
The Potential Role of C-Reactive Protein in Metabolic-Dysfunction-Associated Fatty Liver Disease and Aging
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods
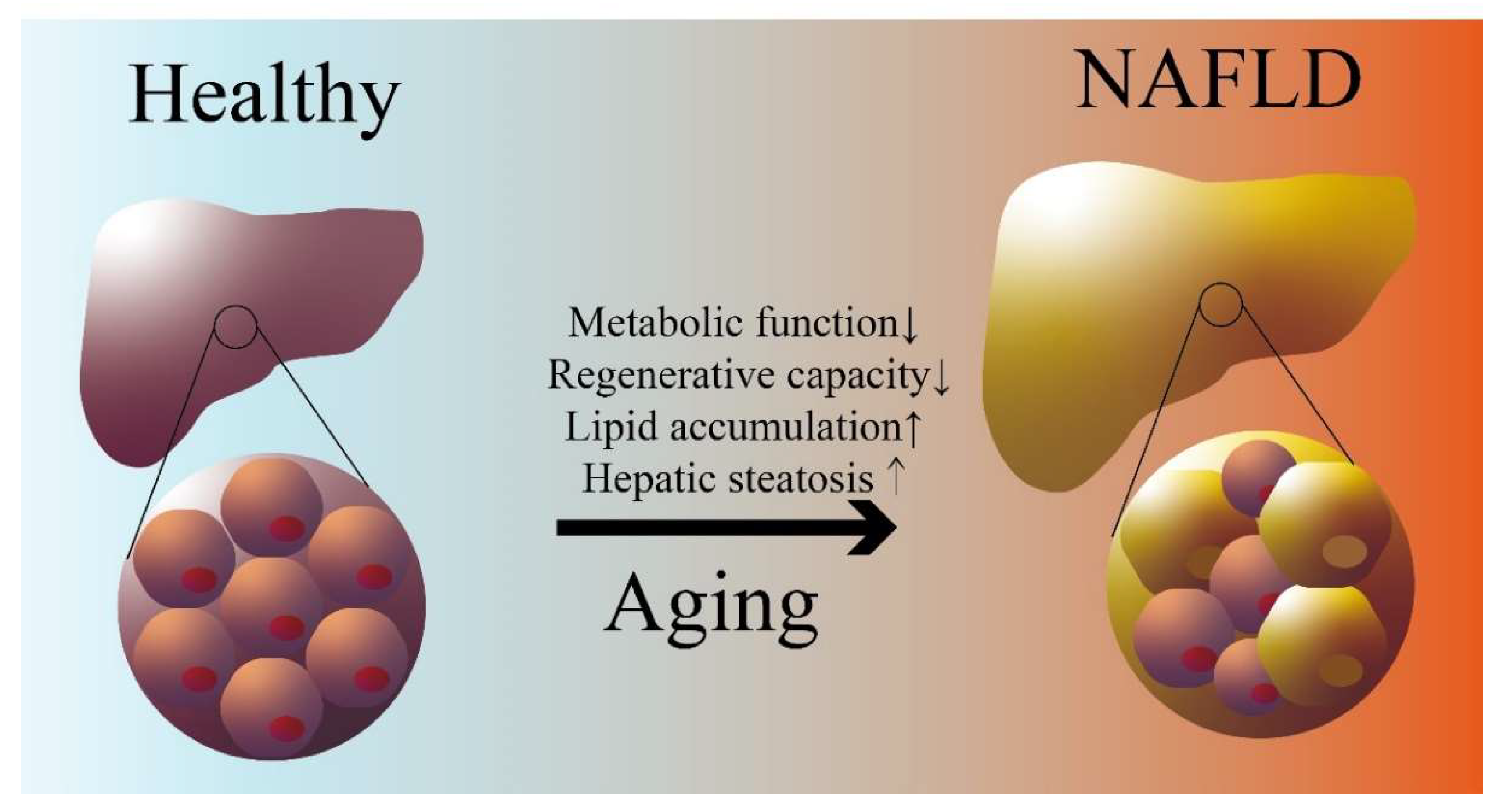
3. Aging Is a Risk Factor for NAFLD/MASLD
3.1. Pathophysiology of NAFLD/MASLD
3.2. Aging-Associated Impaired Lipid Metabolism and NAFLD/MASLD
4. CRP Is a Risk Factor for Both Aging and NAFLD/MASLD
4.1. CRP Definition, Expression Regulation, and Function
4.2. The Link between CRP, NAFLD/MASLD, and Aging
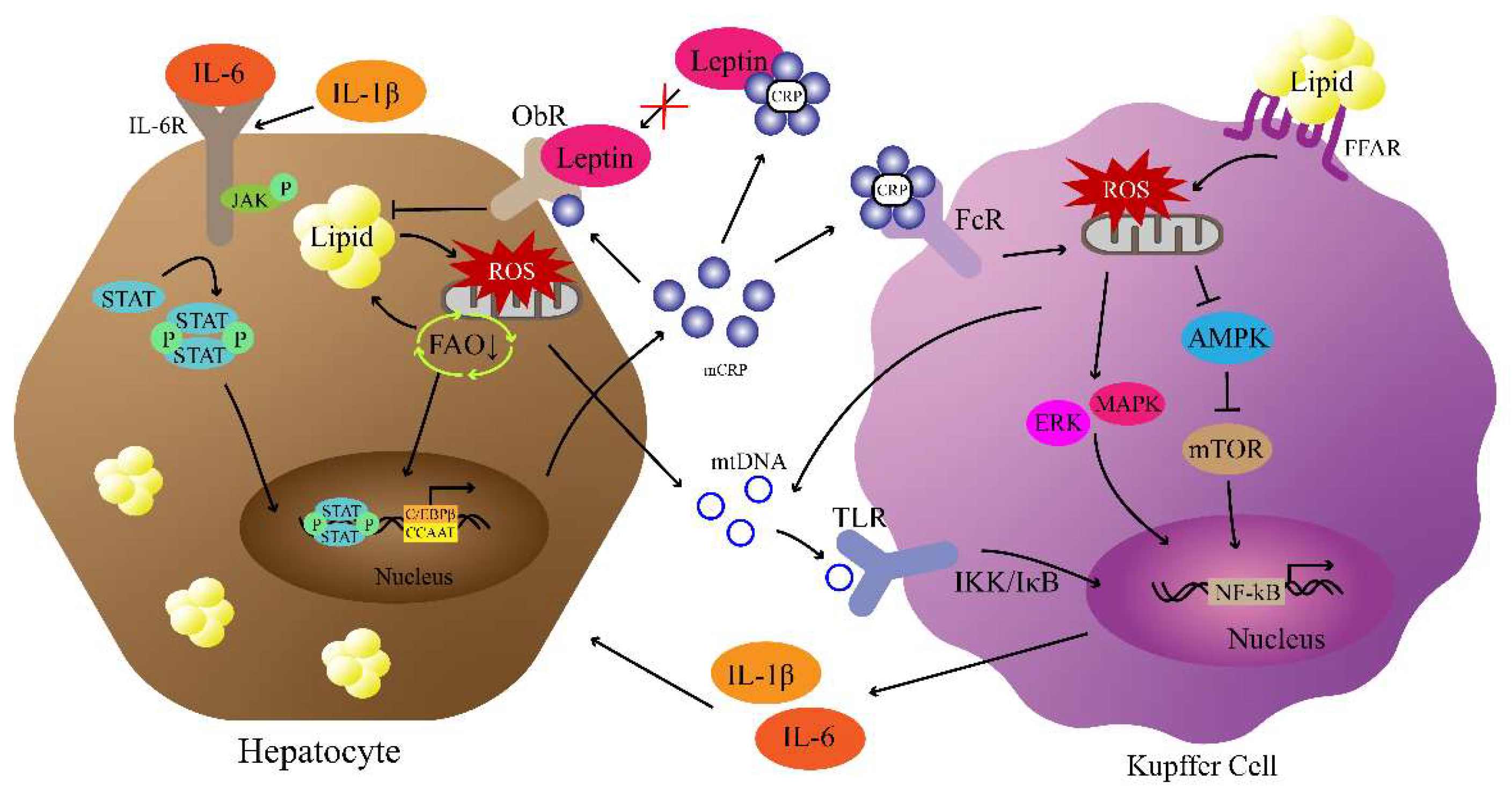
5. CRP Is Involved in the “Multiple-Hit” Mechanisms of NAFLD/MASLD
5.1. CRP, Leptin Signaling Pathway, and NAFLD/MASLD
5.2. CRP, Insulin Signaling Pathway, and NAFLD/MASLD
5.3. CRP, Mitochondrial Dysfunction, and NAFLD/MASLD
5.4. CRP, NF-κB Pathway, and NAFLD/MALSD
6. Potential Strategies to Lower CRP in NAFLD/MASLD Treatment
6.1. Drugs Used in NAFLD/MASLD Treatment and CRP Lowering
6.2. CRP- and CRP-Receptor-Targeted Therapy
6.3. CRP Adsorption Technology
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. Hallmarks of aging: An expanding universe. Cell 2023, 186, 243–278. [Google Scholar] [CrossRef] [PubMed]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed]
- Schneider, K.M.; Schneider, C.V. A new era for steatotic liver disease: Evaluating the novel nomenclature in the UK Biobank. J. Hepatol. 2023, in press. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. WHO European Regional Obesity Report 2022; World Health Organization, Regional Office for Europe: Geneva, Switzerland, 2022. [Google Scholar]
- Harvey, B.E. NASH: Regulatory considerations for clinical drug development and US FDA approval. Acta Pharmacol. Sin. 2022, 43, 1210–1214. [Google Scholar] [CrossRef] [PubMed]
- Nassir, F. NAFLD: Mechanisms, treatments, and biomarkers. Biomolecules 2022, 12, 824. [Google Scholar] [CrossRef] [PubMed]
- Lazarus, J.V.; Mark, H.E.; Anstee, Q.M.; Arab, J.P.; Batterham, R.L.; Castera, L.; Cortez-Pinto, H.; Crespo, J.; Cusi, K.; Dirac, M.A. Advancing the global public health agenda for NAFLD: A consensus statement. Nat. Rev. Gastroenterol. Hepatol. 2022, 19, 60–78. [Google Scholar] [CrossRef] [PubMed]
- Song, S.J.; Lai, J.C.-T.; Wong, G.L.-H.; Wong, V.W.-S.; Yip, T.C.-F. Can we use old NAFLD data under the new MASLD definition? J. Hepatol. 2023, in press. [Google Scholar] [CrossRef] [PubMed]
- Ratziu, V.; Boursier, J.; de Ledinghen, V.; Anty, R.; Costentin, C.; Bureau, C. Confirmatory biomarker diagnostic studies are not needed when transitioning from NAFLD to MASLD. J. Hepatol. 2023, in press. [Google Scholar] [CrossRef]
- Hu, X.; Huang, Y.; Bao, Z.; Wang, Y.; Shi, D.; Liu, F.; Gao, Z.; Yu, X. Prevalence and factors associated with nonalcoholic fatty liver disease in Shanghai work-units. BMC Gastroenterol. 2012, 12, 123. [Google Scholar] [CrossRef]
- Li, W.; Ng, C.H.; Quek, J.; Chan, K.E.; Tan, C.; Zeng, R.W.; Yong, J.N.; Tay, H.; Tan, D.J.H.; Lim, W.H. The growing prevalence of nonalcoholic fatty liver disease (NAFLD), determined by fatty liver index, amongst young adults in the United States. A 20-year experience. Metab. Target Organ Damage 2022, 2, 19. [Google Scholar] [CrossRef]
- Drescher, H.K.; Weiskirchen, S.; Weiskirchen, R. Current status in testing for nonalcoholic fatty liver disease (NAFLD) and nonalcoholic steatohepatitis (NASH). Cells 2019, 8, 845. [Google Scholar] [CrossRef]
- Green, S.; Hillersdal, L. Aging biomarkers and the measurement of health and risk. Hist. Philos. Life Sci. 2021, 43, 28. [Google Scholar] [CrossRef] [PubMed]
- Galkin, F.; Mamoshina, P.; Aliper, A.; de Magalhães, J.P.; Gladyshev, V.N.; Zhavoronkov, A. Biohorology and biomarkers of aging: Current state-of-the-art, challenges and opportunities. Ageing Res. Rev. 2020, 60, 101050. [Google Scholar] [CrossRef] [PubMed]
- Oruc, N.; Ozutemiz, O.; Yuce, G.; Akarca, U.S.; Ersoz, G.; Gunsar, F.; Batur, Y. Serum procalcitonin and CRP levels in non-alcoholic fatty liver disease: A case control study. BMC Gastroenterol. 2009, 9, 16. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Porwal, Y.C.; Dev, N.; Kumar, P.; Chakravarthy, S.; Kumawat, A. Association of high-sensitivity C-reactive protein (hs-CRP) with non-alcoholic fatty liver disease (NAFLD) in Asian Indians: A cross-sectional study. J. Fam. Med. Prim. Care 2020, 9, 390. [Google Scholar]
- Tang, Y.; Fung, E.; Xu, A.; Lan, H.Y. C-reactive protein and ageing. Clin. Exp. Pharmacol. Physiol. 2017, 44, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Lassale, C.; Batty, G.D.; Steptoe, A.; Cadar, D.; Akbaraly, T.N.; Kivimäki, M.; Zaninotto, P. Association of 10-year C-reactive protein trajectories with markers of healthy aging: Findings from the English longitudinal study of aging. J. Gerontol. Ser. A 2019, 74, 195–203. [Google Scholar] [CrossRef]
- Sproston, N.R.; Ashworth, J.J. Role of C-reactive protein at sites of inflammation and infection. Front. Immunol. 2018, 9, 754. [Google Scholar] [CrossRef]
- Eslam, M.; Sanyal, A.J.; George, J.; Sanyal, A.; Neuschwander-Tetri, B.; Tiribelli, C.; Kleiner, D.E.; Brunt, E.; Bugianesi, E.; Yki-Järvinen, H. MAFLD: A consensus-driven proposed nomenclature for metabolic associated fatty liver disease. Gastroenterology 2020, 158, 1999–2014.e1. [Google Scholar] [CrossRef]
- Byrne, C.D.; Targher, G. NAFLD: A multisystem disease. J. Hepatol. 2015, 62, S47–S64. [Google Scholar] [CrossRef]
- Karim, M.F.; Al-Mahtab, M.; Rahman, S.; Debnath, C.R. Non-alcoholic Fatty Liver Disease (NAFLD)—A Review. Mymensingh Med. J. MMJ 2015, 24, 873–880. [Google Scholar] [PubMed]
- Buzzetti, E.; Pinzani, M.; Tsochatzis, E.A. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism 2016, 65, 1038–1048. [Google Scholar] [CrossRef] [PubMed]
- Loomba, R.; Friedman, S.L.; Shulman, G.I. Mechanisms and disease consequences of nonalcoholic fatty liver disease. Cell 2021, 184, 2537–2564. [Google Scholar] [CrossRef] [PubMed]
- Rinella, M.E.; Lazarus, J.V.; Ratziu, V.; Francque, S.M.; Sanyal, A.J.; Kanwal, F.; Romero, D.; Abdelmalek, M.F.; Anstee, Q.M.; Arab, J.P. A multi-society Delphi consensus statement on new fatty liver disease nomenclature. Ann. Hepatol. 2023, 101133. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Huang, J.; Wang, M.; Kumar, R.; Liu, Y.; Liu, S.; Wu, Y.; Wang, X.; Zhu, Y. Comparison of MAFLD and NAFLD diagnostic criteria in real world. Liver Int. 2020, 40, 2082–2089. [Google Scholar] [CrossRef] [PubMed]
- Dodig, S.; Čepelak, I.; Pavić, I. Hallmarks of senescence and aging. Biochem. Med. 2019, 29, 483–497. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Yang, Y.; Li, Q.; Li, J. Understanding the unique microenvironment in the aging liver. Front. Med. 2022, 9, 842024. [Google Scholar] [CrossRef]
- Chung, K.W. Advances in understanding of the role of lipid metabolism in aging. Cells 2021, 10, 880. [Google Scholar] [CrossRef]
- Pibiri, M. Liver regeneration in aged mice: New insights. Aging 2018, 10, 1801–1824. [Google Scholar] [CrossRef]
- He, Y.; Su, Y.; Duan, C.; Wang, S.; He, W.; Zhang, Y.; An, X.; He, M. Emerging role of aging in the progression of NAFLD to HCC. Ageing Res. Rev. 2022, 84, 101833. [Google Scholar] [CrossRef]
- Wong, V.W.-S.; Chu, W.C.-W.; Wong, G.L.-H.; Chan, R.S.-M.; Chim, A.M.-L.; Ong, A.; Yeung, D.K.-W.; Yiu, K.K.-L.; Chu, S.H.-T.; Woo, J.; et al. Prevalence of non-alcoholic fatty liver disease and advanced fibrosis in Hong Kong Chinese: A population study using proton-magnetic resonance spectroscopy and transient elastography. Gut 2012, 61, 409. [Google Scholar] [CrossRef] [PubMed]
- Koehler, E.M.; Schouten, J.N.L.; Hansen, B.E.; van Rooij, F.J.A.; Hofman, A.; Stricker, B.H.; Janssen, H.L.A. Prevalence and risk factors of non-alcoholic fatty liver disease in the elderly: Results from the Rotterdam study. J. Hepatol. 2012, 57, 1305–1311. [Google Scholar] [CrossRef] [PubMed]
- Noureddin, M.; Yates, K.P.; Vaughn, I.A.; Neuschwander-Tetri, B.A.; Sanyal, A.J.; McCullough, A.; Merriman, R.; Hameed, B.; Doo, E.; Kleiner, D.E. Clinical and histological determinants of nonalcoholic steatohepatitis and advanced fibrosis in elderly patients. Hepatology 2013, 58, 1644–1654. [Google Scholar] [CrossRef]
- Houtkooper, R.H.; Argmann, C.; Houten, S.M.; Cantó, C.; Jeninga, E.H.; Andreux, P.A.; Thomas, C.; Doenlen, R.; Schoonjans, K.; Auwerx, J. The metabolic footprint of aging in mice. Sci. Rep. 2011, 1, 134. [Google Scholar] [CrossRef]
- Li, X.; Lu, Y.; Liang, X.; Zhou, X.; Li, D.; Zhang, Z.; Niu, Y.; Liu, S.; Ye, L.; Zhang, R. A new NASH model in aged mice with rapid progression of steatohepatitis and fibrosis. PLoS ONE 2023, 18, e0286257. [Google Scholar] [CrossRef]
- Seo, E.; Kang, H.; Choi, H.; Choi, W.; Jun, H.S. Reactive oxygen species-induced changes in glucose and lipid metabolism contribute to the accumulation of cholesterol in the liver during aging. Aging Cell 2019, 18, e12895. [Google Scholar] [CrossRef]
- Ogrodnik, M.; Miwa, S.; Tchkonia, T.; Tiniakos, D.; Wilson, C.L.; Lahat, A.; Day, C.P.; Burt, A.; Palmer, A.; Anstee, Q.M. Cellular senescence drives age-dependent hepatic steatosis. Nat. Commun. 2017, 8, 15691. [Google Scholar] [CrossRef] [PubMed]
- Tillett, W.S.; Francis, T., Jr. Serological reactions in pneumonia with a non-protein somatic fraction of pneumococcus. J. Exp. Med. 1930, 52, 561. [Google Scholar] [CrossRef]
- Schulze, R.J.; Schott, M.B.; Casey, C.A.; Tuma, P.L.; McNiven, M.A. The cell biology of the hepatocyte: A membrane trafficking machine. J. Cell Biol. 2019, 218, 2096–2112. [Google Scholar] [CrossRef]
- Knolle, P.A. Staying local—Antigen presentation in the liver. Curr. Opin. Immunol. 2016, 40, 36–42. [Google Scholar] [CrossRef]
- Zhou, Z.; Xu, M.-J.; Gao, B. Hepatocytes: A key cell type for innate immunity. Cell. Mol. Immunol. 2016, 13, 301–315. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Sun, M.; Samols, D.; Kushner, I. STAT3 Participates in Transcriptional Activation of the C-reactive Protein Gene by Interleukin-6 (*). J. Biol. Chem. 1996, 271, 9503–9509. [Google Scholar] [CrossRef] [PubMed]
- Ngwa, D.N.; Pathak, A.; Agrawal, A. IL-6 regulates induction of C-reactive protein gene expression by activating STAT3 isoforms. Mol. Immunol. 2022, 146, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, A.; Cha-Molstad, H.; Samols, D.; Kushner, I. Transactivation of C-reactive protein by IL-6 requires synergistic interaction of CCAAT/enhancer binding protein β (C/EBPβ) and Rel p50. J. Immunol. 2001, 166, 2378–2384. [Google Scholar] [CrossRef] [PubMed]
- Kramer, F.; Torzewski, J.; Kamenz, J.; Veit, K.; Hombach, V.; Dedio, J.; Ivashchenko, Y. Interleukin-1β stimulates acute phase response and C-reactive protein synthesis by inducing an NFκB-and C/EBPβ-dependent autocrine interleukin-6 loop. Mol. Immunol. 2008, 45, 2678–2689. [Google Scholar] [CrossRef] [PubMed]
- Ramani, M.; Khechai, F.; Ollivier, V.; Ternisien, C.; Bridey, F.; Hakim, J.; de Prost, D. Interleukin-10 and pentoxifylline inhibit C-reactive protein-induced tissue factor gene expression in peripheral human blood monocytes. FEBS Lett. 1994, 356, 86–88. [Google Scholar] [CrossRef] [PubMed]
- Pathak, A.; Agrawal, A. Evolution of C-reactive protein. Front. Immunol. 2019, 10, 943. [Google Scholar] [CrossRef] [PubMed]
- Macintyre, S.; Samols, D.; Dailey, P. Two carboxylesterases bind C-reactive protein within the endoplasmic reticulum and regulate its secretion during the acute phase response. J. Biol. Chem. 1994, 269, 24496–24503. [Google Scholar] [CrossRef]
- Ansar, W. Multiple Faces of C-Reactive Protein: Structure–Function Relationships. In Clinical Significance of C-Reactive Protein; Springer: Berlin/Heidelberg, Germany, 2020; pp. 1–34. [Google Scholar]
- Torzewski, M. C-reactive protein: Friend or foe? Phylogeny from heavy metals to modified lipoproteins and SARS-CoV-2. Front. Cardiovasc. Med. 2022, 9, 797116. [Google Scholar] [CrossRef]
- Braig, D.; Nero, T.L.; Koch, H.-G.; Kaiser, B.; Wang, X.; Thiele, J.R.; Morton, C.J.; Zeller, J.; Kiefer, J.; Potempa, L.A. Transitional changes in the CRP structure lead to the exposure of proinflammatory binding sites. Nat. Commun. 2017, 8, 14188. [Google Scholar] [CrossRef]
- Pepys, M.B.; Hirschfield, G.M. C-reactive protein: A critical update. J. Clin. Investig. 2003, 111, 1805–1812. [Google Scholar] [CrossRef] [PubMed]
- Forte, L.; Cimmino, G.; Loffredo, F.; De Palma, R.; Abbate, G.; Calabrò, P.; Ingrosso, D.; Galletti, P.; Carangio, C.; Casillo, B. C-reactive protein is released in the coronary circulation and causes endothelial dysfunction in patients with acute coronary syndromes. Int. J. Cardiol. 2011, 152, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Yao, Z.; Zhang, Y.; Wu, H. Regulation of C-reactive protein conformation in inflammation. Inflamm. Res. 2019, 68, 815–823. [Google Scholar] [CrossRef] [PubMed]
- Musolino, A.; Gradishar, W.J.; Rugo, H.S.; Nordstrom, J.L.; Rock, E.P.; Arnaldez, F.; Pegram, M.D. Role of Fcγ receptors in HER2-targeted breast cancer therapy. J. Immunother. Cancer 2022, 10, e003171. [Google Scholar] [CrossRef] [PubMed]
- Akinrinmade, O.A.; Chetty, S.; Daramola, A.K.; Islam, M.-U.; Thepen, T.; Barth, S. CD64: An attractive immunotherapeutic target for M1-type macrophage mediated chronic inflammatory diseases. Biomedicines 2017, 5, 56. [Google Scholar] [CrossRef] [PubMed]
- Luan, Y.-Y.; Yao, Y.-M. The clinical significance and potential role of C-reactive protein in chronic inflammatory and neurodegenerative diseases. Front. Immunol. 2018, 9, 1302. [Google Scholar] [CrossRef] [PubMed]
- Zwaka, T.P.; Hombach, V.; Torzewski, J. C-reactive protein–mediated low density lipoprotein uptake by macrophages: Implications for atherosclerosis. Circulation 2001, 103, 1194–1197. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, I.N.; White, M.; Hoeksema, M.; Deluna, X.; Hartshorn, K. Histone H4 potentiates neutrophil inflammatory responses to influenza A virus: Down-modulation by H4 binding to C-reactive protein and Surfactant protein D. PLoS ONE 2021, 16, e0247605. [Google Scholar] [CrossRef]
- Li, H.-Y.; Tang, Z.-M.; Wang, Z.; Lv, J.-M.; Liu, X.-L.; Liang, Y.-L.; Cheng, B.; Gao, N.; Ji, S.-R.; Wu, Y. C-reactive protein protects against acetaminophen-induced liver injury by preventing complement overactivation. Cell. Mol. Gastroenterol. Hepatol. 2022, 13, 289–307. [Google Scholar] [CrossRef]
- Duan, Y.; Pan, X.; Luo, J.; Xiao, X.; Li, J.; Bestman, P.L.; Luo, M. Association of Inflammatory Cytokines with Non-Alcoholic Fatty Liver Disease. Front. Immunol. 2022, 13, 880298. [Google Scholar] [CrossRef]
- Yoneda, M.; Mawatari, H.; Fujita, K.; Iida, H.; Yonemitsu, K.; Kato, S.; Takahashi, H.; Kirikoshi, H.; Inamori, M.; Nozaki, Y.; et al. High-sensitivity C-reactive protein is an independent clinical feature of nonalcoholic steatohepatitis (NASH) and also of the severity of fibrosis in NASH. J. Gastroenterol. 2007, 42, 573–582. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Huang, D.; Ma, H.; Qian, C.; You, H.; Bu, L.; Qu, S. High-Sensitive CRP Correlates with the Severity of Liver Steatosis and Fibrosis in Obese Patients with Metabolic Dysfunction Associated Fatty Liver Disease. Front. Endocrinol. 2022, 13, 848937. [Google Scholar] [CrossRef] [PubMed]
- Lambrecht, J.; Tacke, F. Controversies and opportunities in the use of inflammatory markers for diagnosis or risk prediction in fatty liver disease. Front. Immunol. 2021, 11, 634409. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, E.; Anty, R.; Tordjman, J.; Verrijken, A.; Gual, P.; Tran, A.; Iannelli, A.; Gugenheim, J.; Bedossa, P.; Francque, S.; et al. C-reactive protein levels in relation to various features of non-alcoholic fatty liver disease among obese patients. J. Hepatol. 2011, 55, 660–665. [Google Scholar] [CrossRef] [PubMed]
- Yeh, E.T.H.; Willerson, J.T. Coming of age of C-reactive protein: Using inflammation markers in cardiology. Circulation 2003, 107, 370–371. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.; Szmitko, P.E.; Ridker, P.M. C-reactive protein comes of age. Nat. Clin. Pract. Cardiovasc. Med. 2005, 2, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Liang, P.; Chen, J.; Fu, S.; Liu, B.; Feng, M.; Lin, B.; Lee, B.; Xu, A.; Lan, H.Y. The baseline levels and risk factors for high-sensitive C-reactive protein in Chinese healthy population. Immun. Ageing 2018, 15, 21. [Google Scholar] [CrossRef] [PubMed]
- Puzianowska-Kuźnicka, M.; Owczarz, M.; Wieczorowska-Tobis, K.; Nadrowski, P.; Chudek, J.; Slusarczyk, P.; Skalska, A.; Jonas, M.; Franek, E.; Mossakowska, M. Interleukin-6 and C-reactive protein, successful aging, and mortality: The PolSenior study. Immun. Ageing 2016, 13, 21. [Google Scholar] [CrossRef]
- McCabe, E.L.; Larson, M.G.; Lunetta, K.L.; Newman, A.B.; Cheng, S.; Murabito, J.M. Association of an Index of Healthy Aging with Incident Cardiovascular Disease and Mortality in a Community-Based Sample of Older Adults. J. Gerontol. Ser. A 2016, 71, 1695–1701. [Google Scholar] [CrossRef]
- Kravitz, B.A.; Corrada, M.M.; Kawas, C.H. High levels of serum C-reactive protein are associated with greater risk of all-cause mortality, but not dementia, in the oldest-old: Results from The 90+ Study. J. Am. Geriatr. Soc. 2009, 57, 641–646. [Google Scholar] [CrossRef]
- Huang, J.; Wang, M.; Wu, Y.; Kumar, R.; Lin, S. Serum high-sensitive C-reactive protein is a simple indicator for all-cause among individuals with MAFLD. Front. Physiol. 2022, 13, 1012887. [Google Scholar] [CrossRef] [PubMed]
- Caruso, A.; Gelsomino, L.; Panza, S.; Accattatis, F.M.; Naimo, G.D.; Barone, I.; Giordano, C.; Catalano, S.; Andò, S. Leptin: A Heavyweight Player in Obesity-Related Cancers. Biomolecules 2023, 13, 1084. [Google Scholar] [CrossRef]
- Jiménez-Cortegana, C.; García-Galey, A.; Tami, M.; Del Pino, P.; Carmona, I.; López, S.; Alba, G.; Sánchez-Margalet, V. Role of leptin in non-alcoholic fatty liver disease. Biomedicines 2021, 9, 762. [Google Scholar] [CrossRef] [PubMed]
- Polyzos, S.A.; Kountouras, J.; Mantzoros, C.S. Leptin in nonalcoholic fatty liver disease: A narrative review. Metabolism 2015, 64, 60–78. [Google Scholar] [CrossRef]
- Chen, K.; Li, F.; Li, J.; Cai, H.; Strom, S.; Bisello, A.; Kelley, D.E.; Friedman-Einat, M.; Skibinski, G.A.; McCrory, M.A. Induction of leptin resistance through direct interaction of C-reactive protein with leptin. Nat. Med. 2006, 12, 425–432. [Google Scholar] [CrossRef] [PubMed]
- Shamsuzzaman, A.S.M.; Winnicki, M.; Wolk, R.; Svatikova, A.; Phillips, B.G.; Davison, D.E.; Berger, P.B.; Somers, V.K. Independent association between plasma leptin and C-reactive protein in healthy humans. Circulation 2004, 109, 2181–2185. [Google Scholar] [CrossRef] [PubMed]
- Sudhakar, M.; Silambanan, S.; Chandran, A.S.; Prabhakaran, A.A.; Ramakrishnan, R. C-reactive protein (CRP) and leptin receptor in obesity: Binding of monomeric CRP to leptin receptor. Front. Immunol. 2018, 9, 1167. [Google Scholar] [CrossRef] [PubMed]
- Marušić, M.; Paić, M.; Knobloch, M.; Liberati Pršo, A.-M. NAFLD, insulin resistance, and diabetes mellitus type 2. Can. J. Gastroenterol. Hepatol. 2021, 2021, 6613827. [Google Scholar] [CrossRef]
- Barzilai, N.; Ferrucci, L. Insulin Resistance and Aging: A Cause or a Protective Response? J. Gerontol. Ser. A 2012, 67, 1329–1331. [Google Scholar] [CrossRef]
- Lee, S.H.; Park, S.Y.; Choi, C.S. Insulin Resistance: From Mechanisms to Therapeutic Strategies. Diabetes Metab. J. 2022, 46, 15–37. [Google Scholar] [CrossRef]
- Santoleri, D.; Titchenell, P.M. Resolving the Paradox of Hepatic Insulin Resistance. Cell. Mol. Gastroenterol. Hepatol. 2019, 7, 447–456. [Google Scholar] [CrossRef] [PubMed]
- Cetin, E.G.; Demir, N.; Sen, I. The Relationship between Insulin Resistance and Liver Damage in non-alcoholic Fatty Liver Patients. Sisli Etfal Hast. Tip Bul 2020, 54, 411–415. [Google Scholar] [CrossRef] [PubMed]
- Alissa, E.M.; Algarni, S.A.; Khaffji, A.J.; Al Mansouri, N.M. Role of inflammatory markers in polycystic ovaries syndrome: In relation to insulin resistance. J. Obstet. Gynaecol. Res. 2021, 47, 1409–1415. [Google Scholar] [CrossRef] [PubMed]
- Tanigaki, K.; Mineo, C.; Yuhanna, I.S.; Chambliss, K.L.; Quon, M.J.; Bonvini, E.; Shaul, P.W. C-reactive protein inhibits insulin activation of endothelial nitric oxide synthase via the immunoreceptor tyrosine-based inhibition motif of FcgammaRIIB and SHIP-1. Circ. Res. 2009, 104, 1275–1282. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Qiu, S.; He, Y.; Li, L.; Wu, T.; Ding, N.; Li, F.; Zhao, A.Z.; Yang, G. Genetic ablation of C-reactive protein gene confers resistance to obesity and insulin resistance in rats. Diabetologia 2021, 64, 1169–1183. [Google Scholar] [CrossRef]
- D’Alessandris, C.; Lauro, R.; Presta, I.; Sesti, G. C-reactive protein induces phosphorylation of insulin receptor substrate-1 on Ser307 and Ser612 in L6 myocytes, thereby impairing the insulin signalling pathway that promotes glucose transport. Diabetologia 2007, 50, 840–849. [Google Scholar] [CrossRef]
- Dąbrowski, R.; Szczubiał, M.; Kostro, K.; Wawron, W.; Ceron, J.J.; Tvarijonaviciute, A. Serum insulin-like growth factor-1 and C-reactive protein concentrations before and after ovariohysterectomy in bitches with pyometra. Theriogenology 2015, 83, 474–477. [Google Scholar] [CrossRef]
- Chen, Z.; Nilsson, E.; Lindholm, B.; Heimbürger, O.; Barany, P.; Stenvinkel, P.; Qureshi, A.R.; Chen, J. Low-plasma insulin-like growth factor-1 associates with increased mortality in chronic kidney disease patients with reduced muscle strength. J. Ren. Nutr. 2023, 33, 298–306. [Google Scholar] [CrossRef]
- Higashi, Y.; Sukhanov, S.; Anwar, A.; Shai, S.-Y.; Delafontaine, P. Aging, Atherosclerosis, and IGF-1. J. Gerontol. Ser. A 2012, 67, 626–639. [Google Scholar] [CrossRef]
- Ashpole, N.M.; Sanders, J.E.; Hodges, E.L.; Yan, H.; Sonntag, W.E. Growth hormone, insulin-like growth factor-1 and the aging brain. Exp. Gerontol. 2015, 68, 76–81. [Google Scholar] [CrossRef]
- Dichtel, L.E.; Corey, K.E.; Misdraji, J.; Bredella, M.A.; Schorr, M.; Osganian, S.A.; Young, B.J.; Sung, J.C.; Miller, K.K. The Association Between IGF-1 Levels and the Histologic Severity of Nonalcoholic Fatty Liver Disease. Clin. Transl. Gastroenterol. 2017, 8, e217. [Google Scholar] [CrossRef] [PubMed]
- Stanley, T.L.; Fourman, L.T.; Zheng, I.; McClure, C.M.; Feldpausch, M.N.; Torriani, M.; Corey, K.E.; Chung, R.T.; Lee, H.; Kleiner, D.E.; et al. Relationship of IGF-1 and IGF-Binding Proteins to Disease Severity and Glycemia in Nonalcoholic Fatty Liver Disease. J. Clin. Endocrinol. Metab. 2020, 106, e520–e533. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, B.D.; Goncalves, M.D.; Cantley, L.C. Insulin–PI3K signalling: An evolutionarily insulated metabolic driver of cancer. Nat. Rev. Endocrinol. 2020, 16, 276–283. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.-J.; Zhong, Y.; You, X.-Y.; Liu, W.-H.; Li, A.-Q.; Liu, S.-M. Insulin-like growth factor 1 opposes the effects of C-reactive protein on endothelial cell activation. Mol. Cell. Biochem. 2014, 385, 199–205. [Google Scholar] [CrossRef] [PubMed]
- Entezari, M.; Hashemi, D.; Taheriazam, A.; Zabolian, A.; Mohammadi, S.; Fakhri, F.; Hashemi, M.; Hushmandi, K.; Ashrafizadeh, M.; Zarrabi, A.; et al. AMPK signaling in diabetes mellitus, insulin resistance and diabetic complications: A pre-clinical and clinical investigation. Biomed. Pharmacother. 2022, 146, 112563. [Google Scholar] [CrossRef] [PubMed]
- Fang, C.; Pan, J.; Qu, N.; Lei, Y.; Han, J.; Zhang, J.; Han, D. The AMPK pathway in fatty liver disease. Front. Physiol. 2022, 13, 970292. [Google Scholar] [CrossRef]
- Garcia, D.; Hellberg, K.; Chaix, A.; Wallace, M.; Herzig, S.; Badur, M.G.; Lin, T.; Shokhirev, M.N.; Pinto, A.F.M.; Ross, D.S.; et al. Genetic Liver-Specific AMPK Activation Protects against Diet-Induced Obesity and NAFLD. Cell Rep. 2019, 26, 192–208.e6. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Wang, L.; Guo, M.; He, J.; Deng, Y.; Liu, J.; Wei, Y.; Wang, C.; Zhou, J.; Ma, L. Clinically relevant high levels of human C-reactive protein induces endothelial dysfunction and hypertension by inhibiting the AMPK-eNOS axis. Clin. Sci. 2020, 134, 1805–1819. [Google Scholar] [CrossRef]
- Salminen, A.; Kaarniranta, K. AMP-activated protein kinase (AMPK) controls the aging process via an integrated signaling network. Ageing Res. Rev. 2012, 11, 230–241. [Google Scholar] [CrossRef]
- Laplante, M.; Sabatini, D.M. mTOR signaling at a glance. J. Cell Sci. 2009, 122, 3589–3594. [Google Scholar] [CrossRef]
- Feng, J.; Qiu, S.; Zhou, S.; Tan, Y.; Bai, Y.; Cao, H.; Guo, J.; Su, Z. mTOR: A potential new target in nonalcoholic fatty liver disease. Int. J. Mol. Sci. 2022, 23, 9196. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Weng, J.; Zhang, Y.; Liang, K.; Fu, G.; Li, Y.; Bai, X.; Gao, Y. mTOR direct crosstalk with STAT5 promotes de novo lipid synthesis and induces hepatocellular carcinoma. Cell Death Dis. 2019, 10, 619. [Google Scholar] [CrossRef] [PubMed]
- You, Y.-K.; Huang, X.-R.; Chen, H.-Y.; Lyu, X.-F.; Liu, H.-F.; Lan, H.Y. C-reactive protein promotes diabetic kidney disease in db/db mice via the CD32b-Smad3-mTOR signaling pathway. Sci. Rep. 2016, 6, 26740. [Google Scholar] [CrossRef] [PubMed]
- Sangwung, P.; Petersen, K.F.; Shulman, G.I.; Knowles, J.W. Mitochondrial Dysfunction, Insulin Resistance, and Potential Genetic Implications: Potential Role of Alterations in Mitochondrial Function in the Pathogenesis of Insulin Resistance and Type 2 Diabetes. Endocrinology 2020, 161, bqaa017. [Google Scholar] [CrossRef] [PubMed]
- Tilg, H.; Moschen, A.R.; Roden, M. NAFLD and diabetes mellitus. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Cheung, E.C.; Vousden, K.H. The role of ROS in tumour development and progression. Nat. Rev. Cancer 2022, 22, 280–297. [Google Scholar] [CrossRef] [PubMed]
- Stefanatos, R.; Sanz, A. The role of mitochondrial ROS in the aging brain. FEBS Lett. 2018, 592, 743–758. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Zhang, D.; Hu, D.; Zhou, X.; Zhou, Y. The role of mitochondria in NLRP3 inflammasome activation. Mol. Immunol. 2018, 103, 115–124. [Google Scholar] [CrossRef]
- Zeller, J.; Bogner, B.; Kiefer, J.; Braig, D.; Winninger, O.; Fricke, M.; Karasu, E.; Peter, K.; Huber-Lang, M.; Eisenhardt, S.U. CRP Enhances the Innate Killing Mechanisms Phagocytosis and ROS Formation in a Conformation and Complement-Dependent Manner. Front. Immunol. 2021, 12, 721887. [Google Scholar] [CrossRef]
- Ryu, J.-W.; Jung, I.-H.; Park, E.-Y.; Kim, K.-H.; Kim, K.; Yeom, J.; Jung, J.; Lee, S.-W. Radiation-induced C-reactive protein triggers apoptosis of vascular smooth muscle cells through ROS interfering with the STAT3/Ref-1 complex. J. Cell. Mol. Med. 2022, 26, 2104–2118. [Google Scholar] [CrossRef]
- Farzanegi, P.; Dana, A.; Ebrahimpoor, Z.; Asadi, M.; Azarbayjani, M.A. Mechanisms of beneficial effects of exercise training on non-alcoholic fatty liver disease (NAFLD): Roles of oxidative stress and inflammation. Eur. J. Sport Sci. 2019, 19, 994–1003. [Google Scholar] [CrossRef] [PubMed]
- Wu, I.C.; Liu, C.-S.; Cheng, W.-L.; Lin, T.-T.; Chen, H.-L.; Chen, P.-F.; Wu, R.-C.; Huang, C.-W.; Hsiung, C.A.; Hsu, C.-C. Association of leukocyte mitochondrial DNA copy number with longitudinal C-reactive protein levels and survival in older adults: A cohort study. Immun. Ageing 2022, 19, 62. [Google Scholar] [CrossRef] [PubMed]
- Knez, J.; Marrachelli, V.G.; Cauwenberghs, N.; Winckelmans, E.; Zhang, Z.; Thijs, L.; Brguljan-Hitij, J.; Plusquin, M.; Delles, C.; Monleon, D. Peripheral blood mitochondrial DNA content in relation to circulating metabolites and inflammatory markers: A population study. PLoS ONE 2017, 12, e0181036. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Liu, B.; Xu, L.; Yu, S.; Fu, J.; Wang, J.; Yan, X.; Su, J. ROS-Induced mtDNA Release: The Emerging Messenger for Communication between Neurons and Innate Immune Cells during Neurodegenerative Disorder Progression. Antioxidants 2021, 10, 1917. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.-C. NF-κB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef] [PubMed]
- Tilstra, J.S.; Clauson, C.L.; Niedernhofer, L.J.; Robbins, P.D. NF-κB in Aging and Disease. Aging Dis. 2011, 2, 449–465. [Google Scholar] [PubMed]
- Kunnumakkara, A.; Shabnam, B.; Girisa, S.; Harsha, C.; Banik, K.; Devi, T.B.; Choudhury, R.; Sahu, H.; Parama, D.; Sailo, B.L. Inflammation, NF-κB, and chronic diseases: How are they linked? Crit. Rev. Immunol. 2020, 40, 1–39. [Google Scholar] [CrossRef]
- Zhang, Q.; Yu, K.; Cao, Y.; Luo, Y.; Liu, Y.; Zhao, C. miR-125b promotes the NF-κB-mediated inflammatory response in NAFLD via directly targeting TNFAIP3. Life Sci. 2021, 270, 119071. [Google Scholar] [CrossRef]
- Devaraj, S.; Jialal, I. C-Reactive Protein Polarizes Human Macrophages to an M1 Phenotype and Inhibits Transformation to the M2 Phenotype. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1397–1402. [Google Scholar] [CrossRef]
- Verma, S.; Badiwala, M.V.; Weisel, R.D.; Li, S.-H.; Wang, C.-H.; Fedak, P.W.M.; Li, R.-K.; Mickle, D.A.G. C-reactive protein activates the nuclear factor-κB signal transduction pathway in saphenous vein endothelial cells: Implications for atherosclerosis and restenosis. J. Thorac. Cardiovasc. Surg. 2003, 126, 1886–1891. [Google Scholar] [CrossRef]
- Tang, P.M.-K.; Zhang, Y.-Y.; Hung, J.S.-C.; Chung, J.Y.-F.; Huang, X.-R.; To, K.-F.; Lan, H.-Y. DPP4/CD32b/NF-ĸB Circuit: A Novel Druggable Target for Inhibiting CRP-Driven Diabetic Nephropathy. Mol. Ther. 2021, 29, 365–375. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Bian, Z.-M.; Yu, W.-Z.; Yan, Z.; Chen, W.-C.; Li, X.-X. Induction of interleukin-8 gene expression and protein secretion by C-reactive protein in ARPE-19 cells. Exp. Eye Res. 2010, 91, 135–142. [Google Scholar] [CrossRef]
- Ruiz-Fernández, C.; Gonzalez-Rodríguez, M.; Francisco, V.; Rajab, I.M.; Gómez, R.; Conde, J.; Lago, F.; Pino, J.; Mobasheri, A.; Gonzalez-Gay, M.A. Monomeric C reactive protein (mCRP) regulates inflammatory responses in human and mouse chondrocytes. Lab. Investig. 2021, 101, 1550–1560. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, W.C.; Sherlock, L.G.; Grayck, M.R.; Zheng, L.; Lacayo, O.A.; Solar, M.; Orlicky, D.J.; Dobrinskikh, E.; Wright, C.J. Innate Immune Zonation in the Liver: NF-κB (p50) Activation and C-Reactive Protein Expression in Response to Endotoxemia Are Zone Specific. J. Immunol. 2023, 210, 1372–1385. [Google Scholar] [CrossRef] [PubMed]
- Liss, K.H.H.; Finck, B.N. PPARs and nonalcoholic fatty liver disease. Biochimie 2017, 136, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Satoh, N.; Ogawa, Y.; Usui, T.; Tagami, T.; Kono, S.; Uesugi, H.; Sugiyama, H.; Sugawara, A.; Yamada, K.; Shimatsu, A. Antiatherogenic effect of pioglitazone in type 2 diabetic patients irrespective of the responsiveness to its antidiabetic effect. Diabetes Care 2003, 26, 2493–2499. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Liu, J.-T.; Li, K.; Wang, S.-Y.; Xu, S. Genistein inhibits Ang II-induced CRP and MMP-9 generations via the ER-p38/ERK1/2-PPARγ-NF-κB signaling pathway in rat vascular smooth muscle cells. Life Sci. 2019, 216, 140–146. [Google Scholar] [CrossRef] [PubMed]
- Nascimbeni, F.; Pellegrini, E.; Lugari, S.; Mondelli, A.; Bursi, S.; Onfiani, G.; Carubbi, F.; Lonardo, A. Statins and nonalcoholic fatty liver disease in the era of precision medicine: More friends than foes. Atherosclerosis 2019, 284, 66–74. [Google Scholar] [CrossRef]
- Kandelouei, T.; Abbasifard, M.; Imani, D.; Aslani, S.; Razi, B.; Fasihi, M.; Shafiekhani, S.; Mohammadi, K.; Jamialahmadi, T.; Reiner, Ž. Effect of statins on serum level of hs-CRP and CRP in patients with cardiovascular diseases: A systematic review and meta-analysis of randomized controlled trials. Mediat. Inflamm. 2022, 2022, 8732360. [Google Scholar] [CrossRef]
- Saboori, S.; Shab-Bidar, S.; Speakman, J.R.; Yousefi Rad, E.; Djafarian, K. Effect of vitamin E supplementation on serum C-reactive protein level: A meta-analysis of randomized controlled trials. Eur. J. Clin. Nutr. 2015, 69, 867–873. [Google Scholar] [CrossRef]
- Nagashimada, M.; Ota, T. Role of vitamin E in nonalcoholic fatty liver disease. IUBMB Life 2019, 71, 516–522. [Google Scholar] [CrossRef] [PubMed]
- Perumpail, B.J.; Li, A.A.; John, N.; Sallam, S.; Shah, N.D.; Kwong, W.; Cholankeril, G.; Kim, D.; Ahmed, A. The Role of Vitamin E in the Treatment of NAFLD. Diseases 2018, 6, 86. [Google Scholar] [CrossRef]
- Escribano-Lopez, I.; Diaz-Morales, N.; Rovira-Llopis, S.; de Marañon, A.M.; Orden, S.; Alvarez, A.; Bañuls, C.; Rocha, M.; Murphy, M.P.; Hernandez-Mijares, A.; et al. The mitochondria-targeted antioxidant MitoQ modulates oxidative stress, inflammation and leukocyte-endothelium interactions in leukocytes isolated from type 2 diabetic patients. Redox Biol. 2016, 10, 200–205. [Google Scholar] [CrossRef] [PubMed]
- Ota, T. Prevention of NAFLD/NASH by Astaxanthin and β-Cryptoxanthin. Carotenoids Biosynthetic Biofunctional Approaches 2021, 1261, 231–238. [Google Scholar]
- Xia, W.; Tang, N.; Kord-Varkaneh, H.; Low, T.Y.; Tan, S.C.; Wu, X.; Zhu, Y. The effects of astaxanthin supplementation on obesity, blood pressure, CRP, glycemic biomarkers, and lipid profile: A meta-analysis of randomized controlled trials. Pharmacol. Res. 2020, 161, 105113. [Google Scholar] [CrossRef] [PubMed]
- Julia, C.; Galan, P.; Touvier, M.; Meunier, N.; Papet, I.; Sapin, V.; Cano, N.; Faure, P.; Hercberg, S.; Kesse-Guyot, E. Antioxidant Status and the Risk of Elevated C-Reactive Protein 12 Years Later. Ann. Nutr. Metab. 2014, 65, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Pepys, M.B.; Hirschfield, G.M.; Tennent, G.A.; Ruth Gallimore, J.; Kahan, M.C.; Bellotti, V.; Hawkins, P.N.; Myers, R.M.; Smith, M.D.; Polara, A.; et al. Targeting C-reactive protein for the treatment of cardiovascular disease. Nature 2006, 440, 1217–1221. [Google Scholar] [CrossRef] [PubMed]
- Kitsis, R.N.; Jialal, I. Limiting myocardial damage during acute myocardial infarction by inhibiting C-reactive protein. N. Engl. J. Med. 2006, 355, 513–515. [Google Scholar] [CrossRef]
- Szalai, A.J.; McCrory, M.A.; Xing, D.; Hage, F.G.; Miller, A.; Oparil, S.; Chen, Y.-F.; Mazzone, M.; Early, R.; Henry, S.P.; et al. Inhibiting C-Reactive Protein for the Treatment of Cardiovascular Disease: Promising Evidence from Rodent Models. Mediat. Inflamm. 2014, 2014, 353614. [Google Scholar] [CrossRef]
- Warren, M.S.; Hughes, S.G.; Singleton, W.; Yamashita, M.; Genovese, M.C. Results of a proof of concept, double-blind, randomized trial of a second generation antisense oligonucleotide targeting high-sensitivity C-reactive protein (hs-CRP) in rheumatoid arthritis. Arthritis Res. Ther. 2015, 17, 80. [Google Scholar] [CrossRef]
- Zhao, C.; Deng, H.; Chen, X. Harnessing immune response using reactive oxygen Species-Generating/Eliminating inorganic biomaterials for disease treatment. Adv. Drug Deliv. Rev. 2022, 188, 114456. [Google Scholar] [CrossRef]
- Liu, H.; Saxena, A.; Sidhu, S.S.; Wu, D. Fc engineering for developing therapeutic bispecific antibodies and novel scaffolds. Front. Immunol. 2017, 8, 38. [Google Scholar] [CrossRef] [PubMed]
- Torzewski, J.; Brunner, P.; Ries, W.; Garlichs, C.D.; Kayser, S.; Heigl, F.; Sheriff, A. Targeting C-reactive protein by selective apheresis in humans: Pros and cons. J. Clin. Med. 2022, 11, 1771. [Google Scholar] [CrossRef] [PubMed]
- Mattecka, S.; Brunner, P.; Hähnel, B.; Kunze, R.; Vogt, B.; Sheriff, A. PentraSorb C-Reactive Protein: Characterization of the Selective C-Reactive Protein Adsorber Resin. Ther. Apher. Dial. 2019, 23, 474–481. [Google Scholar] [CrossRef]
- Ries, W.; Torzewski, J.; Heigl, F.; Pfluecke, C.; Kelle, S.; Darius, H.; Ince, H.; Mitzner, S.; Nordbeck, P.; Butter, C. C-reactive protein apheresis as anti-inflammatory therapy in acute myocardial infarction: Results of the CAMI-1 study. Front. Cardiovasc. Med. 2021, 8, 591714. [Google Scholar] [CrossRef] [PubMed]
- Esposito, F.; Matthes, H.; Schad, F. Seven COVID-19 patients treated with C-reactive protein (CRP) apheresis. J. Clin. Med. 2022, 11, 1956. [Google Scholar] [CrossRef] [PubMed]
- Fendl, B.; Weiss, R.; Eichhorn, T.; Linsberger, I.; Afonyushkin, T.; Puhm, F.; Binder, C.J.; Fischer, M.B.; Weber, V. Extracellular vesicles are associated with C-reactive protein in sepsis. Sci. Rep. 2021, 11, 6996. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ding, Z.; Wei, Y.; Peng, J.; Wang, S.; Chen, G.; Sun, J. The Potential Role of C-Reactive Protein in Metabolic-Dysfunction-Associated Fatty Liver Disease and Aging. Biomedicines 2023, 11, 2711. https://doi.org/10.3390/biomedicines11102711
Ding Z, Wei Y, Peng J, Wang S, Chen G, Sun J. The Potential Role of C-Reactive Protein in Metabolic-Dysfunction-Associated Fatty Liver Disease and Aging. Biomedicines. 2023; 11(10):2711. https://doi.org/10.3390/biomedicines11102711
Chicago/Turabian StyleDing, Zheng, Yuqiu Wei, Jing Peng, Siyu Wang, Guixi Chen, and Jiazeng Sun. 2023. "The Potential Role of C-Reactive Protein in Metabolic-Dysfunction-Associated Fatty Liver Disease and Aging" Biomedicines 11, no. 10: 2711. https://doi.org/10.3390/biomedicines11102711
APA StyleDing, Z., Wei, Y., Peng, J., Wang, S., Chen, G., & Sun, J. (2023). The Potential Role of C-Reactive Protein in Metabolic-Dysfunction-Associated Fatty Liver Disease and Aging. Biomedicines, 11(10), 2711. https://doi.org/10.3390/biomedicines11102711