
Systems Biology and Cytokines Potential Role in Lung Cancer Immunotherapy Targeting Autophagic Axis


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
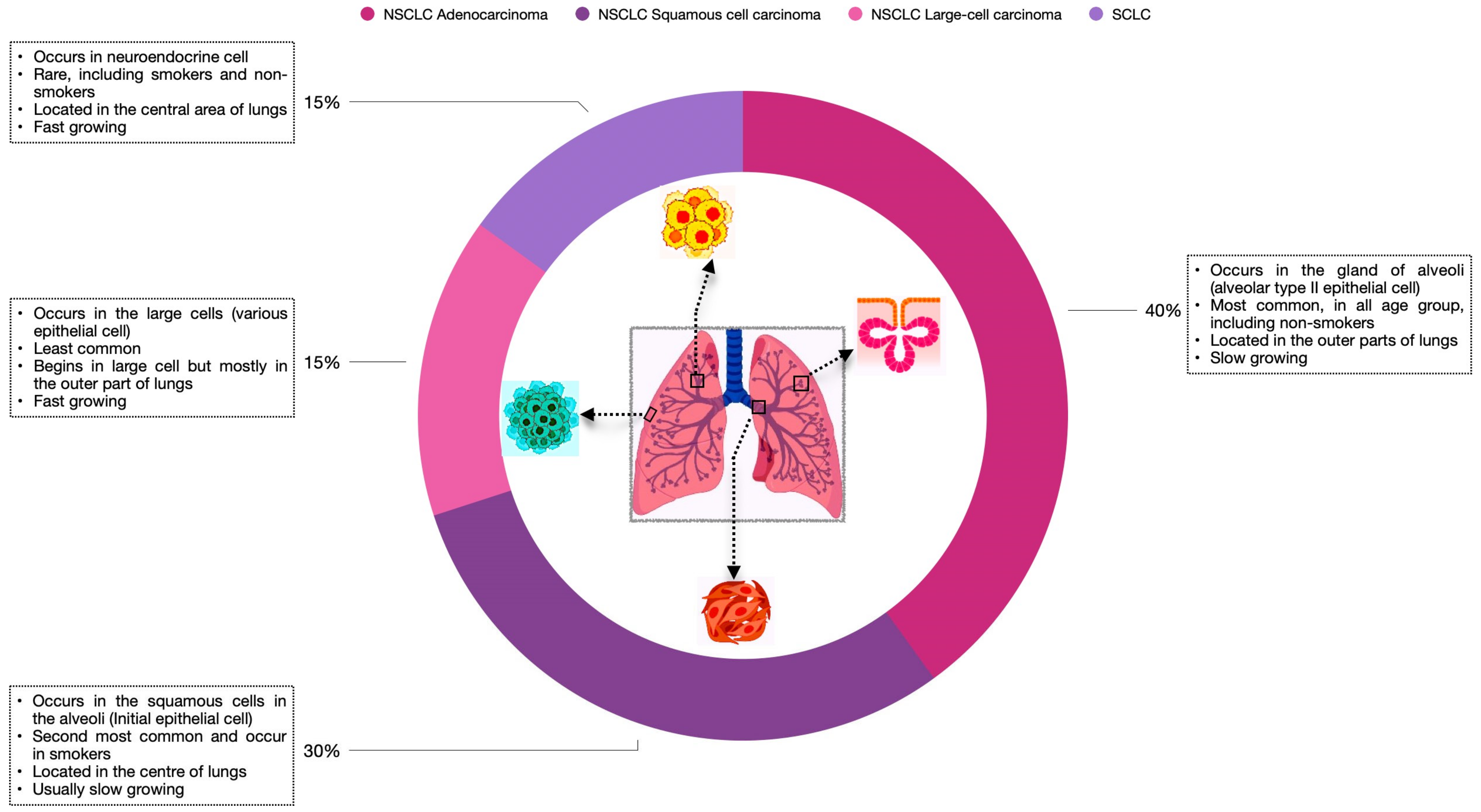
1. Introduction

2. Mechanism of Autophagy
3. Autophagy: An Anomalous Defense in Cancer
3.1. The Role of Autophagy in Tumor Inhibition
3.2. The Role of Autophagy in Tumor Progression
4. Autophagy Describes the Tumor Microenvironment
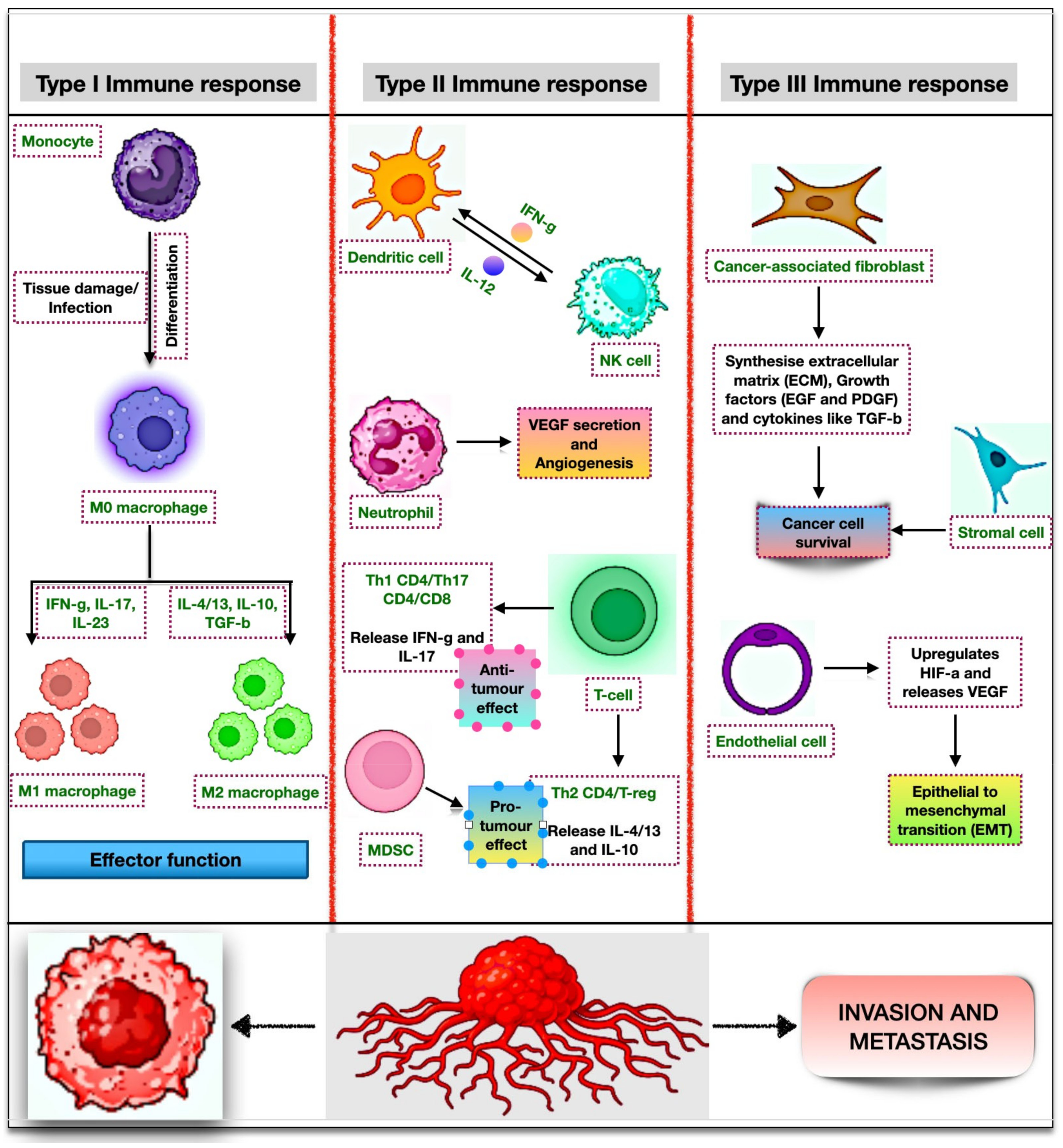
5. Immune Cells as Modulators of the Autophagy Mechanism
5.1. Immune Cells
5.1.1. Macrophages
5.1.2. Dendritic Cell
5.1.3. Natural Killer Cell
5.1.4. Neutrophils
5.1.5. Myeloid-Derived Suppressor Cells
5.1.6. T-Cells
5.1.7. Type III Immune Cells

6. Inflammasome as a Crucial Regulator in Autophagy and Cancer
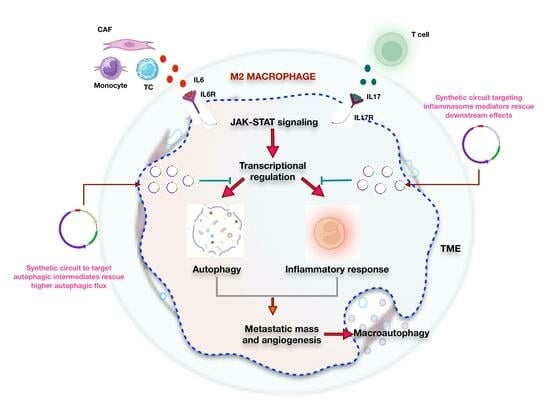
Decisive Role of IL6-IL17-IL23 Axis in Autophagic Cell Death
7. Autophagy and Drug Resistance in Lung Cancer
8. Systems Biology and Its Potential Role in Targeting Autophagy
Network Analysis Using Systems Biology
9. Autophagy as a Therapeutic in Cancer Development
10. Potential Limitations and Challenges of Targeting Autophagy in Lung Cancer Treatment
11. Real World Examples or Case Studies to Illustrate the Practical Application of Systems Biology in Cancer Research
12. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 3-MA | 3-methyladenine |
| AMPK | AMP-activated protein kinase |
| APC | Antigen-presenting cell |
| ASC | Adaptor apoptosis-associated speck-like protein |
| ATG | autophagy-related genes |
| Bif1 | Bax-interacting factor-1 |
| BMP | Bone morphogenetic protein |
| CAF | Cancer-associated fibroblast |
| CARD | Caspase activation and recruitment domain |
| ChIP | Chromatin immunoprecipitation |
| CHMP2A | Chromatin-modifying protein/charged multivesicular body protein 2A |
| CMA | Chaperone-mediated autophagy |
| CQ | Chloroquine |
| DC | Dendritic cell |
| EC | Endothelial cell |
| EGF | Epidermal growth factor |
| EGFR | Epidermal growth factor receptor |
| EMT | Epithelial mesenchymal transition |
| ESCRT | Endosomal complexes required for transport |
| FIP200 | the focal adhesion kinase family interacting protein of 200 kD |
| GABARAP | GABA Type A Receptor-Associated Protein |
| GATE-16 | Golgi-associated ATPase enhancer of 16 kDa |
| GRN | Gene regulatory network |
| GSDMD | Gasdermin D |
| HCC | Hepatocellular carcinoma |
| HIF-1α | hypoxia-inducible factor-1α |
| HIV | Human immune deficiency virus |
| HMGB1 | High mobility group box 1 |
| HQ | Hydroxychloroquine |
| HSC70 | Heat shock cognate 71 KDa protein |
| IBD | Inflammatory bowel disease |
| IFN-γ | Interferon-gamma |
| IL | Interleukin |
| JNK | Jun N-terminal kinase |
| LAMP2A | Lysosomal membrane protein |
| LC | Lung Cancer |
| LC3 | Microtubule-associated protein 1A/1B-light chain 3 |
| LPS | Lipopolysaccharide |
| MAPK | Mitogen-activated protein kinase |
| MAPK | Mitogen-activated protein kinase |
| MCSF | Macrophage colony stimulating factor |
| MDSC | Myeloid-derived suppressor cells |
| MKK | Mitogen-activated protein kinase kinase |
| MMP | Matrix metalloproteinase |
| mTOR | mammalian target of rapamycin |
| NACHT | a central nucleotide binding and oligomerization |
| NK | Natural killer cell |
| NLRP3 | NLR family protein containing a pyrin domain 3 |
| NSCLC | Non-Small Cell Lung Cancer |
| PDGF-b | Plate-derived growth factor |
| PE | Phosphatidyl ethanol amine |
| PI3K | Phosphoinositide-3-kinase |
| PI3KC3 | Phosphatidylinositol 3-phosphate kinase class III |
| PI3P | Phosphatidylinositol-3-phosphate |
| PIP2 | Phosphatidylinositol 4,5-bisphosphate |
| PIP3 | Phosphatidylinositol-3,4,5-trisphosphate |
| PPI | Protein-protein interaction |
| pTEN | a phosphatase and tensin homolog |
| RORγt | Rorc RAR-related orphan receptor gamma |
| ROS | Reactive oxygen species |
| SCLC | Small Cell Lung Cancer |
| scRNA | Single-cell RNA sequencing |
| SNARE | Soluble N-ethylmaleimide-sensitive-factor attachment protein receptor |
| STAT | Signal transducer and activator of transcription |
| TAM | Tumour-associated macrophages |
| TAN | Tumor-associated neutrophil |
| TB | Tuberculosis |
| TC | Tumor Cell |
| TF | Transcription factor |
| TGF-β | Transforming growth factor-b |
| Th | T helper |
| TME | Tumor microenvironment |
| TNF-a | Tumor necrosis factor-a |
| Treg | T-regulatory cell |
| TSC | Tuberous sclerosis |
| ULK1 | unc-51-like kinase 1 |
| UVRAG | UV radiation resistance-associated gene |
| VEGF | Vascular endothelial growth factor |
| VPS | Vacuolar protein sorting |
References
- Viktorsson, K.; Lewensohn, R.; Zhivotovsky, B. Systems biology approaches to develop innovative strategies for lung cancer therapy. Cell Death Dis. 2014, 5, e1260. [Google Scholar] [CrossRef] [PubMed]
- Lan, T.; Chen, L.; Wei, X. Inflammatory Cytokines in Cancer: Comprehensive Understanding and Clinical Progress in Gene Therapy. Cells 2021, 10, 100. [Google Scholar] [CrossRef]
- Monkkonen, T.; Debnath, J. Inflammatory signaling cascades and autophagy in cancer. Autophagy 2018, 14, 190–198. [Google Scholar] [CrossRef] [PubMed]
- Alam, M.; Hasan, G.M.; Eldin, S.M.; Adnan, M.; Riaz, M.B.; Islam, A.; Khan, I.; Hassan, M.I. Investigating regulated signaling pathways in therapeutic targeting of non-small cell lung carcinoma. Biomed. Pharmacother. 2023, 161, 114452. [Google Scholar] [CrossRef] [PubMed]
- Yuan, M.; Zhao, Y.; Arkenau, H.-T.; Lao, T.; Chu, L.; Xu, Q. Signal pathways and precision therapy of small-cell lung cancer. Signal Transduct. Target. Ther. 2022, 7, 187. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Sharma, A.; Boise, L.H.; Shanmugam, M. Cancer Metabolism and the Evasion of Apoptotic Cell Death. Cancers 2019, 11, 1144. [Google Scholar] [CrossRef]
- Feng, H.; Wang, N.; Zhang, N.; Liao, H.H. Alternative autophagy: Mechanisms and roles in different diseases. Cell Commun. Signal. CCS 2022, 20, 43. [Google Scholar] [CrossRef]
- Zada, S.; Hwang, J.S.; Ahmed, M.; Lai, T.H.; Pham, T.M.; Elashkar, O.; Kim, D.R. Cross talk between autophagy and oncogenic signaling pathways and implications for cancer therapy. Biochim. Biophys. Acta Rev. Cancer 2021, 1876, 188565. [Google Scholar] [CrossRef]
- Chavez-Dominguez, R.; Perez-Medina, M.; Lopez-Gonzalez, J.S.; Galicia-Velasco, M.; Aguilar-Cazares, D. The Double-Edge Sword of Autophagy in Cancer: From Tumor Suppression to Pro-tumor Activity. Front. Oncol. 2020, 10, 578418. [Google Scholar] [CrossRef]
- Jiang, G.M.; Tan, Y.; Wang, H.; Peng, L.; Chen, H.T.; Meng, X.J.; Li, L.L.; Liu, Y.; Li, W.F.; Shan, H. The relationship between autophagy and the immune system and its applications for tumor immunotherapy. Mol. Cancer 2019, 18, 17. [Google Scholar] [CrossRef] [PubMed]
- Bhutia, S.K.; Mukhopadhyay, S.; Sinha, N.; Das, D.N.; Panda, P.K.; Patra, S.K.; Maiti, T.K.; Mandal, M.; Dent, P.; Wang, X.Y.; et al. Autophagy: Cancer’s friend or foe? Adv. Cancer Res. 2013, 118, 61–95. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Emr, S.D. Autophagy as a regulated pathway of cellular degradation. Science 2000, 290, 1717–1721. [Google Scholar] [CrossRef] [PubMed]
- Glick, D.; Barth, S.; Macleod, K.F. Autophagy: Cellular and molecular mechanisms. J. Pathol. 2010, 221, 3–12. [Google Scholar] [CrossRef]
- Vargas, J.N.S.; Hamasaki, M.; Kawabata, T.; Youle, R.J.; Yoshimori, T. The mechanisms and roles of selective autophagy in mammals. Nat. Rev. Mol. Cell Biol. 2023, 24, 167–185. [Google Scholar] [CrossRef]
- Chen, R.H.; Chen, Y.H.; Huang, T.Y. Ubiquitin-mediated regulation of autophagy. J. Biomed. Sci. 2019, 26, 80. [Google Scholar] [CrossRef]
- Crotzer, V.L.; Blum, J.S. Autophagy and its role in MHC-mediated antigen presentation. J. Immunol. 2009, 182, 3335–3341. [Google Scholar] [CrossRef]
- Somvanshi, P.R.; Venkatesh, K.V. A conceptual review on systems biology in health and diseases: From biological networks to modern therapeutics. Syst. Synth. Biol. 2014, 8, 99–116. [Google Scholar] [CrossRef]
- Yalcin, G.D.; Danisik, N.; Baygin, R.C.; Acar, A. Systems Biology and Experimental Model Systems of Cancer. J. Pers. Med. 2020, 10, 180. [Google Scholar] [CrossRef]
- Song, M.; Li, D.; Makaryan, S.Z.; Finley, S.D. Quantitative modeling to understand cell signaling in the tumor microenvironment. Curr. Opin. Syst. Biol. 2021, 27, 100345. [Google Scholar] [CrossRef]
- Ohsumi, Y.; Campbell, W.C.; Ōmura, S.; Youyou, T. The Nobel Prize in Physiology or Medicine. Press Release. 2016. Available online: https://www.nobelprize.org/prizes/medicine/2016/press-release/ (accessed on 29 July 2023).
- Yang, Z.; Klionsky, D.J. Eaten alive: A history of macroautophagy. Nat. Cell Biol. 2010, 12, 814–822. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.W.; Klionsky, D.J. The molecular mechanism of autophagy. Mol. Med. 2003, 9, 65–76. [Google Scholar] [CrossRef] [PubMed]
- Hubert, V.; Weiss, S.; Rees, A.J.; Kain, R. Modulating Chaperone-Mediated Autophagy and Its Clinical Applications in Cancer. Cells 2022, 11, 2562. [Google Scholar] [CrossRef] [PubMed]
- Tekirdag, K.; Cuervo, A.M. Chaperone-mediated autophagy and endosomal microautophagy: Joint by a chaperone. J. Biol. Chem. 2018, 293, 5414–5424. [Google Scholar] [CrossRef] [PubMed]
- Reggiori, F.; Komatsu, M.; Finley, K.; Simonsen, A. Autophagy: More than a nonselective pathway. Int. J. Cell Biol. 2012, 2012, 219625. [Google Scholar] [CrossRef]
- Mulcahy Levy, J.M.; Thorburn, A. Autophagy in cancer: Moving from understanding mechanism to improving therapy responses in patients. Cell Death Differ. 2020, 27, 843–857. [Google Scholar] [CrossRef]
- Yun, C.W.; Lee, S.H. The Roles of Autophagy in Cancer. Int. J. Mol. Sci. 2018, 19, 3466. [Google Scholar] [CrossRef]
- Tian, T.; Li, X.; Zhang, J. mTOR Signaling in Cancer and mTOR Inhibitors in Solid Tumor Targeting Therapy. Int. J. Mol. Sci. 2019, 20, 755. [Google Scholar] [CrossRef]
- Masui, K.; Cavenee, W.K.; Mischel, P.S. mTORC2 in the center of cancer metabolic reprogramming. Trends Endocrinol. Metab. 2014, 25, 364–373. [Google Scholar] [CrossRef]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef]
- Jung, C.H.; Jun, C.B.; Ro, S.-H.; Kim, Y.-M.; Otto, N.M.; Cao, J.; Kundu, M.; Kim, D.-H. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol. Biol. Cell 2009, 20, 1992–2003. [Google Scholar] [CrossRef] [PubMed]
- Maes, H.; Rubio, N.; Garg, A.D.; Agostinis, P. Autophagy: Shaping the tumor microenvironment and therapeutic response. Trends Mol. Med. 2013, 19, 428–446. [Google Scholar] [CrossRef] [PubMed]
- Morel, E. Endoplasmic reticulum membrane and contact site dynamics in autophagy regulation and stress response. Front. Cell Dev. Biol. 2020, 8, 343. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Chen, X.; Ji, C.; Zhang, W.; Song, J.; Li, J.; Wang, J. Key Regulators of Autophagosome Closure. Cells 2021, 10, 2814. [Google Scholar] [CrossRef] [PubMed]
- Dikic, I.; Elazar, Z. Mechanism and medical implications of mammalian autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 349–364. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.N.; Padman, B.S.; Usher, J.; Oorschot, V.; Ramm, G.; Lazarou, M. Atg8 family LC3/GABARAP proteins are crucial for autophagosome–lysosome fusion but not autophagosome formation during PINK1/Parkin mitophagy and starvation. J. Cell Biol. 2016, 215, 857–874. [Google Scholar] [CrossRef]
- Weidberg, H.; Shvets, E.; Shpilka, T.; Shimron, F.; Shinder, V.; Elazar, Z. LC3 and GATE-16/GABARAP subfamilies are both essential yet act differently in autophagosome biogenesis. EMBO J. 2010, 29, 1792–1802. [Google Scholar] [CrossRef]
- Li, X.; He, S.; Ma, B. Autophagy and autophagy-related proteins in cancer. Mol. Cancer 2020, 19, 12. [Google Scholar] [CrossRef]
- Towers, C.G.; Wodetzki, D.; Thorburn, A. Autophagy and cancer: Modulation of cell death pathways and cancer cell adaptations. J. Cell Biol. 2019, 219, e201909033. [Google Scholar] [CrossRef]
- Kisen, G.O.; Tessitore, L.; Costelli, P.; Gordon, P.B.; Schwarze, P.E.; Baccino, F.M.; Seglen, P.O. Reduced autophagic activity in primary rat hepatocellular carcinoma and ascites hepatoma cells. Carcinogenesis 1993, 14, 2501–2505. [Google Scholar] [CrossRef]
- Ahmadi-Dehlaghi, F.; Mohammadi, P.; Valipour, E.; Pournaghi, P.; Kiani, S.; Mansouri, K. Autophagy: A challengeable paradox in cancer treatment. Cancer Med. 2023, 12, 11542–11569. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.S.; Vats, S.; Chia, A.Y.; Tan, T.Z.; Deng, S.; Ong, M.S.; Arfuso, F.; Yap, C.T.; Goh, B.C.; Sethi, G.; et al. Dual role of autophagy in hallmarks of cancer. Oncogene 2018, 37, 1142–1158. [Google Scholar] [CrossRef] [PubMed]
- Patergnani, S.; Missiroli, S.; Morciano, G.; Perrone, M.; Mantovani, C.M.; Anania, G.; Fiorica, F.; Pinton, P.; Giorgi, C. Understanding the Role of Autophagy in Cancer Formation and Progression Is a Real Opportunity to Treat and Cure Human Cancers. Cancers 2021, 13, 5622. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Ren, X.; Hait, W.N.; Yang, J.M. Therapeutic targeting of autophagy in disease: Biology and pharmacology. Pharmacol. Rev. 2013, 65, 1162–1197. [Google Scholar] [CrossRef] [PubMed]
- White, E.; DiPaola, R.S. The double-edged sword of autophagy modulation in cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2009, 15, 5308–5316. [Google Scholar] [CrossRef] [PubMed]
- Ávalos, Y.; Canales, J.; Bravo-Sagua, R.; Criollo, A.; Lavandero, S.; Quest, A.F. Tumor suppression and promotion by autophagy. BioMed Res. Int. 2014, 2014, 603980. [Google Scholar] [CrossRef]
- Maehama, T. PTEN: Its deregulation and tumorigenesis. Biol. Pharm. Bull. 2007, 30, 1624–1627. [Google Scholar] [CrossRef]
- Rahman, M.A.; Park, M.N.; Rahman, M.H.; Rashid, M.M.; Islam, R.; Uddin, M.J.; Hannan, M.A.; Kim, B. p53 Modulation of Autophagy Signaling in Cancer Therapies: Perspectives Mechanism and Therapeutic Targets. Front. Cell Dev. Biol. 2022, 10, 761080. [Google Scholar] [CrossRef]
- Tasdemir, E.; Chiara Maiuri, M.; Morselli, E.; Criollo, A.; D’Amelio, M.; Djavaheri-Mergny, M.; Cecconi, F.; Tavernarakis, N.; Kroemer, G. A dual role of p53 in the control of autophagy. Autophagy 2008, 4, 810–814. [Google Scholar] [CrossRef]
- Porta, C.; Paglino, C.; Mosca, A. Targeting PI3K/Akt/mTOR Signaling in Cancer. Front. Oncol. 2014, 4, 64. [Google Scholar] [CrossRef]
- Shaw, R.J.; Cantley, L.C. Ras, PI(3)K and mTOR signalling controls tumour cell growth. Nature 2006, 441, 424–430. [Google Scholar] [CrossRef] [PubMed]
- Warren, C.F.A.; Wong-Brown, M.W.; Bowden, N.A. BCL-2 family isoforms in apoptosis and cancer. Cell Death Dis. 2019, 10, 177. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.; Quinn, B.A.; Das, S.K.; Dash, R.; Emdad, L.; Dasgupta, S.; Wang, X.Y.; Dent, P.; Reed, J.C.; Pellecchia, M.; et al. Targeting the Bcl-2 family for cancer therapy. Expert Opin. Ther. Targets 2013, 17, 61–75. [Google Scholar] [CrossRef] [PubMed]
- Vega-Rubín-de-Celis, S. The Role of Beclin 1-Dependent Autophagy in Cancer. Biology 2019, 9, 4. [Google Scholar] [CrossRef] [PubMed]
- Qiu, D.M.; Wang, G.L.; Chen, L.; Xu, Y.Y.; He, S.; Cao, X.L.; Qin, J.; Zhou, J.M.; Zhang, Y.X.; Qun, E. The expression of beclin-1, an autophagic gene, in hepatocellular carcinoma associated with clinical pathological and prognostic significance. BMC Cancer 2014, 14, 327. [Google Scholar] [CrossRef]
- Huang, X.; Bai, H.M.; Chen, L.; Li, B.; Lu, Y.C. Reduced expression of LC3B-II and Beclin 1 in glioblastoma multiforme indicates a down-regulated autophagic capacity that relates to the progression of astrocytic tumors. J. Clin. Neurosci. Off. J. Neurosurg. Soc. Australas. 2010, 17, 1515–1519. [Google Scholar] [CrossRef]
- Takahashi, Y.; Coppola, D.; Matsushita, N.; Cualing, H.D.; Sun, M.; Sato, Y.; Liang, C.; Jung, J.U.; Cheng, J.Q.; Mulé, J.J.; et al. Bif-1 interacts with Beclin 1 through UVRAG and regulates autophagy and tumorigenesis. Nat. Cell Biol. 2007, 9, 1142–1151. [Google Scholar] [CrossRef]
- Lim, S.M.; Mohamad Hanif, E.A.; Chin, S.F. Is targeting autophagy mechanism in cancer a good approach? The possible double-edge sword effect. Cell Biosci. 2021, 11, 56. [Google Scholar] [CrossRef]
- Takamura, A.; Komatsu, M.; Hara, T.; Sakamoto, A.; Kishi, C.; Waguri, S.; Eishi, Y.; Hino, O.; Tanaka, K.; Mizushima, N. Autophagy-deficient mice develop multiple liver tumors. Genes Dev. 2011, 25, 795–800. [Google Scholar] [CrossRef]
- Scherz-Shouval, R.; Shvets, E.; Fass, E.; Shorer, H.; Gil, L.; Elazar, Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J. 2007, 26, 1749–1760. [Google Scholar] [CrossRef]
- Yue, C.; Yang, X.; Li, J.; Chen, X.; Zhao, X.; Chen, Y.; Wen, Y. Trimethylamine N- oxide prime NLRP3 inflammasome via inhibiting ATG16L1-induced autophagy in colonic epithelial cells. Biochem. Biophys. Res. Commun. 2017, 490, 541–551. [Google Scholar] [CrossRef] [PubMed]
- Majidpoor, J.; Mortezaee, K. Steps in metastasis: An updated review. Med. Oncol. 2021, 38, 3. [Google Scholar] [CrossRef] [PubMed]
- Kenific, C.M.; Thorburn, A.; Debnath, J. Autophagy and metastasis: Another double-edged sword. Curr. Opin. Cell Biol. 2010, 22, 241–245. [Google Scholar] [CrossRef] [PubMed]
- Skrypek, N.; Goossens, S.; De Smedt, E.; Vandamme, N.; Berx, G. Epithelial-to-mesenchymal transition: Epigenetic reprogramming driving cellular plasticity. Trends Genet. 2017, 33, 943–959. [Google Scholar] [CrossRef]
- Polyak, K.; Weinberg, R.A. Transitions between epithelial and mesenchymal states: Acquisition of malignant and stem cell traits. Nat. Rev. Cancer 2009, 9, 265–273. [Google Scholar] [CrossRef]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef]
- Li, J.; Yang, B.; Zhou, Q.; Wu, Y.; Shang, D.; Guo, Y.; Song, Z.; Zheng, Q.; Xiong, J. Autophagy promotes hepatocellular carcinoma cell invasion through activation of epithelial-mesenchymal transition. Carcinogenesis 2013, 34, 1343–1351. [Google Scholar] [CrossRef]
- Catalano, M.; D’Alessandro, G.; Lepore, F.; Corazzari, M.; Caldarola, S.; Valacca, C.; Faienza, F.; Esposito, V.; Limatola, C.; Cecconi, F.; et al. Autophagy induction impairs migration and invasion by reversing EMT in glioblastoma cells. Mol. Oncol. 2015, 9, 1612–1625. [Google Scholar] [CrossRef]
- Zhu, H.; Wang, D.; Zhang, L.; Xie, X.; Wu, Y.; Liu, Y.; Shao, G.; Su, Z. Upregulation of autophagy by hypoxia-inducible factor-1α promotes EMT and metastatic ability of CD133+ pancreatic cancer stem-like cells during intermittent hypoxia. Oncol. Rep. 2014, 32, 935–942. [Google Scholar] [CrossRef]
- Macintosh, R.L.; Timpson, P.; Thorburn, J.; Anderson, K.I.; Thorburn, A.; Ryan, K.M. Inhibition of autophagy impairs tumor cell invasion in an organotypic model. Cell Cycle 2012, 11, 2022–2029. [Google Scholar] [CrossRef]
- Tong, H.; Yin, H.; Hossain, M.A.; Wang, Y.; Wu, F.; Dong, X.; Gao, S.; Zhan, K.; He, W. Starvation- induced autophagy promotes the invasion and migration of human bladder cancer cells via TGF-β1/Smad3-mediated epithelial-mesenchymal transition activation. J. Cell. Biochem. 2019, 120, 5118–5127. [Google Scholar] [CrossRef] [PubMed]
- Lock, R.; Kenific, C.M.; Leidal, A.M.; Salas, E.; Debnath, J. Autophagy-dependent production of secreted factors facilitates oncogenic RAS-driven invasion. Cancer Discov. 2014, 4, 466–479. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.T.; Liu, H.; Mao, M.J.; Tan, Y.; Mo, X.Q.; Meng, X.J.; Cao, M.T.; Zhong, C.Y.; Liu, Y.; Shan, H.; et al. Crosstalk between autophagy and epithelial-mesenchymal transition and its application in cancer therapy. Mol. Cancer 2019, 18, 101. [Google Scholar] [CrossRef] [PubMed]
- Ge, Y.; Huang, M.; Yao, Y.M. Autophagy and proinflammatory cytokines: Interactions and clinical implications. Cytokine Growth Factor Rev. 2018, 43, 38–46. [Google Scholar] [CrossRef]
- Harris, J. Autophagy and cytokines. Cytokine 2011, 56, 140–144. [Google Scholar] [CrossRef]
- Lei, L.; Sun, J.; Han, J.; Jiang, X.; Wang, Z.; Chen, L. Interleukin-17 induces pyroptosis in osteoblasts through the NLRP3 inflammasome pathway in vitro. Int. Immunopharmacol. 2021, 96, 107781. [Google Scholar] [CrossRef]
- Parikh, A.A.; Salzman, A.L.; Kane, C.D.; Fischer, J.E.; Hasselgren, P.O. IL-6 production in human intestinal epithelial cells following stimulation with IL-1β is associated with activation of the transcription factor NF-κB. J. Surg. Res. 1997, 69, 139–144. [Google Scholar] [CrossRef]
- Ngabire, D.; Kim, G.D. Autophagy and Inflammatory Response in the Tumor Microenvironment. Int. J. Mol. Sci. 2017, 18, 2016. [Google Scholar] [CrossRef]
- Yang, X.; Yu, D.D.; Yan, F.; Jing, Y.Y.; Han, Z.P.; Sun, K.; Liang, L.; Hou, J.; Wei, L.X. The role of autophagy induced by tumor microenvironment in different cells and stages of cancer. Cell Biosci. 2015, 5, 14. [Google Scholar] [CrossRef]
- Allam, A.; Yakou, M.; Pang, L.; Ernst, M.; Huynh, J. Exploiting the STAT3 Nexus in Cancer-Associated Fibroblasts to Improve Cancer Therapy. Front. Immunol. 2021, 12, 767939. [Google Scholar] [CrossRef]
- Martinez-Outschoorn, U.E.; Lin, Z.; Trimmer, C.; Flomenberg, N.; Wang, C.; Pavlides, S.; Pestell, R.G.; Howell, A.; Sotgia, F.; Lisanti, M.P. Cancer cells metabolically “fertilize” the tumor microenvironment with hydrogen peroxide, driving the Warburg effect: Implications for PET imaging of human tumors. Cell Cycle 2011, 10, 2504–2520. [Google Scholar] [CrossRef]
- Marino, M.L.; Pellegrini, P.; Di Lernia, G.; Djavaheri-Mergny, M.; Brnjic, S.; Zhang, X.; Hägg, M.; Linder, S.; Fais, S.; Codogno, P.; et al. Autophagy Is a Protective Mechanism for Human Melanoma Cells under Acidic Stress. J. Biol. Chem. 2012, 287, 30664–30676. [Google Scholar] [CrossRef]
- Kroemer, G.; Mariño, G.; Levine, B. Autophagy and the integrated stress response. Mol. Cell 2010, 40, 280–293. [Google Scholar] [CrossRef] [PubMed]
- Luo, N. Editorial: Tumor microenvironment in cancer hallmarks and therapeutics. Front. Mol. Biosci. 2022, 9, 1019830. [Google Scholar] [CrossRef] [PubMed]
- Anderson, N.M.; Simon, M.C. The tumor microenvironment. Curr. Biol. 2020, 30, R921–R925. [Google Scholar] [CrossRef]
- Whiteside, T.L. The tumor microenvironment and its role in promoting tumor growth. Oncogene 2008, 27, 5904–5912. [Google Scholar] [CrossRef]
- Cicchese, J.M.; Evans, S.; Hult, C.; Joslyn, L.R.; Wessler, T.; Millar, J.A.; Marino, S.; Cilfone, N.A.; Mattila, J.T.; Linderman, J.J.; et al. Dynamic balance of pro-andanti-inflammatory signals controls disease and limits pathology. Immunol. Rev. 2018, 285, 147–167. [Google Scholar] [CrossRef] [PubMed]
- Disis, M.L. Immune regulation of cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2010, 28, 4531–4538. [Google Scholar] [CrossRef]
- Lavin, Y.; Mortha, A.; Rahman, A.; Merad, M. Regulation of macrophage development and function in peripheral tissues. Nat. Rev. Immunol. 2015, 15, 731–744. [Google Scholar] [CrossRef]
- Gonzalez, H.; Robles, I.; Werb, Z. Innate and acquired immune surveillance in the post dissemination phase of metastasis. FEBS J. 2018, 285, 654–664. [Google Scholar] [CrossRef]
- Noy, R.; Pollard, J.W. Tumor-associated macrophages: From mechanisms to therapy. Immunity 2014, 41, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Arora, S.; Dev, K.; Agarwal, B.; Das, P.; Syed, M.A. Macrophages: Their role, activation and polarization in pulmonary diseases. Immunobiology 2018, 223, 383–396. [Google Scholar] [CrossRef] [PubMed]
- Galli, F.; Aguilera, J.V.; Palermo, B.; Markovic, S.N.; Nisticò, P.; Signore, A. Relevance of immune cell and tumor microenvironment imaging in the new era of immunotherapy. J. Exp. Clin. Cancer Res. 2020, 39, 89. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, H.; Hagerling, C.; Werb, Z. Roles of the immune system in cancer: From tumor initiation to metastatic progression. Genes Dev. 2018, 32, 1267–1284. [Google Scholar] [CrossRef]
- Ostrand-Rosenberg, S.; Sinha, P.; Beury, D.W.; Clements, V.K. Cross-talk between myeloid-derived suppressor cells (MDSC), macrophages, and dendritic cells enhances tumor- induced immune suppression. Semin. Cancer Biol. 2012, 22, 275–281. [Google Scholar] [CrossRef]
- Sun, Q.; Fan, J.; Billiar, T.R.; Scott, M.J. Inflammasome and autophagy regulation—A two-way street. Mol. Med. 2017, 23, 188–195. [Google Scholar] [CrossRef]
- Yaseen, M.M.; Abuharfeil, N.M.; Darmani, H.; Daoud, A. Mechanisms of immune suppression by myeloid-derived suppressor cells: The role of interleukin-10 as a key immunoregulatory cytokine. Open Biol. 2020, 10, 200111. [Google Scholar] [CrossRef]
- Bi, Y.; Chen, J.; Hu, F.; Liu, J.; Li, M.; Zhao, L. M2 Macrophages as a Potential Target for Antiatherosclerosis Treatment. Neural Plast. 2019, 2019, 6724903. [Google Scholar] [CrossRef]
- Romano, M.; Fanelli, G.; Albany, C.J.; Giganti, G.; Lombardi, G. Past, Present, and Future of Regulatory T Cell Therapy in Transplantation and Autoimmunity. Front. Immunol. 2019, 10, 43. [Google Scholar] [CrossRef]
- O’Sullivan, C.; Lewis, C.E.; Harris, A.L.; McGee, J.O. Secretion of epidermal growth factor by macrophages associated with breast carcinoma. Lancet 1993, 342, 148–149. [Google Scholar] [CrossRef]
- Shojaei, F.; Zhong, C.; Wu, X.; Yu, L.; Ferrara, N. Role of myeloid cells in tumor angiogenesis and growth. Trends Cell Biol. 2008, 18, 372–378. [Google Scholar] [CrossRef] [PubMed]
- Kessenbrock, K.; Plaks, V.; Werb, Z. Matrix metalloproteinases: Regulators of the tumor microenvironment. Cell 2010, 141, 52–67. [Google Scholar] [CrossRef] [PubMed]
- Abraham, D.; Zins, K.; Sioud, M.; Lucas, T.; Schäfer, R.; Stanley, E.R.; Aharinejad, S. Stromal cell-derived CSF-1 blockade prolongs xenograft survival of CSF-1-negative neuroblastoma. Int. J. Cancer 2010, 126, 1339–1352. [Google Scholar] [CrossRef] [PubMed]
- Qian, B.Z.; Li, J.; Zhang, H.; Kitamura, T.; Zhang, J.; Campion, L.R.; Kaiser, E.A.; Snyder, L.A.; Pollard, J.W. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature 2011, 475, 222–225. [Google Scholar] [CrossRef]
- Cho, K.S.; Yoon, Y.H.; Choi, J.A.; Lee, S.J.; Koh, J.Y. Induction of autophagy and cell death by tamoxifen in cultured retinal pigment epithelial and photoreceptor cells. Investig. Ophthalmol. Vis. Sci. 2012, 53, 5344–5353. [Google Scholar] [CrossRef]
- Deci, M.B.; Ferguson, S.W.; Scatigno, S.L.; Nguyen, J. Modulating Macrophage Polarization through CCR2 Inhibition and Multivalent Engagement. Mol. Pharm. 2018, 15, 2721–2731. [Google Scholar] [CrossRef]
- Wisitpongpun, P.; Potup, P.; Usuwanthim, K. Oleamide-Mediated Polarization of M1 Macrophages and IL-1β Production by Regulating NLRP3-Inflammasome Activation in Primary Human Monocyte-Derived Macrophages. Front. Immunol. 2022, 13, 856296. [Google Scholar] [CrossRef]
- Akhtari, M.; Zargar, S.J.; Vojdanian, M.; Jamshidi, A.; Mahmoudi, M. Monocyte- derived and M1 macrophages from ankylosing spondylitis patients released higher TNF-α and expressed more IL1B in response to BzATP than macrophages from healthy subjects. Sci. Rep. 2021, 11, 17842. [Google Scholar] [CrossRef]
- Chen, X.W.; Zhou, S.F. Inflammation, cytokines, the IL-17/IL-6/STAT3/NF-κB axis, and tumorigenesis. Drug Des. Dev. Ther. 2015, 9, 2941–2946. [Google Scholar] [CrossRef]
- Schinocca, C.; Rizzo, C.; Fasano, S.; Grasso, G.; La Barbera, L.; Ciccia, F.; Guggino, G. Role of the IL-23/IL-17 Pathway in Rheumatic Diseases: An Overview. Front. Immunol. 2021, 12, 637829. [Google Scholar] [CrossRef]
- Boutilier, A.J.; Elsawa, S.F. Macrophage Polarization States in the Tumor Microenvironment. Int. J. Mol. Sci. 2021, 22, 6995. [Google Scholar] [CrossRef] [PubMed]
- Jeannin, P.; Paolini, L.; Adam, C.; Delneste, Y. The roles of CSFs on the functional polarization of tumor-associated macrophages. FEBS J. 2018, 285, 680–699. [Google Scholar] [CrossRef] [PubMed]
- Hirano, T. IL-6 in inflammation, autoimmunity and cancer. Int. Immunol. 2021, 33, 127–148. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Sun, X.; Pan, B.; Cao, S.; Cao, J.; Che, D.; Liu, F.; Zhang, S.; Yu, Y. IL-17 induces macrophages to M2-like phenotype via NF-κB. Cancer Manag. Res. 2018, 10, 4217–4228. [Google Scholar] [CrossRef]
- Guo, Y.; Chang, C.; Huang, R.; Liu, B.; Bao, L.; Liu, W. AP1 is essential for generation of autophagosomes from the trans-Golgi network. J. Cell Sci. 2012, 125, 1706–1715. [Google Scholar] [CrossRef]
- Trocoli, A.; Djavaheri-Mergny, M. The complex interplay between autophagy and NF-κB signaling pathways in cancer cells. Am. J. Cancer Res. 2011, 1, 629–649. [Google Scholar]
- Vatner, R.E.; Janssen, E.M. STING, DCs and the link between innate and adaptive tumor immunity. Mol. Immunol. 2019, 110, 13–23. [Google Scholar] [CrossRef]
- Ing, R.; Stevenson, M.M. Dendritic cell and NK cell reciprocal cross talk promotes gamma interferon-dependent immunity to blood-stage Plasmodium chabaudi AS infection in mice. Infect. Immun. 2009, 77, 770–782. [Google Scholar] [CrossRef]
- Wculek, S.K.; Cueto, F.J.; Mujal, A.M.; Melero, I.; Krummel, M.F.; Sancho, D. Dendritic cells in cancer immunology and immunotherapy. Nat. Rev. Immunol. 2020, 20, 7–24. [Google Scholar] [CrossRef]
- Heufler, C.; Koch, F.; Stanzl, U.; Topar, G.; Wysocka, M.; Trinchieri, G.; Enk, A.; Steinman, R.M.; Romani, N.; Schuler, G. Interleukin-12 is produced by dendritic cells and mediates T helper 1 development as well as interferon-gamma production by T helper 1 cells. Eur. J. Immunol. 1996, 26, 659–668. [Google Scholar] [CrossRef]
- Lin, Y.; Kuang, W.; Wu, B.; Xie, C.; Liu, C.; Tu, Z. IL-12 induces autophagy in human breast cancer cells through AMPK and the PI3K/Akt pathway. Mol. Med. Rep. 2017, 16, 4113–4118. [Google Scholar] [CrossRef] [PubMed]
- Cerwenka, A.; Lanier, L.L. Natural killer cell memory in infection, inflammation and cancer. Nature reviews. Immunology 2016, 16, 112–123. [Google Scholar] [CrossRef] [PubMed]
- Zwirner, N.W.; Ziblat, A. Regulation of NK Cell Activation and Effector Functions by the IL-12 Family of Cytokines: The Case of IL-27. Front. Immunol. 2017, 8, 25. [Google Scholar] [CrossRef] [PubMed]
- Ross, M.E.; Caligiuri, M.A. Cytokine-induced apoptosis of human natural killer cells identifies a novel mechanism to regulate the innate immune response. Blood 1997, 89, 910–918. [Google Scholar] [CrossRef] [PubMed]
- Rosales, C. Neutrophil: A Cell with Many Roles in Inflammation or Several Cell Types? Front. Physiol. 2018, 9, 113. [Google Scholar] [CrossRef]
- SenGupta, S.; Hein, L.E.; Parent, C.A. The Recruitment of Neutrophils to the Tumor Microenvironment Is Regulated by Multiple Mediators. Front. Immunol. 2021, 12, 734188. [Google Scholar] [CrossRef]
- Galdiero, M.R.; Varricchi, G.; Loffredo, S.; Mantovani, A.; Marone, G. Roles of neutrophils in cancer growth and progression. J. Leukoc. Biol. 2018, 103, 457–464. [Google Scholar] [CrossRef]
- Wu, Y.; Yi, M.; Niu, M.; Mei, Q.; Wu, K. Myeloid-derived suppressor cells: An emerging target for anticancer immunotherapy. Mol. Cancer 2022, 21, 184. [Google Scholar] [CrossRef]
- Wu, M.Y.; Lu, J.H. Autophagy and Macrophage Functions: Inflammatory Response and Phagocytosis. Cells 2019, 9, 70. [Google Scholar] [CrossRef]
- Speiser, D.E.; Ho, P.C.; Verdeil, G. Regulatory circuits of T cell function in cancer. Nat. Rev. Immunol. 2016, 16, 599–611. [Google Scholar] [CrossRef]
- Fridman, W.H.; Pagès, F.; Sautès-Fridman, C.; Galon, J. The immune contexture in human tumours: Impact on clinical outcome. Nat. Rev. Cancer 2012, 12, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, inflammation, and cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef] [PubMed]
- Ivashkiv, L.B. IFNγ: Signalling, epigenetics and roles in immunity, metabolism, disease and cancer immunotherapy. Nat. Rev. Immunol. 2018, 18, 545–558. [Google Scholar] [CrossRef] [PubMed]
- Bhat, P.; Leggatt, G.; Waterhouse, N.; Frazer, I.H. Interferon-γ derived from cytotoxic lymphocytes directly enhances their motility and cytotoxicity. Cell Death Dis. 2017, 8, e2836. [Google Scholar] [CrossRef]
- Phares, T.W.; Stohlman, S.A.; Hwang, M.; Min, B.; Hinton, D.R.; Bergmann, C.C. CD4 T cells promote CD8 T cell immunity at the priming and effector site during viral encephalitis. J. Virol. 2012, 86, 2416–2427. [Google Scholar] [CrossRef]
- Millington, K.A.; Innes, J.A.; Hackforth, S.; Hinks, T.S.; Deeks, J.J.; Dosanjh, D.P.; Guyot- Revol, V.; Gunatheesan, R.; Klenerman, P.; Lalvani, A. Dynamic relationship between IFN-gamma and IL-2 profile of Mycobacterium tuberculosis-specific T cells and antigen load. J. Immunol. 2007, 178, 5217–5226. [Google Scholar] [CrossRef]
- Egwuagu, C.E. STAT3 in CD4+ T helper cell differentiation and inflammatory diseases. Cytokine 2009, 47, 149–156. [Google Scholar] [CrossRef]
- Kondĕlková, K.; Vokurková, D.; Krejsek, J.; Borská, L.; Fiala, Z.; Ctirad, A. Regulatory T cells (TREG) and their roles in immune system with respect to immunopathological disorders. Acta Medica 2010, 53, 73–77. [Google Scholar] [CrossRef]
- Lillo, S.; Saleh, M. Inflammasomes in Cancer Progression and Anti-Tumor Immunity. Front. Cell Dev. Biol. 2022, 10, 839041. [Google Scholar] [CrossRef]
- Missiroli, S.; Perrone, M.; Boncompagni, C.; Borghi, C.; Campagnaro, A.; Marchetti, F.; Anania, G.; Greco, P.; Fiorica, F.; Pinton, P.; et al. Targeting the NLRP3 Inflammasome as a New Therapeutic Option for Overcoming Cancer. Cancers 2021, 13, 2297. [Google Scholar] [CrossRef]
- Bent, R.; Moll, L.; Grabbe, S.; Bros, M. Interleukin-1 Beta-A Friend or Foe in Malignancies? Int. J. Mol. Sci. 2018, 19, 2155. [Google Scholar] [CrossRef]
- Zhang, J.; Veeramachaneni, N. Targeting interleukin-1β and inflammation in lung cancer. Biomark. Res. 2022, 10, 5. [Google Scholar] [CrossRef]
- Brunetto, E.; De Monte, L.; Balzano, G.; Camisa, B.; Laino, V.; Riba, M.; Heltai, S.; Bianchi, M.; Bordignon, C.; Falconi, M.; et al. The IL-1/IL-1 receptor axis and tumor cell released inflammasome adaptor ASC are key regulators of TSLP secretion by cancer associated fibroblasts in pancreatic cancer. J. Immunother. Cancer 2019, 7, 45. [Google Scholar] [CrossRef]
- Daley, D.; Mani, V.R.; Mohan, N.; Akkad, N.; Pandian, G.S.D.B.; Savadkar, S.; Lee, K.B.; Torres-Hernandez, A.; Aykut, B.; Diskin, B.; et al. NLRP3 signaling drives macrophage-induced adaptive immune suppression in pancreatic carcinoma. J. Exp. Med. 2017, 214, 1711–1724. [Google Scholar] [CrossRef]
- Tengesdal, I.W.; Dinarello, A.; Powers, N.E.; Burchill, M.A.; Joosten, L.A.B.; Marchetti, C.; Dinarello, C.A. Tumor NLRP3-Derived IL-1β Drives the IL-6/STAT3 Axis Resulting in Sustained MDSC-Mediated Immunosuppression. Front. Immunol. 2021, 12, 661323. [Google Scholar] [CrossRef]
- Hamarsheh, S.; Zeiser, R. NLRP3 Inflammasome Activation in Cancer: A Double-Edged Sword. Front. Immunol. 2020, 11, 1444. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.E.; Lee, J.Y.; Yang, G.; Kang, H.C.; Cho, Y.Y.; Lee, H.S.; Lee, J.Y. Inhibition of NLRP3 inflammasome in tumor microenvironment leads to suppression of metastatic potential of cancer cells. Sci. Rep. 2019, 9, 12277. [Google Scholar] [CrossRef]
- Kiss, M.; Vande Walle, L.; Saavedra, P.H.V.; Lebegge, E.; Van Damme, H.; Murgaski, A.; Qian, J.; Ehling, M.; Pretto, S.; Bolli, E.; et al. IL1β Promotes Immune Suppression in the Tumor Microenvironment Independent of the Inflammasome and Gasdermin D. Cancer Immunol. Res. 2021, 9, 309–323. [Google Scholar] [CrossRef] [PubMed]
- Yano, S.; Nokihara, H.; Yamamoto, A.; Goto, H.; Ogawa, H.; Kanematsu, T.; Miki, T.; Uehara, H.; Saijo, Y.; Nukiwa, T.; et al. Multifunctional interleukin-1beta promotes metastasis of human lung cancer cells in SCID mice via enhanced expression of adhesion-, invasion- and angiogenesis-related molecules. Cancer Sci. 2003, 94, 244–252. [Google Scholar] [CrossRef]
- Huang, L.; Duan, S.; Shao, H.; Zhang, A.; Chen, S.; Zhang, P.; Wang, N.; Wang, W.; Wu, Y.; Wang, J.; et al. NLRP3 deletion inhibits inflammation-driven mouse lung tumorigenesis induced by benzo(a)pyrene and lipopolysaccharide. Respir. Res. 2019, 20, 20. [Google Scholar] [CrossRef] [PubMed]
- Boaru, S.G.; Borkham-Kamphorst, E.; Van de Leur, E.; Lehnen, E.; Liedtke, C.; Weiskirchen, R. NLRP3 inflammasome expression is driven by NF-κB in cultured hepatocytes. Biochem. Biophys. Res. Commun. 2015, 458, 700–706. [Google Scholar] [CrossRef]
- Yu, H.; Lin, L.; Zhang, Z.; Zhang, H.; Hu, H. Targeting NF-κB pathway for the therapy of diseases: Mechanism and clinical study. Signal Transduct. Target. Ther. 2020, 5, 209. [Google Scholar] [CrossRef] [PubMed]
- Masclaux-Daubresse, C.; Chen, Q.; Havé, M. Regulation of nutrient recycling via autophagy. Curr. Opin. Plant Biol. 2017, 39, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Chung, C.; Seo, W.; Silwal, P.; Jo, E.K. Crosstalks between inflammasome and autophagy in cancer. J. Hematol. Oncol. 2020, 13, 100. [Google Scholar] [CrossRef] [PubMed]
- Saitoh, T.; Fujita, N.; Jang, M.H.; Uematsu, S.; Yang, B.G.; Satoh, T.; Omori, H.; Noda, T.; Yamamoto, N.; Komatsu, M.; et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature 2008, 456, 264–268. [Google Scholar] [CrossRef]
- Biasizzo, M.; Kopitar-Jerala, N. Interplay Between NLRP3 Inflammasome and Autophagy. Front. Immunol. 2020, 11, 591803. [Google Scholar] [CrossRef]
- Jounai, N.; Kobiyama, K.; Shiina, M.; Ogata, K.; Ishii, K.J.; Takeshita, F. NLRP4 negatively regulates autophagic processes through an association with beclin1. J. Immunol. 2011, 186, 1646–1655. [Google Scholar] [CrossRef]
- Xiao, L.; Li, X.; Cao, P.; Fei, W.; Zhou, H.; Tang, N.; Liu, Y. Interleukin-6 mediated inflammasome activation promotes oral squamous cell carcinoma progression via JAK2/STAT3/Sox4/NLRP3 signaling pathway. J. Exp. Clin. Cancer Res. 2022, 41, 166. [Google Scholar] [CrossRef]
- Le Jan, S.; Muller, C.; Plee, J.; Durlach, A.; Bernard, P.; Antonicelli, F. IL-23/IL-17 Axis Activates IL-1β-Associated Inflammasome in Macrophages and Generates an Auto-Inflammatory Response in a Subgroup of Patients with Bullous Pemphigoid. Front. Immunol. 2019, 10, 1972. [Google Scholar] [CrossRef]
- Budai, M.M.; Tőzsér, J.; Benkő, S. Different dynamics of NLRP3 inflammasome- mediated IL-1β production in GM-CSF- and M-CSF-differentiated human macrophages. J. Leukoc. Biol. 2017, 101, 1335–1347. [Google Scholar] [CrossRef]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef]
- Okada, M.; Matsuzawa, A.; Yoshimura, A.; Ichijo, H. The lysosome rupture-activated TAK1-JNK pathway regulates NLRP3 inflammasome activation. J. Biol. Chem. 2014, 289, 32926–32936. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-κB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef] [PubMed]
- Ghonime, M.G.; Shamaa, O.R.; Das, S.; Eldomany, R.A.; Fernandes-Alnemri, T.; Alnemri, E.S.; Gavrilin, M.A.; Wewers, M.D. Inflammasome priming by lipopolysaccharide is dependent upon ERK signaling and proteasome function. J. Immunol. 2014, 192, 3881–3888. [Google Scholar] [CrossRef] [PubMed]
- Wan, P.; Zhang, S.; Ruan, Z.; Liu, X.; Yang, G.; Jia, Y.; Li, Y.; Pan, P.; Wang, W.; Li, G.; et al. AP-1 signaling pathway promotes pro-IL-1β transcription to facilitate NLRP3 inflammasome activation upon influenza A virus infection. Virulence 2022, 13, 502–513. [Google Scholar] [CrossRef] [PubMed]
- Fields, J.; Ghorpade, A. C/EBPβ regulates multiple IL-1β-induced human astrocyte inflammatory genes. J. Neuroinflamm. 2012, 9, 177. [Google Scholar] [CrossRef]
- Kaur, S.; Bansal, Y.; Kumar, R.; Bansal, G. A panoramic review of IL-6: Structure, pathophysiological roles and inhibitors. Bioorg. Med. Chem. 2020, 28, 115327. [Google Scholar] [CrossRef]
- Simpson, R.J.; Hammacher, A.; Smith, D.K.; Matthews, J.M.; Ward, L.D. Interleukin-6: Structure-function relationships. Protein Sci. Publ. Protein Soc. 1997, 6, 929–955. [Google Scholar] [CrossRef]
- Zenobia, C.; Hajishengallis, G. Basic biology and role of interleukin-17 in immunity and inflammation. Periodontology 2000 2015, 69, 142–159. [Google Scholar] [CrossRef]
- Tang, C.; Chen, S.; Qian, H.; Huang, W. Interleukin-23: As a drug target for autoimmune inflammatory diseases. Immunology 2012, 135, 112–124. [Google Scholar] [CrossRef]
- Gabay, C. Interleukin-6 and chronic inflammation. Arthritis Res. Ther. 2006, 8 (Suppl. S2), S3. [Google Scholar] [CrossRef]
- Mills, K.H.G. IL-17 and IL-17-producing cells in protection versus pathology. Nat. Rev. Immunol. 2023, 23, 38–54. [Google Scholar] [CrossRef]
- Hodes, G.E.; Ménard, C.; Russo, S.J. Integrating Interleukin-6 into depression diagnosis and treatment. Neurobiol. Stress 2016, 4, 15–22. [Google Scholar] [CrossRef]
- Alvarez-Meythaler, J.G.; Garcia-Mayea, Y.; Mir, C.; Kondoh, H.; LLeonart, M.E. Autophagy Takes Center Stage as a Possible Cancer Hallmark. Front. Oncol. 2020, 10, 586069. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.J.; Lei, Y.H.; Yao, N.; Wang, C.R.; Hu, N.; Ye, W.C.; Zhang, D.M.; Chen, Z.S. Autophagy and multidrug resistance in cancer. Chin. J. Cancer 2017, 36, 52. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Rauch, J.; Kolch, W. Targeting MAPK Signaling in Cancer: Mechanisms of Drug Resistance and Sensitivity. Int. J. Mol. Sci. 2020, 21, 1102. [Google Scholar] [CrossRef] [PubMed]
- McCubrey, J.A.; Abrams, S.L.; Fitzgerald, T.L.; Cocco, L.; Martelli, A.M.; Montalto, G.; Steelman, L.S. Roles of signaling pathways in drug resistance, cancer initiating cells and cancer progression and metastasis. Adv. Biol. Regul. 2015, 57, 75–101. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Z.; Virshup, D.M. Wnt Signaling and Drug Resistance in Cancer. Mol. Pharmacol. 2020, 97, 72–89. [Google Scholar] [CrossRef]
- Hientz, K.; Mohr, A.; Bhakta-Guha, D.; Effertth, T. The role of p53 in cancer drug resistance and targeted chemotherapy. Oncotarget 2017, 8, 8921–8946. [Google Scholar] [CrossRef]
- Li, Y.Y.; Lam, S.K.; Mak, J.C.; Zheng, C.Y.; Ho, J.C. Erlotinib-induced autophagy in epidermal growth factor receptor mutated non-small cell lung cancer. Lung Cancer 2013, 81, 354–361. [Google Scholar] [CrossRef]
- Wang, J.; Liu, Q.; Zhao, Y.; Fu, J.; Su, J. Tumor Cells Transmit Drug Resistance via Cisplatin-Induced Extracellular Vesicles. Int. J. Mol. Sci. 2023, 24, 12347. [Google Scholar] [CrossRef]
- Chang, H.; Zou, Z. Targeting autophagy to overcome drug resistance: Further developments. J. Hematol. Oncol. 2020, 13, 159. [Google Scholar] [CrossRef]
- Zupanic, A.; Bernstein, H.C.; Heiland, I. Systems biology: Current status and challenges. Cell. Mol. Life Sci. 2020, 77, 379–380. [Google Scholar] [CrossRef] [PubMed]
- Stelling, J.; Sauer, U.; Szallasi, Z.; Doyle, F.J., 3rd; Doyle, J. Robustness of cellular functions. Cell 2004, 118, 675–685. [Google Scholar] [CrossRef]
- Tu, Y.; Rappel, W.J. Adaptation of Living Systems. Annu. Rev. Condens. Matter Phys. 2018, 9, 183–205. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, H.; Chen, X. Drug resistance and combating drug resistance in cancer. Cancer Drug Resist. 2019, 2, 141–160. [Google Scholar] [CrossRef] [PubMed]
- Rocca, A.; Kholodenko, B.N. Can Systems Biology Advance Clinical Precision Oncology? Cancers 2021, 13, 6312. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, A.C.; Mehmood, A.; Wei, D.Q.; Dai, X. Systems Biology Integration and Screening of Reliable Prognostic Markers to Create Synergies in the Control of Lung Cancer Patients. Front. Mol. Biosci. 2020, 7, 47. [Google Scholar] [CrossRef]
- Verzella, D.; Pescatore, A.; Capece, D.; Vecchiotti, D.; Ursini, M.V.; Franzoso, G.; Alesse, E.; Zazzeroni, F. Life, death, and autophagy in cancer: NF-κB turns up everywhere. Cell Death Dis. 2020, 11, 210. [Google Scholar] [CrossRef]
- Housman, G.; Byler, S.; Heerboth, S.; Lapinska, K.; Longacre, M.; Snyder, N.; Sarkar, S. Drug resistance in cancer: An overview. Cancers 2014, 6, 1769–1792. [Google Scholar] [CrossRef]
- Du, W.; Elemento, O. Cancer systems biology: Embracing complexity to develop better anticancer therapeutic strategies. Oncogene 2015, 34, 3215–3225. [Google Scholar] [CrossRef]
- Naylor, S.; Chen, J.Y. Unraveling human complexity and disease with systems biology and personalized medicine. Pers. Med. 2010, 7, 275–289. [Google Scholar] [CrossRef]
- Koutrouli, M.; Karatzas, E.; Paez-Espino, D.; Pavlopoulos, G.A. A Guide to Conquer the Biological Network Era Using Graph Theory. Front. Bioeng. Biotechnol. 2020, 8, 34. [Google Scholar] [CrossRef]
- Charitou, T.; Bryan, K.; Lynn, D.J. Using biological networks to integrate, visualize and analyze genomics data. Genet. Sel. Evol. 2016, 48, 27. [Google Scholar] [CrossRef] [PubMed]
- Milo, R.; Shen-Orr, S.; Itzkovitz, S.; Kashtan, N.; Chklovskii, D.; Alon, U. Network motifs: Simple building blocks of complex networks. Science 2002, 298, 824–827. [Google Scholar] [CrossRef]
- Przulj, N.; Corneil, D.G.; Jurisica, I. Efficient estimation of graphlet frequency distributions in protein-protein interaction networks. Bioinformatics 2006, 22, 974–980. [Google Scholar] [CrossRef]
- Vidal, M.; Cusick, M.E.; Barabási, A.L. Interactome networks and human disease. Cell 2011, 144, 986–998. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.Y.; Mamidipalli, S.; Huan, T. HAPPI: An online database of comprehensive human annotated and predicted protein interactions. BMC Genom. 2009, 10 (Suppl. S1), S16. [Google Scholar] [CrossRef] [PubMed]
- Huttlin, E.L.; Bruckner, R.J.; Navarrete-Perea, J.; Cannon, J.R.; Baltier, K.; Gebreab, F.; Gygi, M.P.; Thornock, A.; Zarraga, G.; Tam, S.; et al. Dual proteome-scale networks reveal cell-specific remodeling of the human interactome. Cell 2021, 184, 3022–3040.e28. [Google Scholar] [CrossRef]
- Olsen, J.; Gerds, T.A.; Seidelin, J.B.; Csillag, C.; Bjerrum, J.T.; Troelsen, J.T.; Nielsen, O.H. Diagnosis of ulcerative colitis before onset of inflammation by multivariate modeling of genome-wide gene expression data. Inflamm. Bowel Dis. 2009, 15, 1032–1038. [Google Scholar] [CrossRef] [PubMed]
- Armingol, E.; Officer, A.; Harismendy, O.; Lewis, N.E. Deciphering cell-cell interactions and communication from gene expression. Nat. Rev. Genet. 2021, 22, 71–88. [Google Scholar] [CrossRef]
- Türei, D.; Valdeolivas, A.; Gul, L.; Palacio-Escat, N.; Klein, M.; Ivanova, O.; Ölbei, M.; Gábor, A.; Theis, F.; Módos, D.; et al. Integrated intra- and intercellular signaling knowledge for multicellular omics analysis. Mol. Syst. Biol. 2021, 17, e9923. [Google Scholar] [CrossRef]
- Thomas, J.P.; Modos, D.; Korcsmaros, T.; Brooks-Warburton, J. Network Biology Approaches to Achieve Precision Medicine in Inflammatory Bowel Disease. Front. Genet. 2021, 12, 760501. [Google Scholar] [CrossRef]
- Anand, S.; Mukherjee, K.; Padmanabhan, P. An insight to flux-balance analysis for biochemical networks. Biotechnol. Genet. Eng. Rev. 2020, 36, 32–55. [Google Scholar] [CrossRef]
- Thiele, I.; Swainston, N.; Fleming, R.M.; Hoppe, A.; Sahoo, S.; Aurich, M.K.; Haraldsdottir, H.; Mo, M.L.; Rolfsson, O.; Stobbe, M.D.; et al. A community-driven global reconstruction of human metabolism. Nat. Biotechnol. 2013, 31, 419–425. [Google Scholar] [CrossRef] [PubMed]
- Zielinski, D.C.; Jamshidi, N.; Corbett, A.J.; Bordbar, A.; Thomas, A.; Palsson, B.O. Systems biology analysis of drivers underlying hallmarks of cancer cell metabolism. Sci. Rep. 2017, 7, 41241. [Google Scholar] [CrossRef] [PubMed]
- Borer, B.; Magnúsdóttir, S. The media composition as a crucial element in high-throughput metabolic network reconstruction. Interface Focus 2023, 13, 20220070. [Google Scholar] [CrossRef]
- Yeh, C.S.; Wang, Z.; Miao, F.; Ma, H.; Kao, C.T.; Hsu, T.S.; Yu, J.H.; Hung, E.T.; Lin, C.C.; Kuan, C.Y.; et al. A novel synthetic-genetic-array-based yeast one-hybrid system for high discovery rate and short processing time. Genome Res. 2019, 29, 1343–1351. [Google Scholar] [CrossRef]
- Aschenbrenner, D.; Quaranta, M.; Banerjee, S.; Ilott, N.; Jansen, J.; Steere, B.; Chen, Y.H.; Ho, S.; Cox, K.; Arancibia-Cárcamo, C.V.; et al. Deconvolution of monocyte responses in inflammatory bowel disease reveals an IL-1 cytokine network that regulates IL-23 in genetic and acquired IL-10 resistance. Gut 2021, 70, 1023–1036. [Google Scholar] [CrossRef]
- Maloy, K.J.; Kullberg, M.C. IL-23 and Th17 cytokines in intestinal homeostasis. Mucosal Immunol. 2008, 1, 339–349. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Xu, J.; Huang, Q.; Han, J.; Duan, L.; Fan, J.; Lv, Z.; Guo, M.; Hu, G.; Chen, L.; et al. The Role of Interleukin-17 in Lung Cancer. Mediat. Inflamm. 2016, 2016, 8494079. [Google Scholar] [CrossRef]
- Cam, C.; Karagoz, B.; Muftuoglu, T.; Bigi, O.; Emirzeoglu, L.; Celik, S.; Ozgun, A.; Tuncel, T.; Top, C. The inflammatory cytokine interleukin-23 is elevated in lung cancer, particularly small cell type. Contemp. Oncol. 2016, 20, 215–219. [Google Scholar] [CrossRef] [PubMed]
- Borowczak, J.; Szczerbowski, K.; Maniewski, M.; Kowalewski, A.; Janiczek-Polewska, M.; Szylberg, A.; Marszałek, A.; Szylberg, Ł. The Role of Inflammatory Cytokines in the Pathogenesis of Colorectal Carcinoma-Recent Findings and Review. Biomedicines 2022, 10, 1670. [Google Scholar] [CrossRef]
- Zhao, F.; Gong, W.; Song, J.; Shen, Z.; Cui, D. The paradoxical role of MDSCs in inflammatory bowel diseases: From bench to bedside. Front. Immunol. 2022, 13, 1021634. [Google Scholar] [CrossRef]
- Cassetta, L.; Bruderek, K.; Skrzeczynska-Moncznik, J.; Osiecka, O.; Hu, X.; Rundgren, I.M.; Lin, A.; Santegoets, K.; Horzum, U.; Godinho-Santos, A.; et al. Differential expansion of circulating human MDSC subsets in patients with cancer, infection and inflammation. J. Immunother. Cancer 2020, 8, e001223. [Google Scholar] [CrossRef]
- Magcwebeba, T.; Dorhoi, A.; du Plessis, N. The Emerging Role of Myeloid-Derived Suppressor Cells in Tuberculosis. Front. Immunol. 2019, 10, 917. [Google Scholar] [CrossRef]
- Hammoud, Z.; Kramer, F. Multilayer networks: Aspects, implementations, and application in biomedicine. Big Data Anal. 2020, 5, 2. [Google Scholar] [CrossRef]
- Csermely, P.; Korcsmáros, T.; Kiss, H.J.; London, G.; Nussinov, R. Structure and dynamics of molecular networks: A novel paradigm of drug discovery: A comprehensive review. Pharmacol. Ther. 2013, 138, 333–408. [Google Scholar] [CrossRef]
- Morel, P.A.; Lee, R.E.C.; Faeder, J.R. Demystifying the cytokine network: Mathematical models point the way. Cytokine 2017, 98, 115–123. [Google Scholar] [CrossRef]
- Sacco, J.J.; Al-Akhrass, H.; Wilson, C.M. Challenges and Strategies in Precision Medicine for Non-Small-Cell Lung Cancer. Curr. Pharm. Des. 2016, 22, 4374–4385. [Google Scholar] [CrossRef] [PubMed]
- Seyed Tabib, N.S.; Madgwick, M.; Sudhakar, P.; Verstockt, B.; Korcsmaros, T.; Vermeire, S. Big data in IBD: Big progress for clinical practice. Gut 2020, 69, 1520–1532. [Google Scholar] [CrossRef]
- Thomas, J.P.; Divekar, D.; Brooks, J.; Watson, A.J.M. Gut Microbes Drive T-Cell Infiltration into Colorectal Cancers and Influence Prognosis. Gastroenterology 2019, 156, 1926–1928. [Google Scholar] [CrossRef] [PubMed]
- Boya, P.; González-Polo, R.A.; Casares, N.; Perfettini, J.L.; Dessen, P.; Larochette, N.; Métivier, D.; Meley, D.; Souquere, S.; Yoshimori, T.; et al. Inhibition of macroautophagy triggers apoptosis. Mol. Cell. Biol. 2005, 25, 1025–1040. [Google Scholar] [CrossRef] [PubMed]
- Petherick, K.J.; Conway, O.J.; Mpamhanga, C.; Osborne, S.A.; Kamal, A.; Saxty, B.; Ganley, I.G. Pharmacological inhibition of ULK1 kinase blocks mammalian target of rapamycin (mTOR)-dependent autophagy. J. Biol. Chem. 2015, 290, 11376–11383. [Google Scholar] [CrossRef]
- Moon, E.K.; Kim, S.H.; Hong, Y.; Chung, D.I.; Goo, Y.K.; Kong, H.H. Autophagy inhibitors as a potential antiamoebic treatment for Acanthamoeba keratitis. Antimicrob. Agents Chemother. 2015, 59, 4020–4025. [Google Scholar] [CrossRef]
- Akin, D.; Wang, S.K.; Habibzadegah-Tari, P.; Law, B.; Ostrov, D.; Li, M.; Yin, X.M.; Kim, J.S.; Horenstein, N.; Dunn, W.A., Jr. A novel ATG4B antagonist inhibits autophagy and has a negative impact on osteosarcoma tumors. Autophagy 2014, 10, 2021–2035. [Google Scholar] [CrossRef]
- Yamamoto, K.; Venida, A.; Perera, R.M.; Kimmelman, A.C. Selective autophagy of MHC-I promotes immune evasion of pancreatic cancer. Autophagy 2020, 16, 1524–1525. [Google Scholar] [CrossRef]
- Mauthe, M.; Orhon, I.; Rocchi, C.; Zhou, X.; Luhr, M.; Hijlkema, K.J.; Coppes, R.P.; Engedal, N.; Mari, M.; Reggiori, F. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy 2018, 14, 1435–1455. [Google Scholar] [CrossRef]
- Chen, J.; Shen, Y.; Wu, B.; Yang, P.; Sun, G.; Liu, X.; Qiang, P.; Gao, Y.; Sha, F.; Li, Z.; et al. CUR5g, a novel autophagy inhibitor, exhibits potent synergistic anticancer effects with cisplatin against non-small-cell lung cancer. Cell Death Discov. 2022, 8, 435. [Google Scholar] [CrossRef]
- Sun, X.; Ma, X.; Li, Q.; Yang, Y.; Xu, X.; Sun, J.; Yu, M.; Cao, K.; Yang, L.; Yang, G.; et al. Anti-cancer effects of fisetin on mammary carcinoma cells via regulation of the PI3K/Akt/mTOR pathway: In vitro and in vivo studies. Int. J. Mol. Med. 2018, 42, 811–820. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chen, Y.; Chen, X.; Liang, Y.; Yang, D.; Dong, J.; Yang, N.; Liang, Z. Angelicin inhibits the malignant behaviours of human cervical cancer potentially via inhibiting autophagy. Exp. Ther. Med. 2019, 18, 3365–3374. [Google Scholar] [CrossRef]
- Xie, Q.; Chen, Y.; Tan, H.; Liu, B.; Zheng, L.L.; Mu, Y. Targeting Autophagy with Natural Compounds in Cancer: A Renewed Perspective from Molecular Mechanisms to Targeted Therapy. Front. Pharmacol. 2021, 12, 748149. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.; Murthy, A. Targeting Autophagy to Treat Cancer: Challenges and Opportunities. Front. Pharmacol. 2020, 11, 590344. [Google Scholar] [CrossRef]
- Jain, V.; Singh, M.P.; Amaravadi, R.K. Recent advances in targeting autophagy in cancer. Trends Pharmacol. Sci. 2023, 44, 290–302. [Google Scholar] [CrossRef] [PubMed]
- Amaravadi, R.K.; Kimmelman, A.C.; Debnath, J. Targeting Autophagy in Cancer: Recent Advances and Future Directions. Cancer Discov. 2019, 9, 1167–1181. [Google Scholar] [CrossRef]
- Levy, J.M.M.; Towers, C.G.; Thorburn, A. Targeting autophagy in cancer. Nat. Rev. Cancer 2017, 17, 528–542. [Google Scholar] [CrossRef]
- Laubenbacher, R.; Hower, V.; Jarrah, A.; Torti, S.V.; Shulaev, V.; Mendes, P.; Torti, F.M.; Akman, S. A systems biology view of cancer. Biochim. Biophys. Acta 2009, 1796, 129–139. [Google Scholar] [CrossRef]
- Archer, T.C.; Fertig, E.J.; Gosline, S.J.; Hafner, M.; Hughes, S.K.; Joughin, B.A.; Meyer, A.S.; Piccolo, S.R.; Shajahan-Haq, A.N. Systems Approaches to Cancer Biology. Cancer Res. 2016, 76, 6774–6777. [Google Scholar] [CrossRef]
- Kreeger, P.K.; Lauffenburger, D.A. Cancer systems biology: A network modeling perspective. Carcinogenesis 2010, 31, 2–8. [Google Scholar] [CrossRef]
- Wang, Y.C.; Chen, B.S. A network-based biomarker approach for molecular investigation and diagnosis of lung cancer. BMC Med. Genom. 2011, 4, 2. [Google Scholar] [CrossRef] [PubMed]
- Vera, J.; Gupta, S.K.; Wolkenhauer, O.; Schuler, G. Envisioning the Application of Systems Biology in Cancer Immunology. In Cancer Immunology; Rezaei, N., Ed.; Springer: Berlin/Heidelberg, Germany, 2015. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khilwani, R.; Singh, S. Systems Biology and Cytokines Potential Role in Lung Cancer Immunotherapy Targeting Autophagic Axis. Biomedicines 2023, 11, 2706. https://doi.org/10.3390/biomedicines11102706
Khilwani R, Singh S. Systems Biology and Cytokines Potential Role in Lung Cancer Immunotherapy Targeting Autophagic Axis. Biomedicines. 2023; 11(10):2706. https://doi.org/10.3390/biomedicines11102706
Chicago/Turabian StyleKhilwani, Riya, and Shailza Singh. 2023. "Systems Biology and Cytokines Potential Role in Lung Cancer Immunotherapy Targeting Autophagic Axis" Biomedicines 11, no. 10: 2706. https://doi.org/10.3390/biomedicines11102706
APA StyleKhilwani, R., & Singh, S. (2023). Systems Biology and Cytokines Potential Role in Lung Cancer Immunotherapy Targeting Autophagic Axis. Biomedicines, 11(10), 2706. https://doi.org/10.3390/biomedicines11102706