, M.; Futerman, A.H. Gaucher disease: Pathological mechanisms and modern management. Br. J. Haematol. 2005, 129, 178–188. [Google Scholar] [CrossRef]
- Sugie, K. Autophagic vacuolar myopathy: Danon disease and related myopathies. Neurol. Clin. Neurosci. 2022, 10, 273–278. [Google Scholar] [CrossRef]
- Rowland, T.J.; Sweet, M.E.; Mestroni, L.; Taylor, M.R.G. Danon disease—dysregulation of autophagy in a multisystem disorder with cardiomyopathy. J. Cell Sci. 2016, 129, 2135–2143. [Google Scholar] [CrossRef] [Green Version]
- Yadin, D.; Petrover, Z.; Shainberg, A.; Alcalai, R.; Waldman, M.; Seidman, J.; Seidman, C.E.; Abraham, N.G.; Hochhauser, E.; Arad, M. Autophagy guided interventions to modify the cardiac phenotype of Danon disease. Biochem. Pharmacol. 2022, 204, 115229. [Google Scholar] [CrossRef] [PubMed]
- Pryor, P.R.; Reimann, F.; Gribble, F.M.; Luzio, J.P. Mucolipin-1 Is a Lysosomal Membrane Protein Required for Intracellular Lactosylceramide Traffic. Traffic 2006, 7, 1388–1398. [Google Scholar] [CrossRef]
- Chen, C.-S.; Bach, G.; Pagano, R.E. Abnormal transport along the lysosomal pathway in Mucolipidosis, type IV disease. Proc. Natl. Acad. Sci. USA 1998, 95, 6373–6378. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Amaral, O.; Martins, M.; Oliveira, A.R.; Duarte, A.J.; Mondragão-Rodrigues, I.; Macedo, M.F. The Biology of Lysosomes: From Order to Disorder. Biomedicines 2023, 11, 213. https://doi.org/10.3390/biomedicines11010213
Amaral O, Martins M, Oliveira AR, Duarte AJ, Mondragão-Rodrigues I, Macedo MF. The Biology of Lysosomes: From Order to Disorder. Biomedicines. 2023; 11(1):213. https://doi.org/10.3390/biomedicines11010213
Chicago/Turabian StyleAmaral, Olga, Mariana Martins, Ana Rita Oliveira, Ana Joana Duarte, Inês Mondragão-Rodrigues, and M. Fátima Macedo. 2023. "The Biology of Lysosomes: From Order to Disorder" Biomedicines 11, no. 1: 213. https://doi.org/10.3390/biomedicines11010213
APA StyleAmaral, O., Martins, M., Oliveira, A. R., Duarte, A. J., Mondragão-Rodrigues, I., & Macedo, M. F. (2023). The Biology of Lysosomes: From Order to Disorder. Biomedicines, 11(1), 213. https://doi.org/10.3390/biomedicines11010213