
Low-Dose Metformin Treatment Reduces In Vitro Growth of the LL/2 Non-small Cell Lung Cancer Cell Line

 , ,
, , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Metformin Treatment
2.3. Gamma Secretase Inhibition
2.4. Co-Treatment with Metformin and Gamma Secretase Inhibition
2.5. MTT Assay
2.6. Cell Lysate Homogenization and mRNA Quantification
2.7. cDNA and Real-time PCR
2.8. Western Blotting
2.9. Immunofluorescence Staining
2.10. Immunofluorescence Quantification
2.11. Statistical Analyses
3. Results
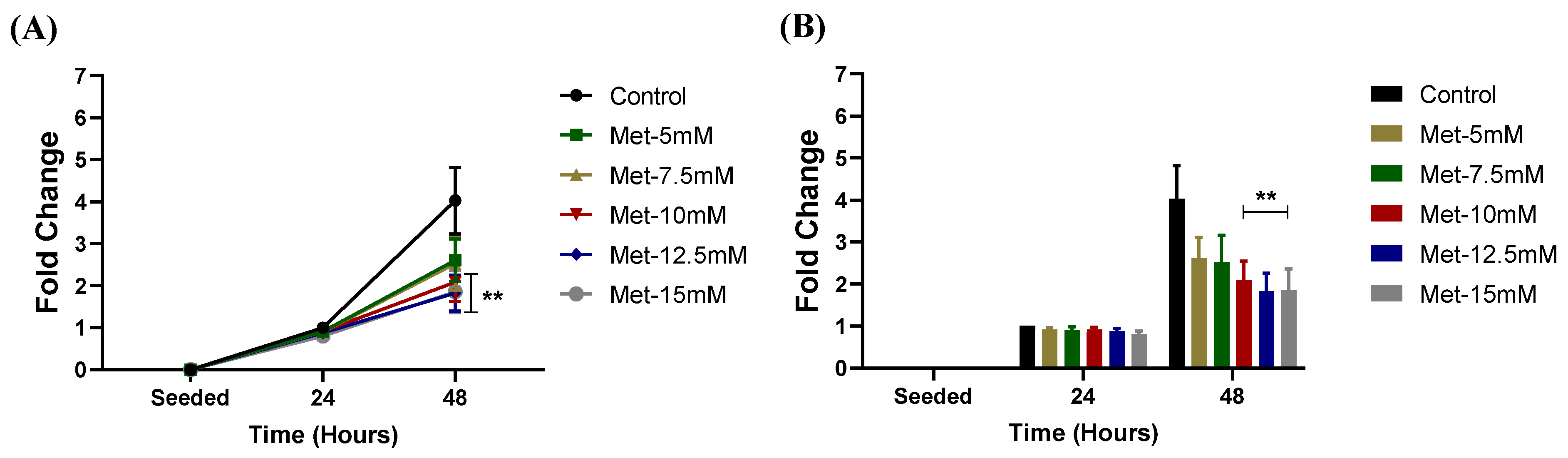
3.1. Metformin Inhibited NSCLC Cell Proliferation
3.2. In Vitro NSCLC Cell Gene Expression after a 48 h Metformin Treatment
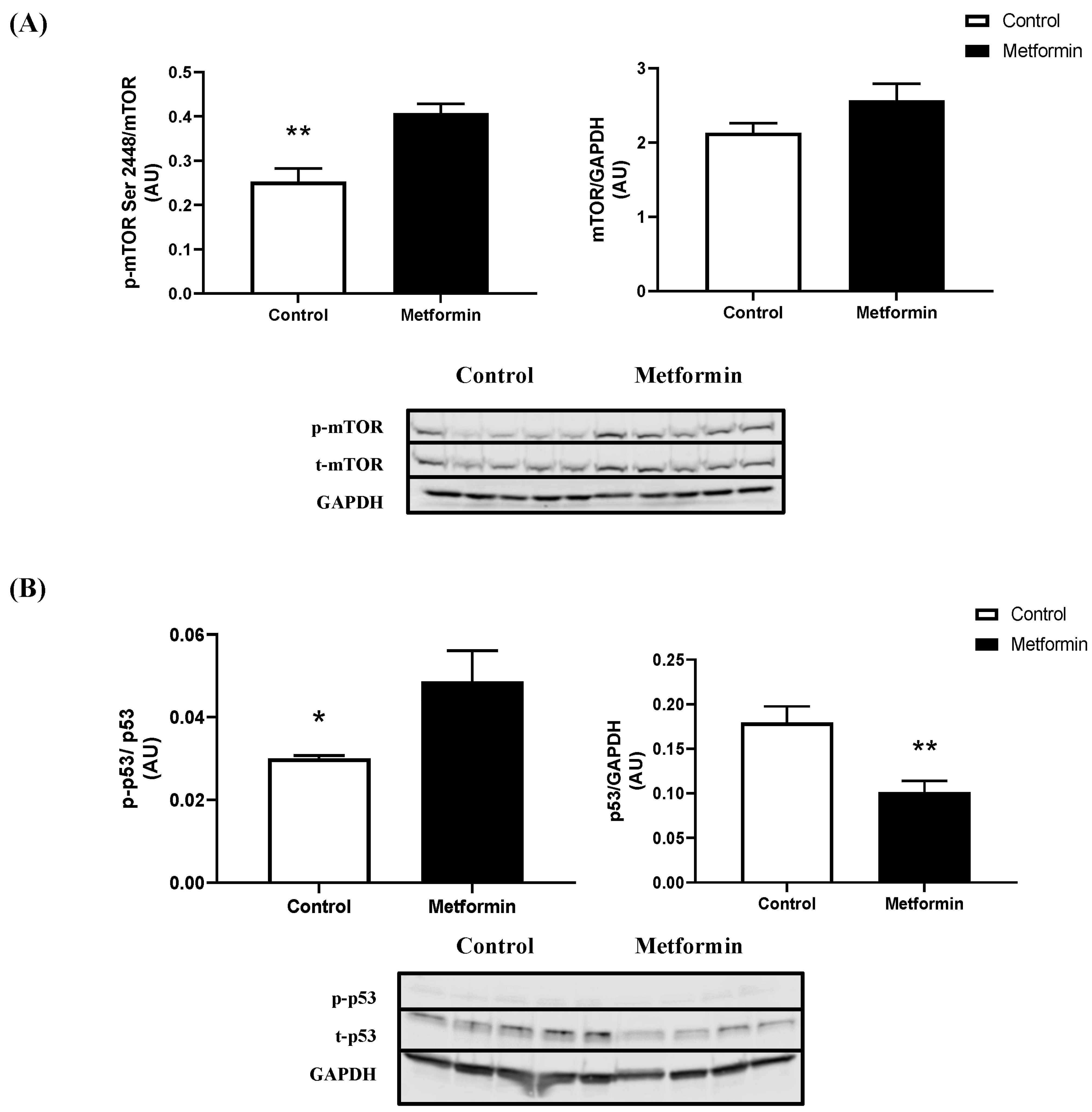
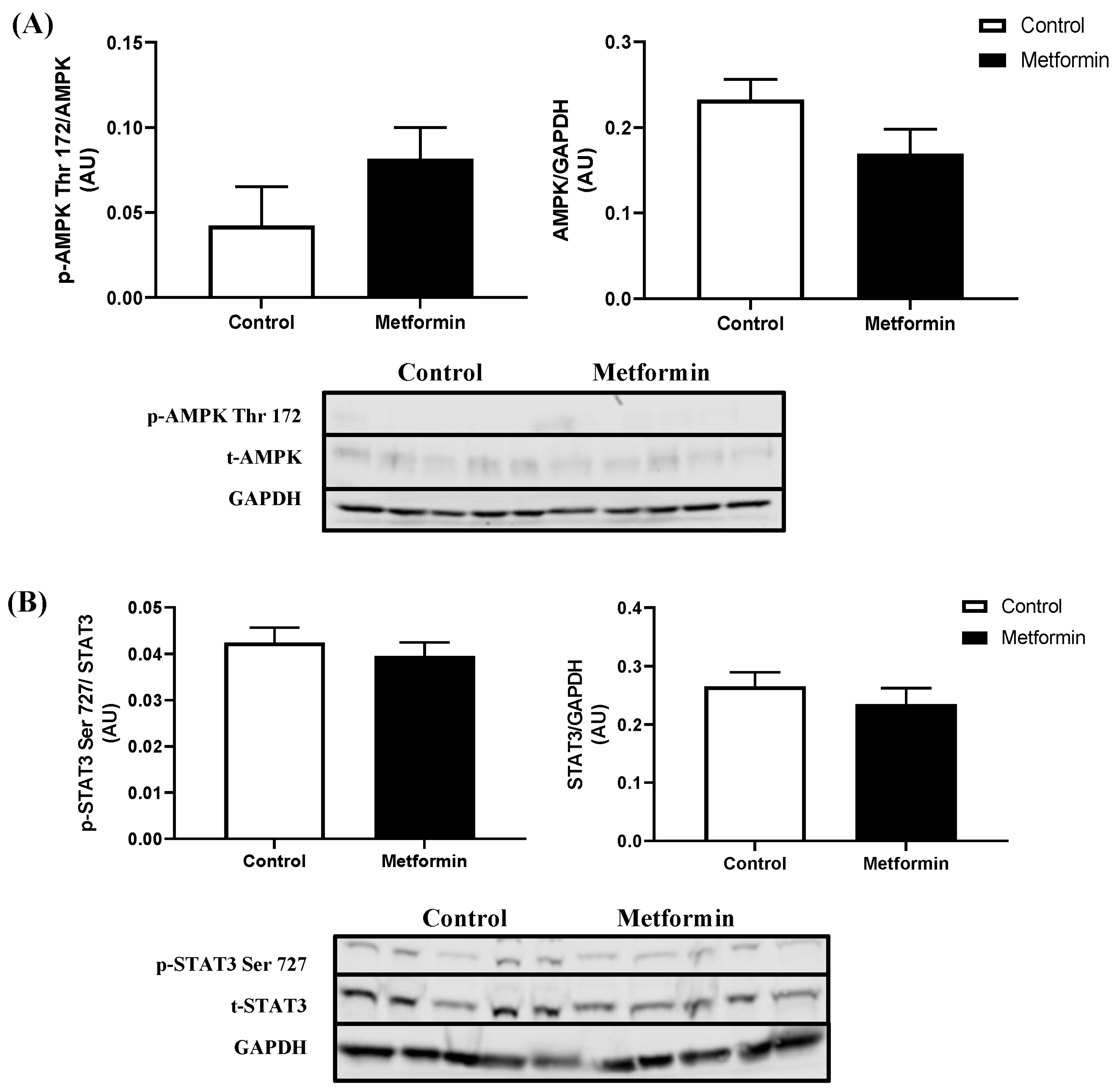
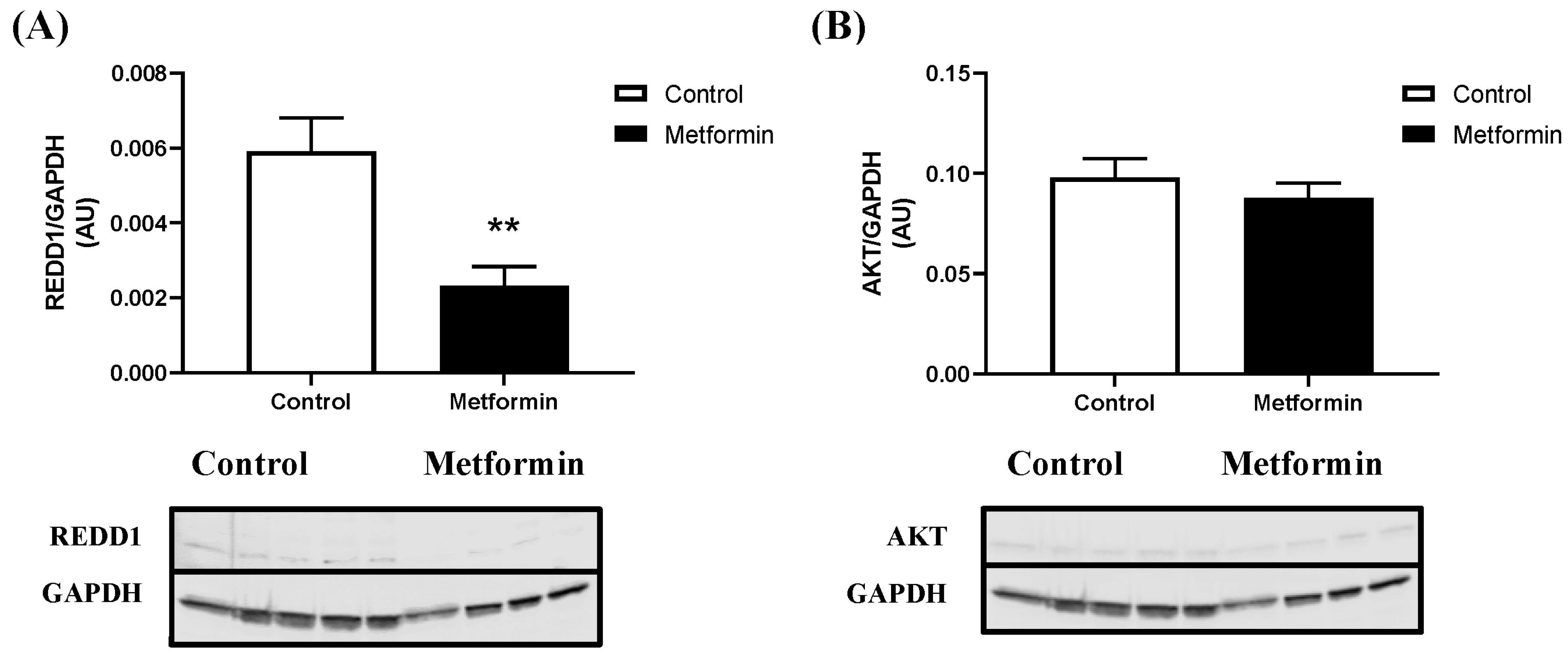
3.3. In Vitro NSCLC Cell Protein Expression following a 48 h Metformin Treatment
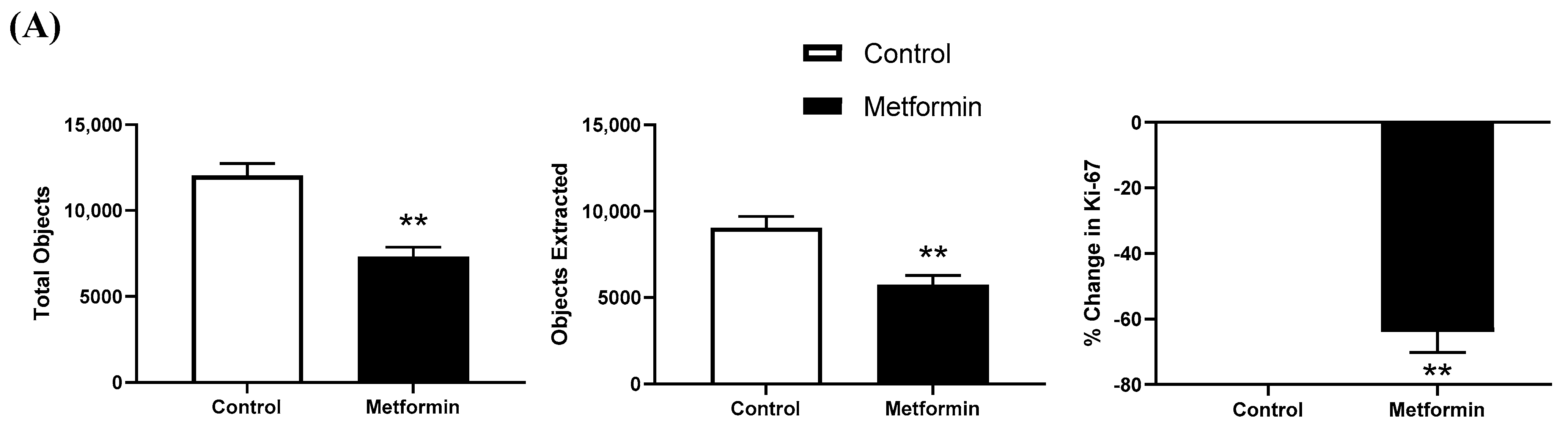
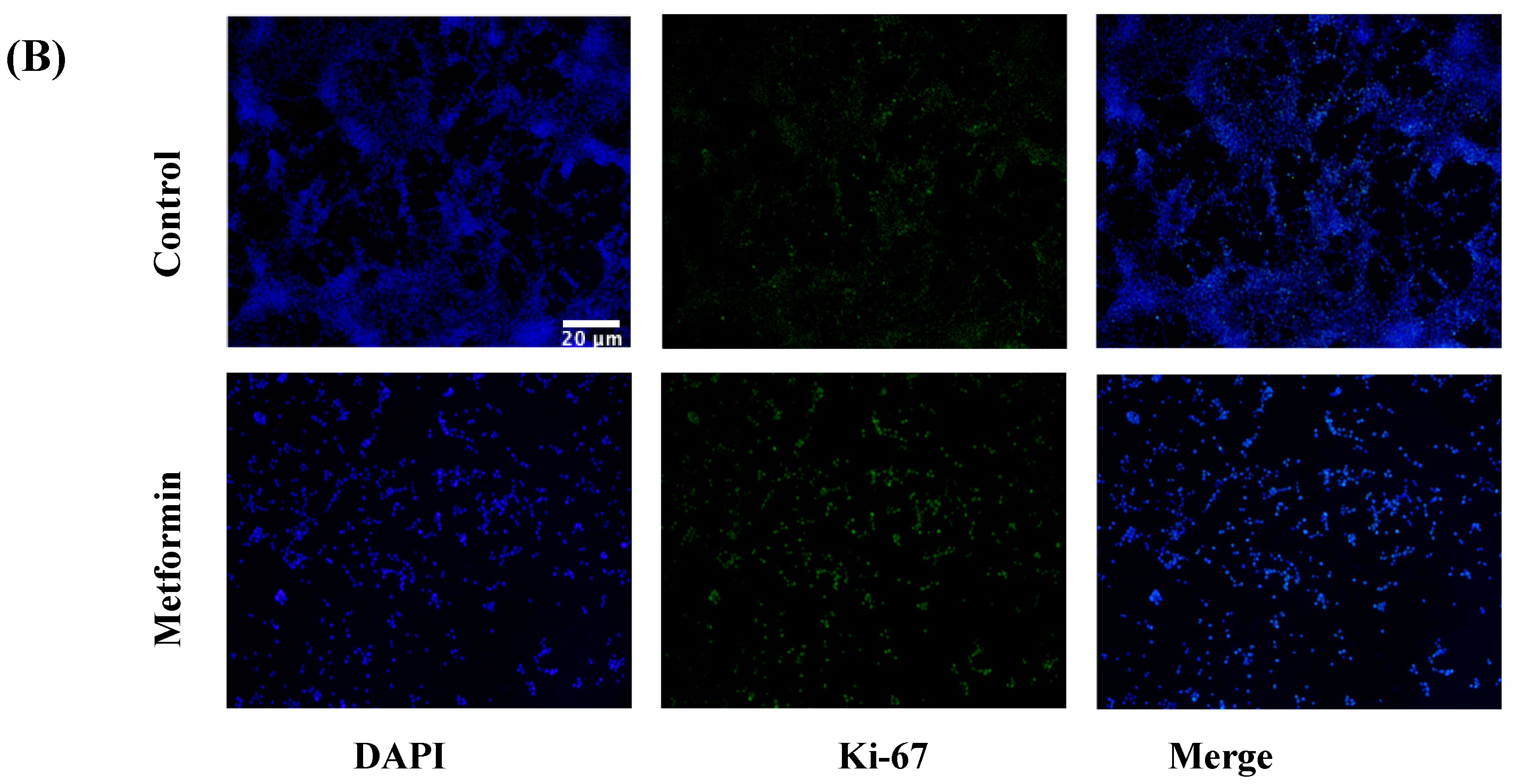
3.4. Ki-67 Immunofluorescence in NSCLC Cells following a 48 h Metformin Treatment
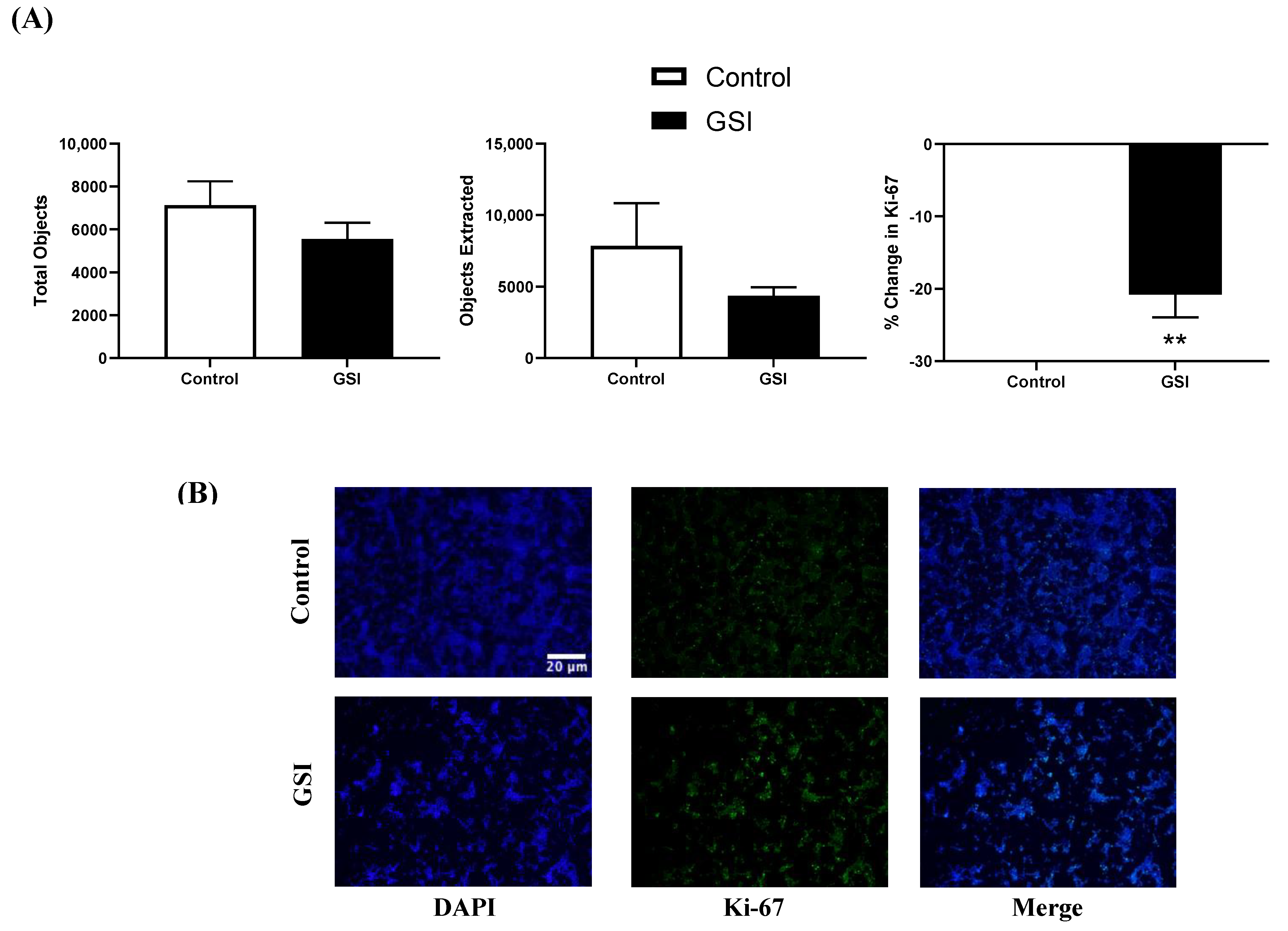
3.5. Ki-67 Immunofluorescence in NSCLC Cells after a 48 h GSI Treatment
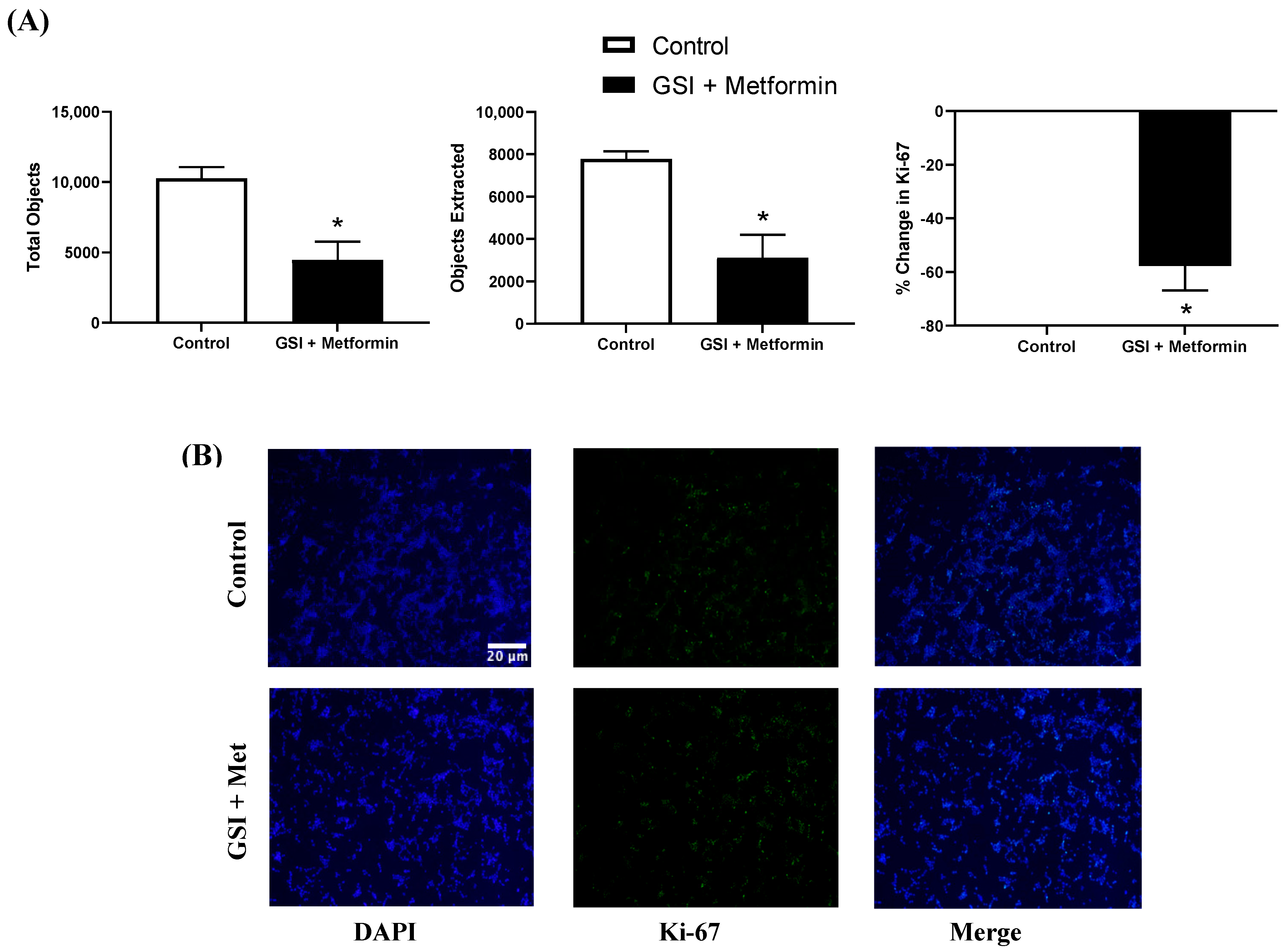
3.6. Ki-67 Immunofluorescence in NSCLC Cells following a 48 h Co-Treatment with GSI and Metformin
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molina, J.R.; Yang, P.; Cassivi, S.D.; Schild, S.E.; Adjei, A.A. Non-small cell lung cancer: Epidemiology, risk factors, treatment, and survivorship. Mayo Clin. Proc. 2008, 83, 584–594. [Google Scholar] [CrossRef] [PubMed]
- Rena, G.; Hardie, D.G.; Pearson, E.R. The mechanisms of action of metformin. Diabetologia 2017, 60, 1577–1585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Yang, S.; Chen, J.; Su, Z. Unraveling the Regulation of Hepatic Gluconeogenesis. Front. Endocrinol. 2018, 9, 802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarmento-Cabral, A.; L-López, F.; Gahete, M.D.; Castano, J.P.; Luque, R.M. Metformin Reduces Prostate Tumor Growth, in a Diet-Dependent Manner, by Modulating Multiple Signaling Pathways. Mol. Cancer Res. 2017, 15, 862–874. [Google Scholar] [CrossRef] [Green Version]
- Checkley, L.A.; Rho, O.; Angel, J.M.; Cho, J.; Blando, J.; Beltran, L.; Hursting, S.D.; DiGiovanni, J. Metformin inhibits skin tumor promotion in overweight and obese mice. Cancer Prev. Res. 2014, 7, 54–64. [Google Scholar] [CrossRef] [Green Version]
- Algire, C.; Amrein, L.; Zakikhani, M.; Panasci, L.; Pollak, M. Metformin blocks the stimulative effect of a high-energy diet on colon carcinoma growth in vivo and is associated with reduced expression of fatty acid synthase. Endocr. Relat. Cancer 2010, 17, 351–360. [Google Scholar] [CrossRef] [Green Version]
- Park, J.; Kim, W.G.; Zhao, L.; Enomoto, K.; Willingham, M.; Cheng, S.-Y. Metformin blocks progression of obesity-activated thyroid cancer in a mouse model. Oncotarget 2016, 7, 34832–34844. [Google Scholar] [CrossRef] [Green Version]
- Queiroz, E.A.; Puukila, S.; Eichler, R.; Sampaio, S.C.; Forsyth, H.L.; Lees, S.J.; Barbosa, A.M.; Dekker, R.F.; Fortes, Z.B.; Khaper, N. Metformin induces apoptosis and cell cycle arrest mediated by oxidative stress, AMPK and FOXO3a in MCF-7 breast cancer cells. PLoS ONE 2014, 9, e98207. [Google Scholar] [CrossRef]
- Lei, Y.; Yi, Y.; Liu, Y.; Liu, X.; Keller, E.T.; Qian, C.N.; Zhang, J.; Lu, Y. Metformin targets multiple signaling pathways in cancer. Chin. J. Cancer 2017, 36, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, X.L.; Bhattacharyya, K.K.; Dutta, S.K.; Bamlet, W.R.; Rabe, K.G.; Wang, E.; Smyrk, T.C.; Oberg, A.L.; Petersen, G.M.; Mukhopadhyay, D. Metformin suppresses pancreatic tumor growth with inhibition of NFkappaB/STAT3 inflammatory signaling. Pancreas 2015, 44, 636–647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Memmott, R.M.; Mercado, J.R.; Maier, C.R.; Kawabata, S.; Fox, S.D.; Dennis, P.A. Metformin prevents tobacco carcinogen--induced lung tumorigenesis. Cancer Prev. Res. 2010, 3, 1066–1076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saisho, Y. Metformin and Inflammation: Its Potential Beyond Glucose-lowering Effect. Endocr. Metab. Immune Disord. Drug Targets 2015, 15, 196–205. [Google Scholar] [CrossRef]
- Willows, R.; Sanders, M.J.; Xiao, B.; Patel, B.R.; Martin, S.R.; Read, J.; Wilson, J.R.; Hubbard, J.; Gamblin, S.J.; Carling, D. Phosphorylation of AMPK by upstream kinases is required for activity in mammalian cells. Biochem. J. 2017, 474, 3059–3073. [Google Scholar] [CrossRef] [Green Version]
- Inoki, K.; Zhu, T.; Guan, K.L. TSC2 mediates cellular energy response to control cell growth and survival. Cell 2003, 115, 577–590. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.W.; Wong, L.L.; Tse, E.Y.; Liu, H.F.; Leong, V.Y.; Lee, J.M.; Hardie, D.G.; Ng, I.O.; Ching, Y.P. AMPK promotes p53 acetylation via phosphorylation and inactivation of SIRT1 in liver cancer cells. Cancer Res. 2012, 72, 4394–4404. [Google Scholar] [CrossRef] [Green Version]
- Irie, H.; Banno, K.; Yanokura, M.; Iida, M.; Adachi, M.; Nakamura, K.; Umene, K.; Nogami, Y.; Masuda, K.; Kobayashi, Y.; et al. Metformin: A candidate for the treatment of gynecological tumors based on drug repositioning. Oncol. Lett. 2016, 11, 1287–1293. [Google Scholar] [CrossRef] [Green Version]
- Karnevi, E.; Said, K.; Andersson, R.; Rosendahl, A.H. Metformin-mediated growth inhibition involves suppression of the IGF-I receptor signalling pathway in human pancreatic cancer cells. BMC Cancer 2013, 13, 235. [Google Scholar] [CrossRef] [Green Version]
- Ikhlas, S.; Ahmad, M. Metformin: Insights into its anticancer potential with special reference to AMPK dependent and independent pathways. Life Sci. 2017, 185, 53–62. [Google Scholar] [CrossRef]
- Cai, S.L.; Tee, A.R.; Short, J.D.; Bergeron, J.M.; Kim, J.; Shen, J.; Guo, R.; Johnson, C.L.; Kiguchi, K.; Walker, C.L. Activity of TSC2 is inhibited by AKT-mediated phosphorylation and membrane partitioning. J. Cell Biol. 2006, 173, 279–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Constantinou, C.; Fontes de Oliveira, C.C.; Mintzopoulos, D.; Busquets, S.; He, J.; Kesarwani, M.; Mindrinos, M.; Rahme, L.G.; Argiles, J.M.; Tzika, A.A. Nuclear magnetic resonance in conjunction with functional genomics suggests mitochondrial dysfunction in a murine model of cancer cachexia. Int. J. Mol. Med. 2011, 27, 15–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hemmings, B.A.; Restuccia, D.F. PI3K-PKB/Akt pathway. Cold Spring Harb. Perspect. Biol. 2012, 4, a011189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arthur, S.T.; Cooley, I.D. The effect of physiological stimuli on sarcopenia; impact of Notch and Wnt signaling on impaired aged skeletal muscle repair. Int. J. Biol. Sci. 2012, 8, 731–760. [Google Scholar] [CrossRef] [Green Version]
- Schroeter, E.H.; Kisslinger, J.A.; Kopan, R. Notch-1 signalling requires ligand-induced proteolytic release of intracellular domain. Nature 1998, 393, 382–386. [Google Scholar] [CrossRef]
- Capaccione, K.M.; Pine, S.R. The Notch signaling pathway as a mediator of tumor survival. Carcinogenesis 2013, 34, 1420–1430. [Google Scholar] [CrossRef] [Green Version]
- Pine, S.R. Rethinking Gamma-secretase Inhibitors for Treatment of Non-small-Cell Lung Cancer: Is Notch the Target? Clin. Cancer Res. 2018, 24, 6136–6141. [Google Scholar] [CrossRef] [Green Version]
- Sharif, A.; Shaji, A.; Chammaa, M.; Pawlik, E.; Fernandez-Valdivia, R. Notch Transduction in Non-Small Cell Lung Cancer. Int. J. Mol. Sci. 2020, 21, 5691. [Google Scholar] [CrossRef]
- Rani, A.; Greenlaw, R.; Smith, R.A.; Galustian, C. HES1 in immunity and cancer. Cytokine Growth Factor Rev. 2016, 30, 113–117. [Google Scholar] [CrossRef] [Green Version]
- Guo, Q.; Liu, Z.; Jiang, L.; Liu, M.; Ma, J.; Yang, C.; Han, L.; Nan, K.; Liang, X. Metformin inhibits growth of human non-small cell lung cancer cells via liver kinase B-1-independent activation of adenosine monophosphate-activated protein kinase. Mol. Med. Rep. 2016, 13, 2590–2596. [Google Scholar] [CrossRef]
- Storozhuk, Y.; Hopmans, S.N.; Sanli, T.; Barron, C.; Tsiani, E.; Cutz, J.C.; Pond, G.; Wright, J.; Singh, G.; Tsakiridis, T. Metformin inhibits growth and enhances radiation response of non-small cell lung cancer (NSCLC) through ATM and AMPK. Br. J. Cancer 2013, 108, 2021–2032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dowling, R.J.; Lam, S.; Bassi, C.; Mouaaz, S.; Aman, A.; Kiyota, T.; Al-Awar, R.; Goodwin, P.J.; Stambolic, V. Metformin Pharmacokinetics in Mouse Tumors: Implications for Human Therapy. Cell Metab. 2016, 23, 567–568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abbastabar, M.; Kheyrollah, M.; Azizian, K.; Bagherlou, N.; Tehrani, S.S.; Maniati, M.; Karimian, A. Multiple functions of p27 in cell cycle, apoptosis, epigenetic modification and transcriptional regulation for the control of cell growth: A double-edged sword protein. DNA Repair 2018, 69, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Chu, I.M.; Hengst, L.; Slingerland, J.M. The Cdk inhibitor p27 in human cancer: Prognostic potential and relevance to anticancer therapy. Nat. Rev. Cancer 2008, 8, 253–267. [Google Scholar] [CrossRef] [PubMed]
- Abbas, T.; Dutta, A. p21 in cancer: Intricate networks and multiple activities. Nat. Rev. Cancer 2009, 9, 400–414. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, E.; Infante, J.R. Targeting CDK4/6 in patients with cancer. Cancer Treat. Rev. 2016, 45, 129–138. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Dai, W.; Chu, X.; Yang, B.; Zhao, M.; Sun, Y. Metformin inhibits lung cancer cells proliferation through repressing microRNA-222. Biotechnol. Lett. 2013, 35, 2013–2019. [Google Scholar] [CrossRef]
- Lengyel, E.; Litchfield, L.M.; Mitra, A.K.; Nieman, K.M.; Mukherjee, A.; Zhang, Y.; Johnson, A.; Bradaric, M.; Lee, W.; Romero, I.L. Metformin inhibits ovarian cancer growth and increases sensitivity to paclitaxel in mouse models. Am. J. Obs. Gynecol. 2015, 212, 479.e1–479.e410. [Google Scholar] [CrossRef] [Green Version]
- Yang, B.; Huang, C.Z.; Yu, T.; Zhou, S.N.; Liu, Q.; Liu, G.J.; Chen, S.; Han, F.H. Metformin depresses overactivated Notch1/Hes1 signaling in colorectal cancer patients with type 2 diabetes mellitus. Anti-Cancer Drugs 2017, 28, 531–539. [Google Scholar] [CrossRef]
- Tendler, S.; Kanter, L.; Lewensohn, R.; Ortiz-Villalón, C.; Viktorsson, K.; De Petris, L. The prognostic implications of Notch1, Hes1, Ascl1, and DLL3 protein expression in SCLC patients receiving platinum-based chemotherapy. PLoS ONE 2020, 15, e0240973. [Google Scholar] [CrossRef]
- Cao, N.; Lu, Y.; Liu, J.; Cai, F.; Xu, H.; Chen, J.; Zhang, X.; Hua, Z.C.; Zhuang, H. Metformin Synergistically Enhanced the Antitumor Activity of Celecoxib in Human Non-Small Cell Lung Cancer Cells. Front. Pharmacol. 2020, 11, 1094. [Google Scholar] [CrossRef] [PubMed]
- Brugarolas, J.; Lei, K.; Hurley, R.L.; Manning, B.D.; Reiling, J.H.; Hafen, E.; Witters, L.A.; Ellisen, L.W.; Kaelin, W.G., Jr. Regulation of mTOR function in response to hypoxia by REDD1 and the TSC1/TSC2 tumor suppressor complex. Genes Dev. 2004, 18, 2893–2904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Britto, F.A.; Dumas, K.; Giorgetti-Peraldi, S.; Ollendorff, V.; Favier, F.B. Is REDD1 a metabolic double agent? Lessons from physiology and pathology. Am. J. Physiol. Cell Physiol. 2020, 319, C807–C824. [Google Scholar] [CrossRef]
- Jin, H.O.; Seo, S.K.; Woo, S.H.; Kim, Y.S.; Hong, S.E.; Yi, J.Y.; Noh, W.C.; Kim, E.K.; Lee, J.K.; Hong, S.I.; et al. Redd1 inhibits the invasiveness of non-small cell lung cancer cells. Biochem. Biophys. Res. Commun. 2011, 407, 507–511. [Google Scholar] [CrossRef]
- Ben Sahra, I.; Regazzetti, C.; Robert, G.; Laurent, K.; Le Marchand-Brustel, Y.; Auberger, P.; Tanti, J.F.; Giorgetti-Peraldi, S.; Bost, F. Metformin, independent of AMPK, induces mTOR inhibition and cell-cycle arrest through REDD1. Cancer Res. 2011, 71, 4366–4372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morelli, A.P.; Tortelli, T.C., Jr.; Pavan, I.C.B.; Silva, F.R.; Granato, D.C.; Peruca, G.F.; Pauletti, B.A.; Domingues, R.R.; Bezerra, R.M.N.; De Moura, L.P.; et al. Metformin impairs cisplatin resistance effects in A549 lung cancer cells through mTOR signaling and other metabolic pathways. Int. J. Oncol. 2021, 58, 28. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Pardoll, D.; Jove, R. STATs in cancer inflammation and immunity: A leading role for STAT3. Nat. Rev. Cancer 2009, 9, 798–809. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.C.; Yeh, H.H.; Huang, W.L.; Yan, J.J.; Lai, W.W.; Su, W.P.; Chen, H.H.; Su, W.C. Metformin enhances cisplatin cytotoxicity by suppressing signal transducer and activator of transcription-3 activity independently of the liver kinase B1-AMP-activated protein kinase pathway. Am. J. Respir. Cell Mol. Biol. 2013, 49, 241–250. [Google Scholar] [CrossRef]
- Deng, X.S.; Wang, S.; Deng, A.; Liu, B.; Edgerton, S.M.; Lind, S.E.; Wahdan-Alaswad, R.; Thor, A.D. Metformin targets Stat3 to inhibit cell growth and induce apoptosis in triple-negative breast cancers. Cell Cycle 2012, 11, 367–376. [Google Scholar] [CrossRef] [Green Version]
- Li, L.T.; Jiang, G.; Chen, Q.; Zheng, J.N. Ki67 is a promising molecular target in the diagnosis of cancer (review). Mol. Med. Rep. 2015, 11, 1566–1572. [Google Scholar] [CrossRef]
- Bond, N.L.S.; Dréau, D.; Marriott, I.; Bennett, J.M.; Turner, M.J.; Arthur, S.T.; Marino, J.S. Low-Dose Metformin as a Monotherapy Does Not Reduce Non-Small-Cell Lung Cancer Tumor Burden in Mice. Biomedicines 2021, 9, 1685. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.; Zhou, Y.S.; Wang, L.C.; Huang, J.B. Advances in metformin-based metabolic therapy for non-small cell lung cancer (Review). Oncol. Rep. 2022, 47, 55. [Google Scholar] [CrossRef] [PubMed]
- Tortelli, T.C.; Tamura, R.E.; de Souza Junqueira, M.; da Silva Mororó, J.; Bustos, S.O.; Natalino, R.J.M.; Russell, S.; Désaubry, L.; Strauss, B.E.; Chammas, R. Metformin-induced chemosensitization to cisplatin depends on P53 status and is inhibited by Jarid1b overexpression in non-small cell lung cancer cells. Aging 2021, 13, 21914–21940. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer | Sequence | |
|---|---|---|
| Cyclin D | Forward | GATGGCGATCGTCCTGTCAT |
| Reverse | ACAGGCCGCTACAAGAAACA | |
| CDK4 | Forward | ATGGCTGCCACTCGATATGAA |
| Reverse | TCCTCCATTAGGAACTCTCACAC | |
| p27 | Forward | TCTCTTCGGCCCGGTCAAT |
| Reverse | AAATTCCACTTGCGCTGACTC | |
| p21 | Forward | TGGTGATGTCCGACCTGTT |
| Reverse | CATGAGCGCATCGCAATC | |
| HES1 | Forward | GGTCCTGGAATAGTGCTACCG |
| Reverse | CACCGGGGAGGAGGAATTTTT | |
| GAPDH | Forward | ATGTTTGTGATGGGTGTGAA |
| Reverse | ATGCCAAAGTTGTCATGGAT | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bond, N.L.S.; Dréau, D.; Marriott, I.; Bennett, J.M.; Turner, M.J.; Arthur, S.T.; Marino, J.S. Low-Dose Metformin Treatment Reduces In Vitro Growth of the LL/2 Non-small Cell Lung Cancer Cell Line. Biomedicines 2023, 11, 65. https://doi.org/10.3390/biomedicines11010065
Bond NLS, Dréau D, Marriott I, Bennett JM, Turner MJ, Arthur ST, Marino JS. Low-Dose Metformin Treatment Reduces In Vitro Growth of the LL/2 Non-small Cell Lung Cancer Cell Line. Biomedicines. 2023; 11(1):65. https://doi.org/10.3390/biomedicines11010065
Chicago/Turabian StyleBond, Nicole L. Stott, Didier Dréau, Ian Marriott, Jeanette M. Bennett, Michael J. Turner, Susan T. Arthur, and Joseph S. Marino. 2023. "Low-Dose Metformin Treatment Reduces In Vitro Growth of the LL/2 Non-small Cell Lung Cancer Cell Line" Biomedicines 11, no. 1: 65. https://doi.org/10.3390/biomedicines11010065
APA StyleBond, N. L. S., Dréau, D., Marriott, I., Bennett, J. M., Turner, M. J., Arthur, S. T., & Marino, J. S. (2023). Low-Dose Metformin Treatment Reduces In Vitro Growth of the LL/2 Non-small Cell Lung Cancer Cell Line. Biomedicines, 11(1), 65. https://doi.org/10.3390/biomedicines11010065