

Preclinical Evidence for the Role of the Yin/Yang Angiotensin System Components in Autism Spectrum Disorder: A Therapeutic Target of Astaxanthin


 , and
, and
Abstract
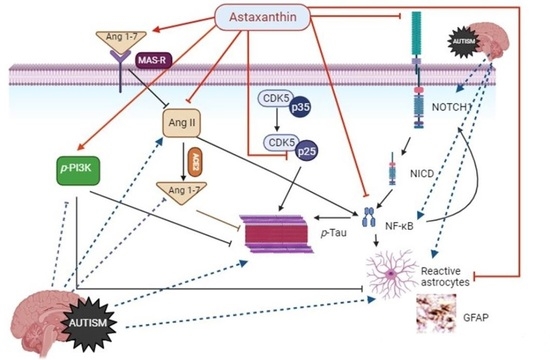
:
1. Introduction
2. Materials and Methods
2.1. Animals and Induction of the ASD Model
2.2. Experimental Design
2.3. Assessment of Autistic-Like Behaviors
2.3.1. Marble-Burying Test (MBT)
2.3.2. Three-Chamber Sociability Test
2.3.3. Open Field Test (OFT)
2.4. Cortical Processing
2.4.1. Assessments of p(S396)-tau, p(Y458/199)-PI3K p85/p55, Notch1, NICD, p25, and p35 Using the Western Blot Technique
2.4.2. Assessments of Cortical Gene Expressions of Mas Receptor with qRT-PCR
2.4.3. Quantification of Cortical Contents of Ang II, ACE2, and Ang1-7 Using the ELISA Technique
2.5. Microscopic Investigation
2.5.1. Hematoxylin and Eosin Staining
2.5.2. Nissl Staining
2.5.3. Cortical GFAP and NF-κB p65 Immunoreactivity
2.5.4. Electron Microscopy
2.6. Statistical Analysis
3. Results
3.1. AST Increases Sociability While Reducing Repetitive/Compulsive Behaviors in Autistic Rats
3.2. AST Modulates Cortical AngII/ACE2/Ang1-7/Mas Receptor/Trajectory and Augments PI3K p85/p55 in Autistic Rats
3.3. AST Deactivates the Cortical Notch1/NICD-1/NF-κB Trajectory and Increases the Cortical p25/p35 Ratio in Autistic Rats
3.4. AST Reduces ASD Hallmarks Astrogliosis/p-tau, Amends Cortical Structure, and Rescues Neurons in Autistic Rats
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| 3-CT | Three chamber test |
| ACE2 | Angiotensin-converting enzyme 2 |
| AD | Alzheimer disease |
| Akt/PKB | Protein kinase B |
| Ang1-7 | Angiotensin 1–7 |
| Ang II | Angiotensin 2 |
| ASD | Autism spectrum disorder |
| AST | Astaxanthin |
| CDK5 | Cyclin-dependent kinase 5 |
| GD | Gestational day |
| GFAP | Glial fibrillary acidic protein |
| MasR | Mas receptor |
| MBT | Marble burying test |
| NF-κB p65 | Nuclear factor Kappa B p65 |
| NICD | Notch intracellular domain |
| Notch1 | Neurogenic locus notch homolog protein 1 |
| OFT | Open field test |
| PI3K p85/p55 | Phosphoinositide 3-kinase p85/p55 |
| PND | Postnatal day |
| p-Tau | Phosphorylated tau protein |
| RAS | Renin angiotensin system |
| SI | Sociability index |
| SNI | Social novelty preference index |
| VPA | Valproic acid |
References
- Wiggins, L.D.; Rice, C.E.; Barger, B.; Soke, G.N.; Lee, L.C.; Moody, E.; Edmondson-Pretzel, R.; Levy, S.E. DSM-5 Criteria for Autism Spectrum Disorder Maximizes Diagnostic Sensitivity and Specificity in Preschool Children. Soc. Psychiatry Psychiatr. Epidemiol. 2019, 54, 693–701. [Google Scholar] [CrossRef]
- Komatsu, H.; Watanabe, E.; Fukuchi, M. Psychiatric Neural Networks and Precision Therapeutics by Machine Learning. Biomedicines 2021, 9, 403. [Google Scholar] [CrossRef] [PubMed]
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders; American Psychiatric Publishing: Washington, DC, USA, 2013. [Google Scholar] [CrossRef]
- Lai, M.C.; Lombardo, M.V.; Baron-Cohen, S. Autism. Lancet 2014, 383, 896–910. [Google Scholar] [CrossRef] [PubMed]
- Stoner, R.; Chow, M.L.; Boyle, M.P.; Sunkin, S.M.; Mouton, P.R.; Roy, S.; Wynshaw-Boris, A.; Colamarino, S.A.; Lein, E.S.; Courchesne, E. Patches of Disorganization in the Neocortex of Children with Autism. N. Engl. J. Med. 2014, 370, 1209–1219. [Google Scholar] [CrossRef] [PubMed]
- Nadeem, M.S.; Hosawi, S.; Alshehri, S.; Ghoneim, M.M.; Imam, S.S.; Murtaza, B.N.; Kazmi, I. Symptomatic, Genetic, and Mechanistic Overlaps between Autism and Alzheimer’s Disease. Biomolecules 2021, 11, 1635. [Google Scholar] [CrossRef]
- Ornoy, A.; Weinstein-Fudim, L.; Ergaz, Z. Prenatal Factors Associated with Autism Spectrum Disorder (ASD). Reprod. Toxicol. 2015, 56, 155–169. [Google Scholar] [CrossRef]
- Christensen, J.; Grnøborg, T.K.; Srøensen, M.J.; Schendel, D.; Parner, E.T.; Pedersen, L.H.; Vestergaard, M. Prenatal Valproate Exposure and Risk of Autism Spectrum Disorders and Childhood Autism. JAMA 2013, 309, 1696. [Google Scholar] [CrossRef]
- Bescoby-Chambers, N.; Forster, P.; Bates, G. Foetal Valproate Syndrome and Autism: Additional Evidence of an Association. Dev. Med. Child Neurol. 2001, 43, 847. [Google Scholar] [CrossRef]
- Christianson, A.L.; Chester, N.; Kromberg, J.G.R. Fetal Valproate Syndrome: Clinical and Neuro-Developmental Features in Two Sibling Pairs. Dev. Med. Child Neurol. 1994, 36, 361–369. [Google Scholar] [CrossRef]
- Ornoy, A. Valproic Acid in Pregnancy: How Much Are We Endangering the Embryo and Fetus? Reprod. Toxicol. 2009, 28, 1–10. [Google Scholar] [CrossRef]
- Vakilzadeh, G.; Martinez-Cerdeño, V. Pathology and Astrocytes in Autism. Neuropsychiatr. Dis. Treat. 2023, 19, 841–850. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Tóth, F.; Polyák, H.; Szabó, Á.; Mándi, Y.; Vécsei, L. Immune Influencers in Action: Metabolites and Enzymes of the Tryptophan-Kynurenine Metabolic Pathway. Biomedicines 2021, 9, 734. [Google Scholar] [CrossRef] [PubMed]
- Hedley, D.; Uljarević, M.; Foley, K.R.; Richdale, A.; Trollor, J. Risk and Protective Factors Underlying Depression and Suicidal Ideation in Autism Spectrum Disorder. Depress. Anxiety 2018, 35, 648–657. [Google Scholar] [CrossRef] [PubMed]
- Viscidi, E.W.; Triche, E.W.; Pescosolido, M.F.; McLean, R.L.; Joseph, R.M.; Spence, S.J.; Morrow, E.M. Clinical Characteristics of Children with Autism Spectrum Disorder and Co-Occurring Epilepsy. PLoS ONE 2013, 8, e67797. [Google Scholar] [CrossRef] [PubMed]
- Sundelin, H.E.K.; Larsson, H.; Lichtenstein, P.; Almqvist, C.; Hultman, C.M.; Tomson, T.; Ludvigsson, J.F. Autism and Epilepsy: A Population-Based Nationwide Cohort Study. Neurology 2016, 87, 192–197. [Google Scholar] [CrossRef]
- Zeidán-Chuliá, F.; de Oliveira, B.H.N.; Salmina, A.B.; Casanova, M.F.; Gelain, D.P.; Noda, M.; Verkhratsky, A.; Moreira, J.C. Altered Expression of Alzheimer’s Disease-Related Genes in the Cerebellum of Autistic Patients: A Model for Disrupted Brain Connectome and Therapy. Cell Death Dis. 2014, 5, e1250. [Google Scholar] [CrossRef]
- Lahiri, D.K.; Maloney, B.; Wang, R.; Sokol, D.K.; Rogers, J.T.; Westmark, C.J. How Autism and Alzheimer’s Disease Are TrAPPed. Mol. Psychiatry 2021, 26, 26–29. [Google Scholar] [CrossRef]
- Gouveia, F.; Camins, A.; Ettcheto, M.; Bicker, J.; Falcão, A.; Cruz, M.T.; Fortuna, A. Targeting Brain Renin-Angiotensin System for the Prevention and Treatment of Alzheimer’s Disease: Past, Present and Future. Ageing Res. Rev. 2022, 77, 101612. [Google Scholar] [CrossRef]
- Jeon, S.J.; Seo, J.E.; Yang, S.-I.; Choi, J.W.; Wells, D.; Shin, C.Y.; Ko, K.H. Cellular Stress-Induced up-Regulation of FMRP Promotes Cell Survival by Modulating PI3K-Akt Phosphorylation Cascades. J. Biomed. Sci. 2011, 18, 17. [Google Scholar] [CrossRef]
- Waite, K.; Eickholt, B.J. The Neurodevelopmental Implications of PI3K Signaling. Curr. Top. Microbiol. Immunol. 2010, 346, 245–265. [Google Scholar] [CrossRef]
- Wu, Z.T.; Ren, C.Z.; Yang, Y.H.; Zhang, R.W.; Sun, J.C.; Wang, Y.K.; Su, D.F.; Wang, W.Z. The PI3K Signaling-Mediated Nitric Oxide Contributes to Cardiovascular Effects of Angiotensin-(1-7) in the Nucleus Tractus Solitarii of Rats. Nitric Oxide 2016, 52, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.; Sharma, A.; Sharma, L. A Comprehensive Review of Alzheimer’s Association with Related Proteins: Pathological Role and Therapeutic Significance. Curr. Neuropharmacol. 2020, 18, 674–695. [Google Scholar] [CrossRef] [PubMed]
- Grigg, I.; Ivashko-Pachima, Y.; Hait, T.A.; Korenková, V.; Touloumi, O.; Lagoudaki, R.; Van Dijck, A.; Marusic, Z.; Anicic, M.; Vukovic, J.; et al. Tauopathy in the Young Autistic Brain: Novel Biomarker and Therapeutic Target. Transl. Psychiatry 2020, 10, 228. [Google Scholar] [CrossRef] [PubMed]
- Gassowska-Dobrowolska, M.; Kolasa-Wołosiuk, A.; Cieślik, M.; Dominiak, A.; Friedland, K.; Adamczyk, A. Alterations in Tau Protein Level and Phosphorylation State in the Brain of the Autistic-Like Rats Induced by Prenatal Exposure to Valproic Acid. Int. J. Mol. Sci. 2021, 22, 3209. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.W.; Shao, E.; Mucke, L. Tau: Enabler of Diverse Brain Disorders and Target of Rapidly Evolving Therapeutic Strategies. Science 2021, 371, eabb8255. [Google Scholar] [CrossRef] [PubMed]
- Engmann, O.; Giese, K.P. Crosstalk between Cdk5 and GSK3beta: Implications for Alzheimer’s Disease. Front. Mol. Neurosci. 2009, 2, 2. [Google Scholar] [CrossRef]
- Gassowska, M.; Baranowska-Bosiacka, I.; Moczydłowska, J.; Tarnowski, M.; Pilutin, A.; Gutowska, I.; Struzyńska, L.; Chlubek, D.; Adamczyk, A. Perinatal Exposure to Lead (Pb) Promotes Tau Phosphorylation in the Rat Brain in a GSK-3β and CDK5 Dependent Manner: Relevance to Neurological Disorders. Toxicology 2016, 347–349, 17–28. [Google Scholar] [CrossRef]
- Mishiba, T.; Tanaka, M.; Mita, N.; He, X.; Sasamoto, K.; Itohara, S.; Ohshima, T. Cdk5/P35 Functions as a Crucial Regulator of Spatial Learning and Memory. Mol. Brain 2014, 7, 82. [Google Scholar] [CrossRef]
- Brai, E.; Alina Raio, N.; Alberi, L. Notch1 Hallmarks Fibrillary Depositions in Sporadic Alzheimer’s Disease. Acta Neuropathol. Commun. 2016, 4, 64. [Google Scholar] [CrossRef]
- Nagarsheth, M.H.; Viehman, A.; Lippa, S.M.; Lippa, C.F. Notch-1 Immunoexpression Is Increased in Alzheimer’s and Pick’s Disease. J. Neurol. Sci. 2006, 244, 111–116. [Google Scholar] [CrossRef]
- AbdAlla, S.; El Hakim, A.; Abdelbaset, A.; Elfaramawy, Y.; Quitterer, U. Inhibition of ACE Retards Tau Hyperphosphorylation and Signs of Neuronal Degeneration in Aged Rats Subjected to Chronic Mild Stress. Biomed. Res. Int. 2015, 2015, 917156. [Google Scholar] [CrossRef] [PubMed]
- De Dios, L.; Collazo, C.; Inostroza-Nieves, Y. Renin-Angiotensin-System Increases Phosphorylated Tau and Reactive Oxygen Species in Human Cortical Neuron Cell Line. Biochem. Biophys. Rep. 2022, 32, 101355. [Google Scholar] [CrossRef] [PubMed]
- Varshney, V.; Garabadu, D. Ang (1-7)/Mas Receptor-Axis Activation Promotes Amyloid Beta-Induced Altered Mitochondrial Bioenergetics in Discrete Brain Regions of Alzheimer’s Disease-like Rats. Neuropeptides 2021, 86, 102122. [Google Scholar] [CrossRef] [PubMed]
- Rabie, M.A.; Abd El Fattah, M.A.; Nassar, N.N.; El-Abhar, H.S.; Abdallah, D.M. Angiotensin 1-7 Ameliorates 6-Hydroxydopamine Lesions in Hemiparkinsonian Rats through Activation of MAS Receptor/PI3K/Akt/BDNF Pathway and Inhibition of Angiotensin II Type-1 Receptor/NF-ΚB Axis. Biochem. Pharmacol. 2018, 151, 126–134. [Google Scholar] [CrossRef]
- Kehoe, P.G.; Wong, S.; Al Mulhim, N.; Palmer, L.E.; Miners, J.S. Angiotensin-Converting Enzyme 2 Is Reduced in Alzheimer’s Disease in Association with Increasing Amyloid-β and Tau Pathology. Alzheimers Res. Ther. 2016, 8, 50. [Google Scholar] [CrossRef]
- Firouzabadi, N.; Ghazanfari, N.; Shoushtari, A.A.; Erfani, N.; Fathi, F.; Bazrafkan, M.; Bahramali, E. Genetic Variants of Angiotensin-Converting Enzyme Are Linked to Autism: A Case-Control Study. PLoS ONE 2016, 11, e0153667. [Google Scholar] [CrossRef]
- Raciti, M.; Salma, J.; Spulber, S.; Gaudenzi, G.; Khalajzeyqami, Z.; Conti, M.; Anderlid, B.M.; Falk, A.; Hermanson, O.; Ceccatelli, S. NRXN1 Deletion and Exposure to Methylmercury Increase Astrocyte Differentiation by Different Notch-Dependent Transcriptional Mechanisms. Front. Genet. 2019, 10, 593. [Google Scholar] [CrossRef]
- Simone, M.; De Giacomo, A.; Palumbi, R.; Palazzo, C.; Lucisano, G.; Pompamea, F.; Micella, S.; Pascali, M.; Gabellone, A.; Marzulli, L.; et al. Serum Neurofilament Light Chain and Glial Fibrillary Acidic Protein as Potential Diagnostic Biomarkers in Autism Spectrum Disorders: A Preliminary Study. Int. J. Mol. Sci. 2023, 24, 3057. [Google Scholar] [CrossRef]
- Ahlsén, G.; Rosengren, L.; Belfrage, M.; Palm, A.; Haglid, K.; Hamberger, A.; Gillberg, C. Glial Fibrillary Acidic Protein in the Cerebrospinal Fluid of Children with Autism and Other Neuropsychiatric Disorders. Biol. Psychiatry 1993, 33, 734–743. [Google Scholar] [CrossRef]
- Bailey, A.R.; Hou, H.; Song, M.; Obregon, D.F.; Portis, S.; Barger, S.; Shytle, D.; Stock, S.; Mori, T.; Sanberg, P.G.; et al. GFAP Expression and Social Deficits in Transgenic Mice Overexpressing Human SAPPα. Glia 2013, 61, 1556–1569. [Google Scholar] [CrossRef]
- Grandbarbe, L.; Bouissac, J.; Rand, M.; Hrabé de Angelis, M.; Artavanis-Tsakonas, S.; Mohier, E. Delta-Notch Signaling Controls the Generation of Neurons/Glia from Neural Stem Cells in a Stepwise Process. Development 2003, 130, 1391–1402. [Google Scholar] [CrossRef] [PubMed]
- Nagao, M.; Sugimori, M.; Nakafuku, M. Cross Talk between Notch and Growth Factor/Cytokine Signaling Pathways in Neural Stem Cells. Mol. Cell. Biol. 2007, 27, 3982–3994. [Google Scholar] [CrossRef] [PubMed]
- Alzarea, S.I.; Alhassan, H.H.; Alzarea, A.I.; Al-Onazi, Z.H.; Afzal, M. Antidepressant-like Effects of Renin Inhibitor Aliskiren in an Inflammatory Mouse Model of Depression. Brain Sci. 2022, 12, 655. [Google Scholar] [CrossRef] [PubMed]
- Yao, M.; Wang, X.; Wang, X.; Zhang, T.; Chi, Y.; Gao, F. The Notch Pathway Mediates the Angiotensin II-Induced Synthesis of Extracellular Matrix Components in Podocytes. Int. J. Mol. Med. 2015, 36, 294–300. [Google Scholar] [CrossRef]
- Forrester, S.J.; Booz, G.W.; Sigmund, C.D.; Coffman, T.M.; Kawai, T.; Rizzo, V.; Scalia, R.; Eguchi, S. Angiotensin II Signal Transduction: An Update on Mechanisms of Physiology and Pathophysiology. Physiol. Rev. 2018, 98, 1627–1738. [Google Scholar] [CrossRef]
- Vitiello, B. Editorial: Targeting the Core Symptoms of Autism Spectrum Disorder With Mechanism-Based Medications. J. Am. Acad. Child Adolesc. Psychiatry 2021, 60, 816–817. [Google Scholar] [CrossRef] [PubMed]
- Niizawa, I.; Espinaco, B.Y.; Zorrilla, S.E.; Sihufe, G.A. Natural Astaxanthin Encapsulation: Use of Response Surface Methodology for the Design of Alginate Beads. Int. J. Biol. Macromol. 2019, 121, 601–608. [Google Scholar] [CrossRef]
- Bahbah, E.I.; Ghozy, S.; Attia, M.S.; Negida, A.; Emran, T.B.; Mitra, S.; Albadrani, G.M.; Abdel-Daim, M.M.; Uddin, M.S.; Simal-Gandara, J. Molecular Mechanisms of Astaxanthin as a Potential Neurotherapeutic Agent. Mar. Drugs 2021, 19, 201. [Google Scholar] [CrossRef]
- Al-Amin, M.M.; Rahman, M.M.; Khan, F.R.; Zaman, F.; Mahmud Reza, H. Astaxanthin Improves Behavioral Disorder and Oxidative Stress in Prenatal Valproic Acid-Induced Mice Model of Autism. Behav. Brain Res. 2015, 286, 112–121. [Google Scholar] [CrossRef]
- Thomas, S.D.; Jha, N.K.; Ojha, S.; Sadek, B. MTOR Signaling Disruption and Its Association with the Development of Autism Spectrum Disorder. Molecules 2023, 28, 1889. [Google Scholar] [CrossRef]
- Thabault, M.; Turpin, V.; Maisterrena, A.; Jaber, M.; Egloff, M.; Galvan, L. Cerebellar and Striatal Implications in Autism Spectrum Disorders: From Clinical Observations to Animal Models. Int. J. Mol. Sci. 2022, 23, 2294. [Google Scholar] [CrossRef] [PubMed]
- Rebik, A.; Broshevitskaya, N.; Kuzhuget, S.; Aleksandrov, P.; Abbasova, K.; Zaichenko, M.; Midzyanovskaya, I. Audiogenic Seizures and Social Deficits: No Aggravation Found in Krushinsky-Molodkina Rats. Biomedicines 2023, 11, 2566. [Google Scholar] [CrossRef]
- Cremone, I.M.; Nardi, B.; Amatori, G.; Palego, L.; Baroni, D.; Casagrande, D.; Massimetti, E.; Betti, L.; Giannaccini, G.; Dell’Osso, L.; et al. Unlocking the Secrets: Exploring the Biochemical Correlates of Suicidal Thoughts and Behaviors in Adults with Autism Spectrum Conditions. Biomedicines 2023, 11, 1600. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Spekker, E.; Szabó, Á.; Polyák, H.; Vécsei, L. Modelling the Neurodevelopmental Pathogenesis in Neuropsychiatric Disorders. Bioactive Kynurenines and Their Analogues as Neuroprotective Agents-in Celebration of 80th Birthday of Professor Peter Riederer. J. Neural. Transm. 2022, 129, 627–642. [Google Scholar] [CrossRef] [PubMed]
- Kerr, D.M.; Downey, L.; Conboy, M.; Finn, D.P.; Roche, M. Alterations in the Endocannabinoid System in the Rat Valproic Acid Model of Autism. Behav. Brain Res. 2013, 249, 124–132. [Google Scholar] [CrossRef]
- Zamberletti, E.; Gabaglio, M.; Woolley-Roberts, M.; Bingham, S.; Rubino, T.; Parolaro, D. Cannabidivarin Treatment Ameliorates Autism-Like Behaviors and Restores Hippocampal Endocannabinoid System and Glia Alterations Induced by Prenatal Valproic Acid Exposure in Rats. Front. Cell. Neurosci. 2019, 13, 367. [Google Scholar] [CrossRef]
- Kim, H.Y.; Lee, Y.J.; Kim, S.J.; Lee, J.D.; Kim, S.; Ko, M.J.; Kim, J.W.; Shin, C.Y.; Kim, K.B. Metabolomics Profiling of Valproic Acid-Induced Symptoms Resembling Autism Spectrum Disorders Using 1H NMR Spectral Analysis in Rat Model. J. Toxicol. Environ. Health A 2022, 85, 1–13. [Google Scholar] [CrossRef]
- Mabunga, D.F.N.; Gonzales, E.L.T.; Kim, J.; Kim, K.C.; Shin, C.Y. Exploring the Validity of Valproic Acid Animal Model of Autism. Exp. Neurobiol. 2015, 24, 285–300. [Google Scholar] [CrossRef]
- Bambini-Junior, V.; Zanatta, G.; Della Flora Nunes, G.; Mueller de Melo, G.; Michels, M.; Fontes-Dutra, M.; Nogueira Freire, V.; Riesgo, R.; Gottfried, C. Resveratrol Prevents Social Deficits in Animal Model of Autism Induced by Valproic Acid. Neurosci. Lett. 2014, 583, 176–181. [Google Scholar] [CrossRef]
- Kim, K.C.; Kim, P.; Go, H.S.; Choi, C.S.; Yang, S.-I.; Cheong, J.H.; Shin, C.Y.; Ko, K.H. The Critical Period of Valproate Exposure to Induce Autistic Symptoms in Sprague-Dawley Rats. Toxicol. Lett. 2011, 201, 137–142. [Google Scholar] [CrossRef]
- Weinstock, M. Prenatal Stressors in Rodents: Effects on Behavior. Neurobiol. Stress 2016, 6, 3–13. [Google Scholar] [CrossRef]
- Ying, C.-J.; Zhang, F.; Zhou, X.Y.; Hu, X.T.; Chen, J.; Wen, X.R.; Sun, Y.; Zheng, K.Y.; Tang, R.X.; Song, Y.J. Anti-Inflammatory Effect of Astaxanthin on the Sickness Behavior Induced by Diabetes Mellitus. Cell. Mol. Neurobiol. 2015, 35, 1027–1037. [Google Scholar] [CrossRef] [PubMed]
- McKinnell, Z.E.; Maze, T.; Ramos, A.; Challans, B.; Plakke, B. Valproic Acid Treated Female Long-Evans Rats Are Impaired on Attentional Set-Shifting. Behav. Brain Res. 2021, 397, 112966. [Google Scholar] [CrossRef] [PubMed]
- Deacon, R.M.J. Digging and Marble Burying in Mice: Simple Methods for in Vivo Identification of Biological Impacts. Nat. Protoc. 2006, 1, 122–124. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.; Han, Y.; Ren, S.; Zhang, B.; Zhao, Y.; Wang, X.; Zhang, S.; Qin, J. Correlation among Gut Microbiota, Fecal Metabolites and Autism-like Behavior in an Adolescent Valproic Acid-Induced Rat Autism Model. Behav. Brain Res. 2022, 417, 113580. [Google Scholar] [CrossRef]
- Elnahas, E.M.; Abuelezz, S.A.; Mohamad, M.I.; Nabil, M.M.; Abdelraouf, S.M.; Bahaa, N.; Hassan, G.A.; Ibrahim, E.A.; Ahmed, A.I.; Aboul-Fotouh, S. Validation of Prenatal versus Postnatal Valproic Acid Rat Models of Autism: A Behavioral and Neurobiological Study. Prog. Neuropsychopharmacol. Biol. Psychiatry 2021, 108, 110185. [Google Scholar] [CrossRef]
- Degroote, S.; Hunting, D.; Takser, L. Periconceptional Folate Deficiency Leads to Autism-like Traits in Wistar Rat Offspring. Neurotoxicol. Teratol. 2018, 66, 132–138. [Google Scholar] [CrossRef]
- Muhammad, R.N.; Ahmed, L.A.; Abdul Salam, R.M.; Ahmed, K.A.; Attia, A.S. Crosstalk Among NLRP3 Inflammasome, ETBR Signaling, and MiRNAs in Stress-Induced Depression-Like Behavior: A Modulatory Role for SGLT2 Inhibitors. Neurotherapeutics 2021, 18, 2664–2681. [Google Scholar] [CrossRef]
- Gao, H.L.; Yu, X.J.; Liu, K.L.; Zuo, Y.Y.; Fu, L.Y.; Chen, Y.M.; Zhang, D.D.; Shi, X.L.; Qi, J.; Li, Y.; et al. Chronic Infusion of Astaxanthin Into Hypothalamic Paraventricular Nucleus Modulates Cytokines and Attenuates the Renin-Angiotensin System in Spontaneously Hypertensive Rats. J. Cardiovasc. Pharmacol. 2021, 77, 170–181. [Google Scholar] [CrossRef]
- Wang, X.L.; Iwanami, J.; Min, L.J.; Tsukuda, K.; Nakaoka, H.; Bai, H.Y.; Shan, B.S.; Kan-No, H.; Kukida, M.; Chisaka, T.; et al. Deficiency of Angiotensin-Converting Enzyme 2 Causes Deterioration of Cognitive Function. npj Aging Mech. Dis. 2016, 2, 16024. [Google Scholar] [CrossRef]
- Freund, M.; Walther, T.; Von Bohlen Und Halbach, O. Immunohistochemical Localization of the Angiotensin-(1-7) Receptor Mas in the Murine Forebrain. Cell Tissue Res. 2012, 348, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Shen, A.; Wu, X.; Shen, Z.; Chen, X.; Li, J.; Liu, L.; Lin, X.; Wu, M.; Chen, Y.; et al. Qingda Granule Attenuates Angiotensin II-Induced Cardiac Hypertrophy and Apoptosis and Modulates the PI3K/AKT Pathway. Biomed. Pharmacother. 2021, 133, 111022. [Google Scholar] [CrossRef]
- Gironacci, M.M.; Cerniello, F.M.; Longo Carbajosa, N.A.; Goldstein, J.; Cerrato, B.D. Protective Axis of the Renin-Angiotensin System in the Brain. Clin. Sci. 2014, 127, 295–306. [Google Scholar] [CrossRef]
- Kostenis, E.; Milligan, G.; Christopoulos, A.; Sanchez-Ferrer, C.F.; Heringer-Walther, S.; Sexton, P.M.; Gembardt, F.; Kellett, E.; Martini, L.; Vanderheyden, P.; et al. G-Protein-Coupled Receptor Mas Is a Physiological Antagonist of the Angiotensin II Type 1 Receptor. Circulation 2005, 111, 1806–1813. [Google Scholar] [CrossRef] [PubMed]
- Nicolini, C.; Ahn, Y.; Michalski, B.; Rho, J.M.; Fahnestock, M. Decreased MTOR Signaling Pathway in Human Idiopathic Autism and in Rats Exposed to Valproic Acid. Acta Neuropathol. Commun. 2015, 3, 3. [Google Scholar] [CrossRef] [PubMed]
- Luo, T.; Ou, J.N.; Cao, L.F.; Peng, X.Q.; Li, Y.M.; Tian, Y.Q. The Autism-Related LncRNA MSNP1AS Regulates Moesin Protein to Influence the RhoA, Rac1, and PI3K/Akt Pathways and Regulate the Structure and Survival of Neurons. Autism Res. 2020, 13, 2073–2082. [Google Scholar] [CrossRef]
- Zhang, Y.; Xiang, Z.; Jia, Y.; He, X.; Wang, L.; Cui, W. The Notch Signaling Pathway Inhibitor Dapt Alleviates Autism-like Behavior, Autophagy and Dendritic Spine Density Abnormalities in a Valproic Acid-Induced Animal Model of Autism. Prog. Neuropsychopharmacol. Biol. Psychiatry 2019, 94, 109644. [Google Scholar] [CrossRef]
- Young, A.M.H.; Campbell, E.; Lynch, S.; Suckling, J.; Powis, S.J. Aberrant NF-KappaB Expression in Autism Spectrum Condition: A Mechanism for Neuroinflammation. Front. Psychiatry 2011, 2, 27. [Google Scholar] [CrossRef]
- Ang, H.L.; Tergaonkar, V. Notch and NFkappaB Signaling Pathways: Do They Collaborate in Normal Vertebrate Brain Development and Function? Bioessays 2007, 29, 1039–1047. [Google Scholar] [CrossRef]
- Li, Y.J.; Zhang, X.; Li, Y.M. Antineuroinflammatory Therapy: Potential Treatment for Autism Spectrum Disorder by Inhibiting Glial Activation and Restoring Synaptic Function. CNS Spectr. 2020, 25, 493–501. [Google Scholar] [CrossRef]
- Zhou, X.; Zhang, J.; Li, Y.; Cui, L.; Wu, K.; Luo, H. Astaxanthin Inhibits Microglia M1 Activation against Inflammatory Injury Triggered by Lipopolysaccharide through Down-Regulating MiR-31-5p. Life Sci. 2021, 267, 118943. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Lu, Y.; Wu, Q.; Dai, H.; Li, W.; Lv, S.; Zhou, X.; Zhang, X.; Hang, C.; Wang, J. Astaxanthin Mitigates Subarachnoid Hemorrhage Injury Primarily by Increasing Sirtuin 1 and Inhibiting the Toll-like Receptor 4 Signaling Pathway. FASEB J. 2019, 33, 722–737. [Google Scholar] [CrossRef] [PubMed]
- Szczepanska-Sadowska, E.; Wsol, A.; Cudnoch-Jedrzejewska, A.; Czarzasta, K.; Żera, T. Multiple Aspects of Inappropriate Action of Renin-Angiotensin, Vasopressin, and Oxytocin Systems in Neuropsychiatric and Neurodegenerative Diseases. J. Clin. Med. 2022, 11, 908. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.Q.; Wei, X.B.; Sun, R.; Cai, Y.W.; Lou, H.Y.; Wang, J.W.; Chen, A.F.; Zhang, X.M. Angiotensin II Stimulates Intercellular Adhesion Molecule-1 via an AT1 Receptor/Nuclear Factor-KappaB Pathway in Brain Microvascular Endothelial Cells. Life Sci. 2006, 78, 1293–1298. [Google Scholar] [CrossRef] [PubMed]
- Tai, C.; Chang, C.W.; Yu, G.Q.; Lopez, I.; Yu, X.; Wang, X.; Guo, W.; Mucke, L. Tau Reduction Prevents Key Features of Autism in Mouse Models. Neuron 2020, 106, 421–437.e11. [Google Scholar] [CrossRef]
- Patrick, G.N.; Zukerberg, L.; Nikolic, M.; De La Monte, S.; Dikkes, P.; Tsai, L.H. Conversion of P35 to P25 Deregulates Cdk5 Activity and Promotes Neurodegeneration. Nature 1999, 402, 615–622. [Google Scholar] [CrossRef]
- Yang, X.B.; Zu, H.B.; Zhao, Y.F.; Yao, K. Agomelatine Prevents Amyloid Plaque Deposition, Tau Phosphorylation, and Neuroinflammation in APP/PS1 Mice. Front. Aging Neurosci. 2022, 13, 766410. [Google Scholar] [CrossRef]
- Codagnone, M.G.; Podestá, M.F.; Uccelli, N.A.; Reinés, A. Differential Local Connectivity and Neuroinflammation Profiles in the Medial Prefrontal Cortex and Hippocampus in the Valproic Acid Rat Model of Autism. Dev. Neurosci. 2015, 37, 215–231. [Google Scholar] [CrossRef]
- Abd El-Fatah, I.M.; Abdelrazek, H.M.A.; Ibrahim, S.M.; Abdallah, D.M.; El-Abhar, H.S. Dimethyl Fumarate Abridged Tauo-/Amyloidopathy in a D-Galactose/Ovariectomy-Induced Alzheimer’s-like Disease: Modulation of AMPK/SIRT-1, AKT/CREB/BDNF, AKT/GSK-3β, Adiponectin/Adipo1R, and NF-ΚB/IL-1β/ROS Trajectories. Neurochem. Int. 2021, 148, 105082. [Google Scholar] [CrossRef]
- Effect of Astaxanthin on Neuron Damage, Inflammatory Factors Expressions and Oxidative Stress in Mice with Subarachnoid Hemorrhage—PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/34956522/ (accessed on 11 October 2023).
- Ongali, B.; Nicolakakis, N.; Tong, X.K.; Aboulkassim, T.; Papadopoulos, P.; Rosa-Neto, P.; Lecrux, C.; Imboden, H.; Hamel, E. Angiotensin II Type 1 Receptor Blocker Losartan Prevents and Rescues Cerebrovascular, Neuropathological and Cognitive Deficits in an Alzheimer’s Disease Model. Neurobiol. Dis. 2014, 68, 126–136. [Google Scholar] [CrossRef]
- Messiha, B.A.S.; Ali, M.R.A.; Khattab, M.M.; Abo-Youssef, A.M. Perindopril Ameliorates Experimental Alzheimer’s Disease Progression: Role of Amyloid β Degradation, Central Estrogen Receptor and Hyperlipidemic-Lipid Raft Signaling. Inflammopharmacology 2020, 28, 1343–1364. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Primer Sequence (5′–3′) | Accession Number |
|---|---|---|
| MAS receptor | F: TGACCATTGAACAGATTGCCA R: TGTAGTTTGTGACGGCTGGTG | NM_153722 |
| β-Actin | F: CCCATCTATGAGGGTTACGC R: TTTAATGTCACGCACGATTTC | NM_031144.3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Samra, A.I.; Kamel, A.S.; Abdallah, D.M.; El Fattah, M.A.A.; Ahmed, K.A.; El-Abhar, H.S. Preclinical Evidence for the Role of the Yin/Yang Angiotensin System Components in Autism Spectrum Disorder: A Therapeutic Target of Astaxanthin. Biomedicines 2023, 11, 3156. https://doi.org/10.3390/biomedicines11123156
Samra AI, Kamel AS, Abdallah DM, El Fattah MAA, Ahmed KA, El-Abhar HS. Preclinical Evidence for the Role of the Yin/Yang Angiotensin System Components in Autism Spectrum Disorder: A Therapeutic Target of Astaxanthin. Biomedicines. 2023; 11(12):3156. https://doi.org/10.3390/biomedicines11123156
Chicago/Turabian StyleSamra, Ayat I., Ahmed S. Kamel, Dalaal M. Abdallah, Mai A. Abd El Fattah, Kawkab A. Ahmed, and Hanan S. El-Abhar. 2023. "Preclinical Evidence for the Role of the Yin/Yang Angiotensin System Components in Autism Spectrum Disorder: A Therapeutic Target of Astaxanthin" Biomedicines 11, no. 12: 3156. https://doi.org/10.3390/biomedicines11123156
APA StyleSamra, A. I., Kamel, A. S., Abdallah, D. M., El Fattah, M. A. A., Ahmed, K. A., & El-Abhar, H. S. (2023). Preclinical Evidence for the Role of the Yin/Yang Angiotensin System Components in Autism Spectrum Disorder: A Therapeutic Target of Astaxanthin. Biomedicines, 11(12), 3156. https://doi.org/10.3390/biomedicines11123156