
Rodent Models of Huntington’s Disease: An Overview
 , , , , , ,
, , , , , ,  and
and 
Abstract
:1. Introduction
2. Utility and Validity of Animal Models of Huntington’s Disease
3. Non-genetic Models of Huntington’s Disease
3.1. Excitotoxin Models
3.2. Metabolic Models

{kind=link}
{kind=link}
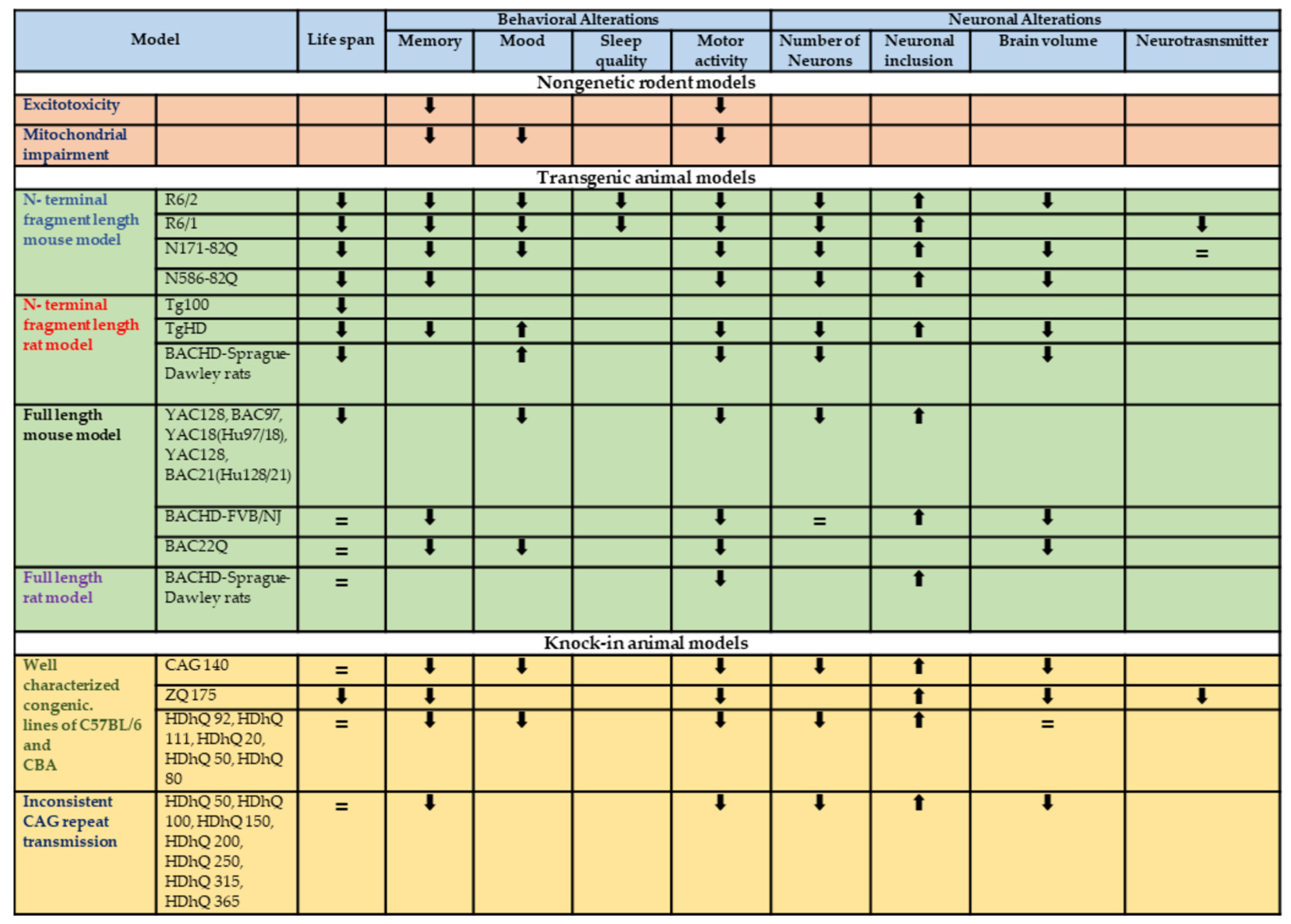
| Animal Model | Strain | Behavioral and Neuronal Changes | Reference | |
|---|---|---|---|---|
| Excitotoxin model: Quinolinic acid | Rat (Sprague-Dawley and Fischer), Mouse and non-human primate | Excitotoxicity | Hyperkinesia, apomorphine-induced dystonia and dyskinesia, spontaneous dyskinesia with higher doses. Poor memory recall Visual–spatial deficiencies Procedural memory deficits | [32,33,34,35,36] |
| Metabolic models: 3-Nitropropionic acid | Rat (all except Fischer), mouse, non-human primates | Mitochondrial impairment by irreversibly inhibiting succinate dehydrogenase | Hyperkinesia (low dose), hypokinesia (high dose), apomorphine-induced dystonia and dyskinesia, spontaneous dyskinesia with long-term administration. Defects in ORDT in non-human primates Rats’ working memory and reference memory tested with radial arm water mazes Open-field apparatus habituation impairments | [30,37,38] |
4. Genetic Rodent Models of HD
4.1. N-Terminal Fragment Mouse Models
4.2. N-Terminal Rat Length Fragment Models
4.3. Full Length Mouse Models
4.4. Full-Length Rat Models
| Transgenic Model | ||||||
|---|---|---|---|---|---|---|
| Animal Model | Strain | CAG Repeat Length | Life Span | Behavioral and Neuronal Alterations | Reference | |
| N-terminal fragment length mouse model | R6/2 | C57BL/6J | 116 | Reduced life span (10–13 weeks) | Progressive decline from 5 weeks rotarod (5–6 weeks). In the open field, anxiety-like behavior (8 weeks). Learning and memory deficits worsen over time. Sleep disturbances (9 weeks). Significant reduction in neurons and neuronal atrophy in the striatum within 3 months. At 4 weeks, neurons had intranuclear inclusions in the cortex. Reduced brain volume. | [43,44,47,92,93,94] |
| DBA/2J | 116 | |||||
| B6CBA/Ca Mixed | 128 | |||||
| B6CBA/Ca Mixed | 160 | |||||
| C57BL/6J | 168 | |||||
| DBA/2J | 168 | |||||
| C57BL/6J | 251 | |||||
| DBA/2J | 251 | |||||
| C57BL/6J | 293 | |||||
| DBA/2J | 293 | |||||
| R6/1 | C57BL/6J, BALB/cByJ, B6CBA/Ca mixed | 116 | Reduced life span (32–40 weeks) | Progressive decline rotarod (8 weeks). Reduced activity open field (23 weeks) Anxiety-like behavior open field. Spatial learning deficits in Morris water maze. Sleep disturbance. Decrease in striatal volume. Presence of cellular inclusions at 5 months of age. Neuronal intranuclear inclusions. Neurotransmitter receptor level changes. | [42,54,95] | |
| N171-82Q | C57BL/6× B6C3H/He J mixed | 82 | Reduced life span (Line77—2.5 months, lines 81 and 100—5–6 months, Line 6—8–11 months) | Progressive decline, gait abnormalities, hypoactivity, depressive-like behavior, and working and reference memory deficits at 14 weeks of age. Shrinkage and neuronal loss in the striatum, hippocampus, and frontal cortex at 4 months of age. Neuronal intranuclear inclusions. Reduced brain volume. Dopamine and serotonin levels remain unchanged. | [47,57,68] | |
| N586-82Q | C57BL/6J B6C3H | 82 | Reduced life span (8–9 months for N586-82Q-63C) | Progressive decline in rotarod, and open field consecutively from 3 and 4 months. Asymmetric dyskinesia (4 months onwards). Fear conditioning causes contextual and cue-dependent memory deficits at 8 months. Large inclusions in every part of the brain, astrogliosis in cerebellum, striatum, and cortex. Granule cells in the cerebellum are gradually degenerating. Total brain capacity is reduced. Cerebellar and hippocampal atrophy occurs. | [55,57,96] | |
| Tg100 | B6SJL mixed | 100 | Reduced life Expectancy (10–12 month) | Cortical changes, formation of dysmorphic dendrites, enhanced intracellular Ca2+ levels. | [59,60,61] | |
| N-terminal fragment length rat model | TgHD | Sprague-Dawley rats | 51 | Increased mortality at 24 months of age. | Around the age of 6 months, the development of aggregate becomes visible. Anxiolytic behavior can emerge as early as 2 months of age, while motor and cognitive phenotypes typically appear around 6–9 months. In specific brain regions, structural changes, neuronal loss, and atrophy. Impaired spatial working. Reference memory, attentional deficiencies | [97] |
| BACHD | Sprague-Dawley rats | 97 | Reduced life Expectancy (16–20 weeks) | Alterations in structure and the presence of decaying neurons, gait anomalies, and rotarod performance. Reduced exploration anxiety. At around 3 months of age, visible aggregate development begins. The onset of behavioral phenotypes differs, with rotarod deficits visible at 1 month and anxiolytic behavior visible at 4 months. Frequently reported smaller brain volume. | [72,87,89] | |
| Full length mouse model | YAC128 | C57BL/6 FVB/N | 128 | Reduced life expectancy in male | Deficits on T-maze. At 3 months of age on an open-field test, they display hyperkinesia, a 50% reduction in body weight when compared to wild-type littermates. Circling behavior, gait problems, ataxia, and hindlimb clasping are all common symptoms. The rotarod test shows a steady deterioration. Hyperactivity (3 months) followed by hypoactivity (12 months). Depressive-like phenotype (3 months) and sucrose consumption. Cell loss ranges from 18% to 40% primarily in the lateral striatum. Increased HTT staining in the nucleus. The striatum, nucleus accumbens, cortex, and cerebellum neurons show intranuclear inclusions (15 months). | [68,70] |
| BAC97 YAC18 (Hu97/18) | FVB/N | 18, 97 | ||||
| YAC128, BAC21 (Hu128/21) | FVB/N | 125 | ||||
| BACHD | FVB/NJ | 97 | Normal life span | Decreased motor learning over time, the rotarod (2 months): progressive deterioration Catwalk examination of gait impairments (9–10 months). Object recognition memory impaired (6 months). Deficits in reversal learning and strategy shift water (9–10 months) T maze and cross maze. Startle response is disrupted by sensorimotor gating abnormalities (9 months), inhibition of the prepulse (7 months). Decrease in activity in the open field (from 6 months). Inclusions of mHtt in the brain and striatum (12 months) There is no neuronal loss. A reduction in cortical and striatal volume (12 months). | [68,71] | |
| BAC22Q | mouse model | - | Normal life span | BAC226Q mice gradually showed HD-like psychiatric and cognitive phenotypes at 2 months. BAC226Q mice presented motor deficits from 3 to 4 months. At 11 months, BAC226Q mice presented neuropathology like brain atrophy, particularly in the cortex and striatum. | [86] | |
| Full length rat model | BACHD | Sprague-Dawley rats | 97 | Normal life span | Transgenic rats perform better on the rotarod test than wild-type littermates, but their performance deteriorates over time. At 10–15 months: head dyskinesias and gait abnormalities. 12 months: Traveling over an elevated beam takes longer. Compared to wild-type rats, at the age of 24 months, lost 20% of their weight. Deficiencies on the radial arm and high plus mazes in the age of 12 months. At 8 months of age, the lateral ventricles are enlarged. At 12 months: Inclusions in the brain and 2 months. | [60,87,89] |
5. Knock-In Mouse Models
6. The Need for Large Animal Models of Huntington’s Disease
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Cepeda, C.; Levine, M.S. Synaptic Dysfunction in Huntington’s Disease: Lessons from Genetic Animal Models. Neuroscientist 2022, 28, 20–40. [Google Scholar] [CrossRef] [PubMed]
- Jia, Q.; Li, S.; Li, X.J.; Yin, P. Neuroinflammation in Huntington’s disease: From animal models to clinical therapeutics. Front. Immunol. 2022, 13, 1088124. [Google Scholar] [CrossRef] [PubMed]
- Walker, F.O. Huntington’s disease. Lancet 2007, 369, 218–228. [Google Scholar] [CrossRef] [PubMed]
- Craufurd, D.; Snowden, J. Neuropsychological and neuropsychiatric aspects of Huntington’s disease. In Huntington’s Disease; Bates, G., Harper, P., Jones, L., Eds.; Oxford University Press: New York, NY, USA, 2002; pp. 62–94. [Google Scholar]
- Baliko, L.; Csala, B.; Czopf, J. Suicide in Hungarian Huntington’s disease patients. Neuroepidemiology 2004, 23, 258–260. [Google Scholar] [CrossRef] [PubMed]
- St-Cyr, S.; Child, D.D.; Giaime, E.; Smith, A.R.; Pascua, C.J.; Hahm, S.; Saiah, E.; Davidson, B.L. Huntington’s disease phenotypes are improved via mTORC1 modulation by small molecule therapy. PLoS ONE 2022, 17, 0273710. [Google Scholar] [CrossRef] [PubMed]
- Robins Wahlin, T.B.; Backman, L.; Lundin, A.; Haegermark, A.; Winblad, B.; Anvret, M. High suicidal ideation in persons testing for Huntington’s disease. Acta Neurol. Scand. 2000, 102, 150–161. [Google Scholar] [CrossRef] [PubMed]
- Gutekunst, C.A.; Norflus, F.; Hersch, S.M. The neuropathology of Huntington’s disease. In Huntington’s Disease; Bates, G., Harper, P., Jones, L., Eds.; Oxford University Press: New York, NY, USA, 2002; pp. 251–275. [Google Scholar]
- Rubinsztein, D.C. Molecular biology of Huntington’s disease (HD) and HD-like disorders. In Genetics of Movement Disorders; Pulst, S., Ed.; Academic Press: Cambridge, MA, USA, 2003; pp. 365–377. [Google Scholar]
- Koch, E.T.; Sepers, M.D.; Cheng, J.; Raymond, L.A. Early Changes in Striatal Activity and Motor Kinematics in a Huntington’s Disease Mouse Model. Mov. Disord. 2022, 37, 2021–2032. [Google Scholar] [CrossRef]
- Wakida, N.M.; Lau, A.L.; Nguyen, J.; Cruz, G.M.S.; Fote, G.M.; Steffan, J.S.; Thompson, L.M.; Berns, M.W. Diminished LC3-Associated Phagocytosis by Huntington’s Disease Striatal Astrocytes. J. Huntingt. Dis. 2022, 11, 25–33. [Google Scholar] [CrossRef]
- Paulsen, J.S.; Hoth, K.F.; Nehl, C.; Stierman, L. Critical periods of suicide risk in Huntington’s disease. Am. J. Psychiatry 2005, 162, 725–731. [Google Scholar] [CrossRef]
- Spargo, E.; Everall, I.P.; Lantos, P.L. Neuronal loss in the hippocampus in Huntington’s disease: A comparison with HIV infection. J. Neurol. Neurosurg. Psychiatry 1993, 56, 487–491. [Google Scholar] [CrossRef]
- Macdonald, V.; Halliday, G.M.; Trent, R.J.; McCusker, E.A. Significant loss of pyramidal neurons in the angular gyrus of patients with Huntington’s disease. Neuropathol. Appl. Neurobiol. 1997, 23, 492–495. [Google Scholar] [CrossRef]
- Macdonald, V.; Halliday, G. Pyramidal cell loss in motor cortices in Huntington’s disease. Neurobiol. Dis. 2002, 10, 378–386. [Google Scholar] [CrossRef] [PubMed]
- Kremer, H.P.; Roos, R.A.; Dingjan, G.M.; Bots, G.T.; Bruyn, G.W.; Hofman, M.A. The hypothalamic lateral tuberal nucleus and the characteristics of neuronal loss in Huntington’s disease. Neurosci. Lett. 1991, 132, 101–104. [Google Scholar] [CrossRef] [PubMed]
- Kremer, H.P. The hypothalamic lateral tuberal nucleus: Normal anatomy and changes in neurological diseases. Prog. Brain Res. 1992, 93, 249–261. [Google Scholar] [PubMed]
- Heinsen, H.; Rüb, U.; Bauer, M.; Ulmar, G.; Bethke, B.; Schüler, M.; Böcker, F.; Eisenmenger, W.; Götz, M.; Korr, H.; et al. Nerve cell loss in the thalamic mediodorsal nucleus in Huntington’s disease. Acta Neuropathol. 1999, 97, 613–622. [Google Scholar] [CrossRef] [PubMed]
- Takano, H.; Gusella, J.F. The predominantly HEAT-like motif structure of huntingtin and its association and coincidentnuclear entry with dorsal, an NF-kB/Rel/dorsal family transcription factor. BMC Neurosci. 2002, 14, 15. [Google Scholar]
- Cattaneo, E.; Zuccato, C.; Tartari, M. Normal huntingtin function: An alternative approach to Huntington’s disease. Nat. Rev. Neurosci. 2005, 6, 919–930. [Google Scholar] [CrossRef]
- Yanai, A.; Huang, K.; Kang, R.; Singaraja, R.R.; Arstikaitis, P.; Gan, L.; Orban, P.C.; Mullard, A.; Cowan, C.M.; Raymond, L.A.; et al. Palmitoylation of huntingtin by HIP14 is essential for its trafficking and function. Nat. Neurosci. 2006, 9, 824–831. [Google Scholar] [CrossRef]
- Sadri-Vakili, G.; Cha, J.H. Mechanisms of disease: Histone modifications in Huntington’s disease. Nat. Clin. Pract. Neurol. 2006, 2, 330–338. [Google Scholar] [CrossRef]
- Savas, J.N.; Ma, B.; Deinhardt, K.; Culver, B.P.; Restituito, S.; Wu, L.; Belasco, J.G.; Chao, M.V.; Tanese, N. A role for Huntington disease protein in dendritic RNA granules. J. Biol. Chem. 2010, 285, 13142–13153. [Google Scholar] [CrossRef]
- Brignull, H.R.; Morley, J.F.; Garcia, S.M.; Morimoto, R.I. Modeling polyglutamine pathogenesis in C. elegans. Methods Enzymol. 2006, 412, 256–282. [Google Scholar] [PubMed]
- Coyle, J.T.; Puttfarcken, P. Oxidative stress, glutamate, and neurodegenerative disorders. Science 1993, 262, 689–695. [Google Scholar] [CrossRef] [PubMed]
- Ramaswamy, S.; McBride, J.L.; Kordower, J.H. Animal models of Huntington’s disease. ILAR J. 2007, 48, 356–373. [Google Scholar] [CrossRef] [PubMed]
- Hargreaves, K.M.; Pardridge, W.M. Neutral amino acid transport at the human blood-brain barrier. J. Biol. Chem. 1988, 263, 19392–19397. [Google Scholar] [CrossRef] [PubMed]
- Schwarcz, R.; Okuno, E.; White, R.J.; Bird, E.D.; Whetsell, W.O., Jr. 3-Hydroxyanthranilate oxygenase activity is increased in the brains of Huntington disease victims. Proc. Natl. Acad. Sci. USA 1988, 85, 4079–4081. [Google Scholar] [CrossRef] [PubMed]
- Bordelon, Y.M.; Chesselet, M.F.; Nelson, D.; Welsh, F.; Erecińska, M. Energetic dysfunction in quinolinic acid-lesioned rat striatum. J. Neurochem. 1997, 69, 1629–1639. [Google Scholar] [CrossRef] [PubMed]
- Ouary, S.; Bizat, N.; Altairac, S.; Ménétrat, H.; Mittoux, V.; Condé, F.; Hantraye, P.; Brouillet, E. Major strain differences in response to chronic systemic administration of the mitochondrial toxin 3-nitropropionic acid in rats: Implications for neuroprotection studies. Neuroscience 2000, 97, 521–530. [Google Scholar] [CrossRef]
- Blum, D.; Gall, D.; Cuvelier, L.; Schiffmann, S.N. Topological analysis of striatal lesions induced by 3-nitropropionic acid in the Lewis rat. Neuro Rep. 2001, 12, 1769–1772. [Google Scholar] [CrossRef]
- Kendall, A.L.; David, F.; Rayment, G.; Torres, E.M.; Annett, L.E.; Dunnett, S.B. The influence of excitotoxic basal ganglia lesions on motor performance in the common marmoset. Brain 2000, 123, 1442–1458. [Google Scholar] [CrossRef]
- Ribeiro, C.A.; Grando, V.; Dutra Filho, C.S.; Wannmacher, C.M.; Wajner, M. Evidence that quinolinic acid severely impairs energy metabolism through activation of NMDA receptors in striatum from developing rats. J. Neurochem. 2006, 99, 1531–1542. [Google Scholar] [CrossRef]
- McLin, J.P.; Thompson, L.M.; Steward, O. Differential susceptibility to striatal neurodegeneration induced by quinolinic acid and kainate in inbred, outbred and hybrid mouse strains. Eur. J. Neurosci. 2006, 24, 3134–3140. [Google Scholar] [CrossRef] [PubMed]
- Emerich, D.F.; Thanos, C.G.; Goddard, M.; Skinner, S.J.; Geany, M.S.; Bell, W.J.; Bintz, B.; Schneider, P.; Chu, Y.; Babu, R.S.; et al. Extensive neuroprotection by choroid plexus transplants in excitotoxin lesioned monkeys. Neurobiol. Dis. 2006, 23, 471–480. [Google Scholar] [CrossRef] [PubMed]
- Vazey, E.M.; Chen, K.; Hughes, S.M.; Connor, B. Transplanted adult neural progenitor cells survive, differentiate and reduce motor function impairment in a rodent model of Huntington’s disease. Exp. Neurol. 2006, 199, 384–396. [Google Scholar] [CrossRef] [PubMed]
- Palfi, S.; Leventhal, L.; Goetz, C.G.; Hantraye, T.; Roitberg, B.Z.; Sramek, J.; Emborg, M.; Kordower, J.H. Delayed onset of progressive dystonia following subacute 3-nitropropionic acid treatment in Cebus apella monkeys. Mov. Disord. 2000, 15, 524–530. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Calingasan, N.Y.; Chen, J.; Ley, J.J.; Becker, D.A.; Beal, M.F. A novel azulenyl nitrone antioxidant protects against MPTP and 3-nitropropionic acid neurotoxicity’s. Exp. Neurol. 2005, 191, 86–93. [Google Scholar] [CrossRef]
- Menalled, L.B.; Chesselet, M.F. Mouse models of Huntington’s disease. Trends Pharmacol. Sci. 2002, 23, 32–39. [Google Scholar] [CrossRef]
- Mangiarini, L.; Sathasivam, K.; Seller, M.; Cozens, B.; Harper, A.; Hetherington, C.; Lawton, M.; Trottier, Y.; Lehrach, H.; Davies, S.W.; et al. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell 1996, 87, 493–506. [Google Scholar] [CrossRef]
- Davies, S.W.; Turmaine, M.; Cozens, B.A.; DiFiglia, M.; Sharp, A.H.; Ross, C.A.; Scherzinger, E.; Wanker, E.E.; Mangiarini, L.; Bates, G.P. Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell 1997, 90, 537–548. [Google Scholar] [CrossRef]
- Bayram-Weston, Z.; Jones, L.; Dunnett, S.B.; Brooks, S.P. Light and electron microscopic characterization of the evolution of cellular pathology in the R6/1 Huntington’s disease transgenic mice. Brain Res. Bull. 2012, 88, 104–112. [Google Scholar] [CrossRef]
- Cummings, D.M.; Alaghband, Y.; Hickey, M.A.; Joshi, P.R.; Hong, S.C.; Zhu, C.; Ando, T.K.; André, V.M.; Cepeda, C.; Watson, J.B.; et al. A critical window of CAG repeat-length correlates with phenotype severity in the R6/2 mouse model of Huntington’s disease. J. Neurophysiol. 2012, 107, 677–691. [Google Scholar] [CrossRef]
- Laforet, G.A.; Sapp, E.; Chase, K.; McIntyre, C.; Boyce, F.M.; Campbell, M.; Cadigan, B.A.; Warzecki, L.; Tagle, D.A.; Reddy, P.H.; et al. Changes in cortical and striatal neurons predict behavioral and electrophysiological abnormalities in a transgenic murine model of Huntington’s disease. J. Neurosci. 2001, 21, 9112–9123. [Google Scholar] [CrossRef] [PubMed]
- Farshim, P.P.; Bates, G.P. Mouse Models of Huntington’s Disease. Methods Mol. Biol. 2018, 1780, 97–120. [Google Scholar] [PubMed]
- Cepeda, C.; Hurst, R.S.; Calvert, C.R.; Hernández-Echeagaray, E.; Nguyen, O.K.; Jocoy, E.; Christian, L.J.; Ariano, M.A.; Levine, M.S. Transient and progressive electrophysiological alterations in the corticostriatal pathway in a mouse model of Huntington’s disease. J. Neurosci. 2003, 23, 961–969. [Google Scholar] [CrossRef] [PubMed]
- Stack, E.C.; Kubilus, J.K.; Smith, K.; Cormier, K.; Del Signore, S.J.; Guelin, E.; Ryu, H.; Hersch, S.M.; Ferrante, R.J. Chronology of behavioral symptoms and neuropathological sequela in R6/2 Huntington’s disease transgenic mice. J. Comp. Neurol. 2005, 490, 354–370. [Google Scholar] [CrossRef] [PubMed]
- Menalled, L.; El-Khodor, B.F.; Patry, M.; Suárez-Fariñas, M.; Orenstein, S.J.; Zahasky, B.; Leahy, C.; Wheeler, V.; Yang, X.W.; MacDonald, M.; et al. Systematic behavioral evaluation of Huntington’s disease transgenic and knock-in mouse models. Neurobiol. Dis. 2009, 35, 319–336. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Del Mar, N.; Meade, C.; Goldowitz, D.; Reiner, A. Differential changes in striatal projection neurons in R6/2 transgenic mice for Huntington’s disease. Neurobiol. Dis. 2002, 11, 369–385. [Google Scholar] [CrossRef] [PubMed]
- Levine, M.S.; Klapstein, G.J.; Koppel, A.; Gruen, E.; Cepeda, C.; Vargas, M.E.; Jokel, E.S.; Carpenter, E.M.; Zanjani, H.; Hurst, R.S.; et al. Enhanced sensitivity to N-methyl-D-aspartate receptor activation in transgenic and knockin mouse models of Huntington’s disease. J. Neurosci. Res. 1999, 58, 515–532. [Google Scholar] [CrossRef]
- Chesselet, M.F. Mapping the basal ganglia. In Brain Mapping: The Applications; Toga, A.W., Mazziotta, J.C., Eds.; Academic Press: Cambridge, MA, USA, 2000; pp. 177–206. [Google Scholar]
- Petersén, A.; Gil, J.; Maat-Schieman, M.L.; Björkqvist, M.; Tanila, H.; Araújo, I.M.; Smith, R.; Popovic, N.; Wierup, N.; Norlén, P.; et al. Orexin loss in Huntington’s disease. Hum. Mol. Genet. 2005, 14, 39–47. [Google Scholar] [CrossRef]
- Naver, B.; Stub, C.; Møller, M.; Fenger, K.; Hansen, A.K.; Hasholt, L.; Sørensen, S.A. Molecular and behavioral analysis of the R6/1 Huntington’s disease transgenic mouse. Neuroscience 2003, 122, 1049–1057. [Google Scholar] [CrossRef]
- Van Dellen, A.; Welch, J.; Dixon, R.M.; Cordery, P.; York, D.; Styles, P.; Blakemore, C.; Hannan, A.J. N-Acetylaspartate and DARPP-32 levels decrease in the corpus striatum of Huntington’s disease mice. Neuroreport 2000, 11, 751–757. [Google Scholar]
- Schilling, G.; Klevytska, A.; Tebbenkamp, A.T.; Juenemann, K.; Cooper, J.; Gonzales, V.; Slunt, H.; Poirer, M.; Ross, C.A.; Borchelt, D.R. Characterization of huntingtin pathologic fragments in human Huntington disease, transgenic mice, and cell models. J. Neuropathol. Exp. Neurol. 2007, 66, 313–320. [Google Scholar] [CrossRef] [PubMed]
- McBride, J.L.; Ramaswamy, S.; Gasmi, M.; Bartus, R.T.; Herzog, C.D.; Brandon, E.P.; Zhou, L.; Pitzer, M.R.; Berry-Kravis, E.M.; Kordower, J.H. Viral delivery of glial cell line-derived neurotrophic factor improves behavior and protects striatal neurons in a mouse model of Huntington’s disease. Proc. Natl. Acad. Sci. USA 2006, 103, 9345–9350. [Google Scholar] [CrossRef] [PubMed]
- Tebbenkamp, A.T.; Green, C.; Xu, G.; Denovan-Wright, E.M.; Rising, A.C.; Fromholt, S.E.; Brown, H.H.; Swing, D.; Mandel, R.J.; Tessarollo, L.; et al. Transgenic mice expressing caspase-6-derived N-terminal fragments of mutant huntingtin develop neurologic abnormalities with predominant cytoplasmic inclusion pathology composed largely of a smaller proteolytic derivative. Hum. Mol. Genet. 2011, 20, 2770–2782. [Google Scholar] [CrossRef] [PubMed]
- Vlamings, R.; Zeef, D.H.; Janssen, M.L.; Oosterloo, M.; Schaper, F.; Jahanshahi, A.; Temel, Y. Lessons learned from the transgenic Huntington’s disease rats. Neural Plast. 2012, 2012, 682712. [Google Scholar] [CrossRef] [PubMed]
- Von Hörsten, S.; Schmitt, I.; Nguyen, H.P.; Holzmann, C.; Schmidt, T.; Walther, T.; Bader, M.; Pabst, R.; Kobbe, P.; Krotova, J.; et al. Transgenic rat model of Huntington’s disease. Hum. Mol. Genet. 2003, 12, 617–624. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.P.; Kobbe, P.; Rahne, H.; Wörpel, T.; Jäger, B.; Stephan, M.; Pabst, R.; Holzmann, C.; Riess, O.; Korr, H.; et al. Behavioral abnormalities precede neuropathological markers in rats transgenic for Huntington’s disease. Hum. Mol. Genet. 2006, 15, 3177–3194. [Google Scholar] [CrossRef] [PubMed]
- Petrasch-Parwez, E.; Nguyen, H.P.; Löbbecke-Schumacher, M.; Habbes, H.W.; Wieczorek, S.; Riess, O.; Andres, K.H.; Dermietzel, R.; Von Hörsten, S. Cellular and subcellular localization of Huntingtin [corrected] aggregates in the brain of a rat transgenic for Huntington disease. J. Comp. Neurol. 2007, 501, 716–730. [Google Scholar] [CrossRef]
- Kirch, R.D.; Meyer, P.T.; Geisler, S.; Braun, F.; Gehrig, S.; Langen, K.J.; von Hörsten, S.; Nikkhah, G.; Cassel, J.C.; Döbrössy, M.D. Early deficits in declarative and procedural memory dependent behavioral function in a transgenic rat model of Huntington’s disease. Behav. Brain Res. 2013, 239, 15–26. [Google Scholar] [CrossRef]
- Kántor, O.; Temel, Y.; Holzmann, C.; Raber, K.; Nguyen, H.P.; Cao, C.; Türkoglu, H.O.; Rutten, B.P.; Visser-Vandewalle, V.; Steinbusch, H.W.; et al. Selective striatal neuron loss and alterations in behavior correlate with impaired striatal function in Huntington’s disease transgenic rats. Neurobiol. Dis. 2006, 22, 538–547. [Google Scholar] [CrossRef]
- Fink, K.D.; Rossignol, J.; Crane, A.T.; Davis, K.K.; Bavar, A.M.; Dekorver, N.W.; Lowrance, S.A.; Reilly, M.P.; Sandstrom, M.I.; von Hörsten, S.; et al. Early cognitive dysfunction in the HD 51 CAG transgenic rat model of Huntington’s disease. Behav. Neurosci. 2012, 126, 479–487. [Google Scholar] [CrossRef]
- Winkler, C.; Gil, J.M.; Araújo, I.M.; Riess, O.; Skripuletz, T.; von Hörsten, S.; Petersén, A. Normal sensitivity to excitotoxicity in a transgenic Huntington’s disease rat. Brain Res. Bull. 2006, 69, 306–310. [Google Scholar] [CrossRef] [PubMed]
- Yu-Taeger, L.; Petrasch-Parwez, E.; Osmand, A.P.; Redensek, A.; Metzger, S.; Clemens, L.E.; Park, L.; Howland, D.; Calaminus, C.; Gu, X.; et al. A novel BACHD transgenic rat exhibits characteristic neuropathological features of Huntington disease. J. Neurosci. 2012, 32, 15426–15438. [Google Scholar] [CrossRef] [PubMed]
- Novati, A.; Manfré, G.; Flunkert, S.; Van der Harst, J.E.; Homberg, J.R.; Wronski, R.; Nguyen, H.P. Validation of behavioral phenotypes in the BACHD rat model. Behav. Brain Res. 2020, 393, 112783. [Google Scholar] [CrossRef]
- Slow, E.J.; van Raamsdonk, J.; Rogers, D.; Coleman, S.H.; Graham, R.K.; Deng, Y.; Oh, R.; Bissada, N.; Hossain, S.M.; Yang, Y.Z.; et al. Selective striatal neuronal loss in a YAC128 mouse model of Huntington disease. Hum. Mol. Genet. 2003, 12, 1555–1567. [Google Scholar] [CrossRef] [PubMed]
- Pouladi, M.A.; Stanek, L.M.; Xie, Y.; Franciosi, S.; Southwell, A.L.; Deng, Y.; Butland, S.; Zhang, W.; Cheng, S.H.; Shihabuddin, L.S.; et al. Marked differences in neurochemistry and aggregates despite similar behavioural and neuropathological features of Huntington disease in the full-length BACHD and YAC128 mice. Hum. Mol. Genet. 2012, 21, 2219–2232. [Google Scholar] [CrossRef] [PubMed]
- Van Raamsdonk, J.M.; Metzler, M.; Slow, E.; Pearson, J.; Schwab, C.; Carroll, J.; Graham, R.K.; Leavitt, B.R.; Hayden, M.R. Phenotypic abnormalities in the YAC128 mouse model of Huntington disease are penetrant on multiple genetic backgrounds and modulated by strain. Neurobiol. Dis. 2007, 26, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Van Raamsdonk, J.M.; Murphy, Z.; Slow, E.J.; Leavitt, B.R.; Hayden, M.R. Selective degeneration and nuclear localization of mutant huntingtin in the YAC128 mouse model of Huntington disease. Hum. Mol. Genet. 2005, 14, 3823–3835. [Google Scholar] [CrossRef]
- Gray, M.; Shirasaki, D.I.; Cepeda, C.; Andre, V.M.; Wilburn, B.; Lu, X.H.; Tao, J.; Yamazaki, I.; Li, S.H.; Sun, Y.E.; et al. Full-length human mutant huntingtin with a stable polyglutamine repeat can elicit progressive and selective neuropathogenesis in BACHD mice. J. Neurosci. 2008, 28, 6182–6195. [Google Scholar] [CrossRef]
- Wegrzynowicz, M.; Bichell, T.J.; Soares, B.D.; Loth, M.K.; McGlothan, J.S.; Mori, S.; Alikhan, F.S.; Hua, K.; Coughlin, J.M.; Holt, H.K.; et al. Novel BAC mouse model of Huntington’s disease with 225 CAG repeats exhibits an early widespread and stable degenerative phenotype. J. Huntingt. Dis. 2015, 4, 17–36. [Google Scholar] [CrossRef]
- Chen, X.; Wu, J.; Lvovskaya, S.; Herndon, E.; Supnet, C.; Bezprozvanny, I. Dantrolene is neuroprotective in Huntington’s disease transgenic mouse model. Mol. Neurodegener. 2011, 6, 81. [Google Scholar] [CrossRef]
- Mantovani, S.; Gordon, R.; Li, R.; Christie, D.C.; Kumar, V.; Woodruff, T.M. Motor deficits associated with Huntington’s disease occur in the absence of striatal degeneration in BACHD transgenic mice. Hum. Mol. Genet. 2016, 25, 1780–1791. [Google Scholar] [CrossRef] [PubMed]
- Lichter, D.G.; Hershey, L.A. Before chorea: Pre-Huntington mild cognitive impairment. Neurology 2010, 75, 490–491. [Google Scholar] [CrossRef] [PubMed]
- Giralt, A.; Saavedra, A.; Alberch, J.; Pérez-Navarro, E. Cognitive Dysfunction in Huntington’s Disease: Humans, Mouse Models and Molecular Mechanisms. J. Huntingt. Dis. 2012, 1, 155–173. [Google Scholar] [CrossRef]
- Southwell, A.L.; Ko, J.; Patterson, P.H. Intrabody gene therapy ameliorates motor, cognitive, and neuropathological symptoms in multiple mouse models of Huntington’sdisease. J. Neurosci. 2009, 29, 13589–13602. [Google Scholar] [CrossRef] [PubMed]
- Doria, J.G.; Silva, F.R.; de Souza, J.M.; Vieira, L.B.; Carvalho, T.G.; Reis, H.J.; Pereira, G.S.; Dobransky, T.; Ribeiro, F.M. Metabotropic glutamate receptor 5 positive allosteric modulators are neuroprotective in a mouse model of Huntington’s disease. Br. J. Pharmacol. 2013, 169, 909–921. [Google Scholar] [CrossRef] [PubMed]
- Aziz, N.A.; van der Burg, J.M.; Landwehrmeyer, G.B.; Brundin, P.; Stijnen, T.; EHDI Study Group; Roos, R.A. Weight loss in Huntington disease increases with higher CAG repeat number. Neurology 2008, 1, 1506–1513. [Google Scholar] [CrossRef] [PubMed]
- Van Raamsdonk, J.M.; Pearson, J.; Rogers, D.A.; Bissada, N.; Vogl, A.W.; Hayden, M.R.; Leavitt, B.R. Loss of wild-type huntingtin influences motor dysfunction and survival in the YAC128 mouse model of Huntington disease. Hum. Mol. Genet. 2005, 14, 1379–1392. [Google Scholar] [CrossRef]
- Van Raamsdonk, J.M.; Pearson, J.; Slow, E.J.; Hossain, S.M.; Leavitt, B.R.; Hayden, M.R. Cognitive dysfunction precedes neuropathology and motor abnormalities in the YAC128 mouse model of Huntington’s disease. J. Neurosci. 2005, 25, 4169–4180. [Google Scholar] [CrossRef]
- Abada, Y.S.; Nguyen, H.P.; Schreiber, R.; Ellenbroek, B. Assessment of motor function, sensory motor gating and recognition memory in a novel BACHD transgenic rat model for huntington disease. PLoS ONE 2013, 8, e68584. [Google Scholar] [CrossRef]
- Aharony, I.; Ehrnhoefer, D.E.; Shruster, A.; Qiu, X.; Franciosi, S.; Hayden, M.R.; Offen, D. A Huntingtin-based peptide inhibitor of caspase-6 provides protection from mutant Huntingtin-induced motor and behavioral deficits. Hum. Mol. Genet. 2015, 24, 2604–2614. [Google Scholar] [CrossRef]
- Brooks, S.P.; Janghra, N.; Higgs, G.V.; Bayram-Weston, Z.; Heuer, A.; Jones, L.; Dunnett, S.B. Selective cognitive impairment in the YAC128 Huntington’s disease mouse. Brain Res. Bull. 2012, 88, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Shenoy, S.A.; Zheng, S.; Liu, W.; Dai, Y.; Liu, Y.; Hou, Z.; Mori, S.; Tang, Y.; Cheng, J.; Duan, W.; et al. A novel and accurate full-length HTT mouse model for Huntington’s disease. Elife 2022, 11, e70217. [Google Scholar] [CrossRef] [PubMed]
- Adjeroud, N.; Yagüe, S.; Yu-Taeger, L.; Bozon, B.; Leblanc-Veyrac, P.; Riess, O.; Allain, P.; Nguyen, H.P.; Doyère, V.; El Massioui, N. Reduced impact of emotion on choice behavior in presymptomatic BACHD rats, a transgenic rodent model for Huntington Disease. Neurobiol. Learn. Mem. 2015, 125, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Yu-Taeger, L.; Bonin, M.; Stricker-Shaver, J.; Riess, O.; Nguyen, H.H.P. Dysregulation of gene expression in the striatum of BACHD rats expressing full-length mutant huntingtin and associated abnormalities on molecular and protein levels. Neuropharmacology 2017, 117, 260–272. [Google Scholar] [CrossRef] [PubMed]
- Clemens, L.E.; Weber, J.J.; Wlodkowski, T.T.; Yu-Taeger, L.; Michaud, M.; Calaminus, C.; Eckert, S.H.; Gaca, J.; Weiss, A.; Magg, J.C.; et al. Olesoxime suppresses calpain activation and mutant huntingtin fragmentation in the BACHD rat. Brain 2015, 138, 3632–3653. [Google Scholar] [CrossRef] [PubMed]
- Abada, Y.S.; Schreiber, R.; Ellenbroek, B. Motor, emotional and cognitive deficits in adult BACHD mice: A model for Huntington’s disease. Behav. Brain Res. 2013, 238, 243–251. [Google Scholar] [CrossRef] [PubMed]
- Manfré, G.; Doyère, V.; Bossi, S.; Riess, O.; Nguyen, H.P.; El Massioui, N. Impulsivity trait in the early symptomatic BACHD transgenic rat model of Huntington disease. Behav. Brain Res. 2016, 299, 6–10. [Google Scholar] [CrossRef]
- Woodman, B.; Butler, R.; Landles, C.; Lupton, M.K.; Tse, J.; Hockly, E.; Moffitt, H.; Sathasivam, K.; Bates, G.P. The Hdh (Q150/Q150) knock-in mouse model of HD and the R6/2 exon 1 model develop comparable and widespread molecular phenotypes. Brain Res. Bull. 2007, 72, 83–97. [Google Scholar] [CrossRef]
- Morton, A.J.; Glynn, D.; Leavens, W.; Zheng, Z.; Faull, R.L.; Skepper, J.N.; Wight, J.M. Paradoxical delay in the onset of disease caused by super-long CAG repeat expansions in R6/2 mice. Neurobiol. Dis. 2009, 33, 331–341. [Google Scholar] [CrossRef]
- Ciamei, A.; Detloff, P.J.; Morton, A.J. Progression of behavioural despair in R6/2 and Hdh knock-in mouse models recapitulates depression in Huntington’s disease. Behav. Brain Res. 2015, 291, 140–146. [Google Scholar] [CrossRef]
- Brooks’, S.; Higgs, G.; Janghra, N.; Jones, L.; Dunnett, S.B. Longitudinal analysis of the behavioural phenotype in YAC128 (C57BL/6J) Huntington’s disease transgenic mice. Brain Res. Bull. 2012, 88, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Lunkes, A.; Lindenberg, K.S.; Ben-Haïem, L.; Weber, C.; Devys, D.; Landwehrmeyer, G.B.; Mandel, J.L.; Trottier, Y. Proteases acting on mutant huntingtin generate cleaved products that differentially build up cytoplasmic and nuclear inclusions. Mol. Cell 2002, 10, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Yanai, S.; Endo, S. Functional Aging in Male C57BL/6J Mice Across the Lifespan: A Systematic Behavioral Analysis of Motor, Emotional, and Memory Function to Define an Aging Phenotype. Front. Aging Neurosci. 2021, 13, 697621. [Google Scholar] [CrossRef] [PubMed]
- Sakakibara, Y.; Sekiya, M.; Saito, T.; Saido, T.C.; Iijima, K.M. Cognitive and emotional alterations in App knock-in mouse models of Aβ amyloidosis. BMC Neurosci. 2018, 19, 46. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.H.; Tallaksen-Greene, S.; Chien, W.M.; Cearley, J.A.; Jackson, W.S.; Crouse, A.B.; Ren, S.; Li, X.J.; Albin, R.L.; Detloff, P.J. Neurological abnormalities in a knock-in mouse model of Huntington’s disease. Hum. Mol. Genet. 2001, 10, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Heng, M.Y.; Duong, D.K.; Albin, R.L.; Tallaksen-Greene, S.J.; Hunter, J.M.; Lesort, M.J.; Osmand, A.; Paulson, H.L.; Detloff, P.J. Early autophagic response in a novel knock-in model of Huntington disease. Hum. Mol. Genet. 2010, 19, 3702–3720. [Google Scholar] [CrossRef]
- Jin, J.; Peng, Q.; Hou, Z.; Jiang, M.; Wang, X.; Langseth, A.J.; Tao, M.; Barker, P.B.; Mori, S.; Bergles, D.E.; et al. Early white matter abnormalities, progressive brain pathology and motor deficits in a novel knock-in mouse model of Huntington’s disease. Hum. Mol. Genet. 2015, 24, 2508–2527. [Google Scholar] [CrossRef]
- Wheeler, V.C.; Auerbach, W.; White, J.K.; Srinidhi, J.; Auerbach, A.; Ryan, A.; Duyao, M.P.; Vrbanac, V.; Weaver, M.; Gusella, J.F.; et al. Length-dependent gametic CAG repeat instability in the Huntington’s disease knock-in mouse. Hum. Mol. Genet. 1999, 8, 115–122. [Google Scholar] [CrossRef]
- Menalled, L.B.; Sison, J.D.; Dragatsis, I.; Zeitlin, S.; Chesselet, M.F. Time course of early motor and neuropathological anomalies in a knock-in mouse model of Huntington’s disease with 140 CAG repeats. J. Comp. Neurol. 2003, 465, 11–26. [Google Scholar] [CrossRef]
- Menalled, L.B.; Kudwa, A.E.; Miller, S.; Fitzpatrick, J.; Watson-Johnson, J.; Keating, N.; Ruiz, M.; Mushlin, R.; Alosio, W.; McConnell, K.; et al. Comprehensive behavioral and molecular characterization of a new knock-in mouse model of Huntington’s disease: zQ175. PLoS ONE 2012, 7, e49838. [Google Scholar] [CrossRef]
- Stricker-Shaver, J.; Novati, A.; Yu-Taeger, L.; Nguyen, H.P. Genetic Rodent Models of Huntington Disease. Adv. Exp. Med. Biol. 2018, 1049, 29–57. [Google Scholar] [PubMed]
- Heikkinen, T.; Lehtimäki, K.; Vartiainen, N.; Puoliväli, J.; Hendricks, S.J.; Glaser, J.R.; Bradaia, A.; Wadel, K.; Touller, C.; Kontkanen, O.; et al. Characterization of neurophysiological and behavioral changes, MRI brain volumetry and 1H MRS in zQ175 knock-in mouse model of Huntington’s disease. PLoS ONE 2012, 7, e50717. [Google Scholar] [CrossRef] [PubMed]
- Menalled, L.; Lutz, C.; Ramboz, S.; Brunner, D.; Lager, B.; Noble, S.; Park, L.; Howland, D. A Field Guide to Working with Mouse Models of Huntington’s Disease; Psychogenics Inc., The Jackson Laboratory, CHDI Foundation: New York, NY, USA, 2014. [Google Scholar]
- Rattray, I.; Smith, E.J.; Crum, W.R.; Walker, T.A.; Gale, R.; Bates, G.P.; Modo, M. Correlations of Behavioral Deficits with Brain Pathology Assessed through Longitudinal MRI, and Histopathology in the HdhQ150/Q150 Mouse Model of Huntington’s Disease. PLoS ONE 2017, 12, e0168556. [Google Scholar] [CrossRef] [PubMed]
- Bayram-Weston, Z.; Torres, E.M.; Jones, L.; Dunnett, S.B.; Brooks, S.P. Light and electron microscopic characterization of the evolution of cellular pathology in the Hdh(CAG)150 Huntington’s disease knock-in mouse. Brain Res. Bull. 2012, 88, 189–198. [Google Scholar] [CrossRef] [PubMed]
- Orvoen, S.; Pla, P.; Gardier, A.M.; Saudou, F.; David, D.J. Huntington’s disease knock-in male mice show specific anxiety-like behaviour and altered neuronal maturation. Neurosci. Lett. 2012, 507, 127–132. [Google Scholar] [CrossRef] [PubMed]
- Hickey, M.A.; Kosmalska, A.; Enayati, J.; Cohen, R.; Zeitlin, S.; Levine, M.S.; Chesselet, M.F. Extensive early motor and non-motor behavioral deficits are followed by striatal neuronal loss in knock-in Huntington’s disease mice. Neuroscience 2008, 157, 280–295. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, V.C.; Gutekunst, C.A.; Vrbanac, V.; Lebel, L.A.; Schilling, G.; Hersch, S.; Friedlander, R.M.; Gusella, J.F.; Vonsattel, J.P.; Borchelt, D.R.; et al. Early phenotypes that presage late-onset neurodegenerative disease allow testing of modifiers in Hdh CAG knock-in mice. Hum. Mol. Genet. 2002, 11, 633–640. [Google Scholar] [CrossRef]
- Smith, G.A.; Rocha, E.M.; McLean, J.R.; Hayes, M.A.; Izen, S.C.; Isacson, O.; Hallett, P.J. Progressive axonal transport and synaptic protein changes correlate with behavioral and neuropathological abnormalities in the heterozygous Q175 KI mouse model of Huntington’s disease. Hum. Mol. Genet. 2014, 23, 4510–4527. [Google Scholar] [CrossRef]
- Heng, M.Y.; Tallaksen-Greene, S.J.; Detloff, P.J.; Albin, R.L. Longitudinal evaluation of the Hdh(CAG)150 knock-in murine model of Huntington’s disease. J. Neurosci. 2007, 27, 8989–8998. [Google Scholar] [CrossRef]
- Trueman, R.C.; Brooks, S.P.; Jones, L.; Dunnett, S.B. Time course of choice reaction time deficits in the Hdh(Q92) knock-in mouse model of Huntington’s disease in the operant serial implicit learning task (SILT). Behav. Brain Res. 2008, 189, 317–324. [Google Scholar] [CrossRef]
- Heng, M.Y.; Detloff, P.J.; Paulson, H.L.; Albin, R.L. Early alterations of autophagy in Huntington disease-like mice. Autophagy 2010, 6, 1206–1208. [Google Scholar] [CrossRef] [PubMed]
- Li, X.J.; Li, S. Large Animal Models of Huntington’s Disease. Curr. Top. Behav. Neurosci. 2015, 22, 149–160. [Google Scholar] [PubMed]
- Yang, S.H.; Cheng, P.H.; Banta, H.; Piotrowska-Nitsche, K.; Yang, J.J.; Cheng, E.C.; Snyder, B.; Larkin, K.; Liu, J.; Orkin, J.; et al. Towards a transgenic model of Huntington’s disease in a non-human primate. Nature 2008, 453, 921–924. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, E.; Suemizu, H.; Shimada, A.; Hanazawa, K.; Oiwa, R.; Kamioka, M.; Tomioka, I.; Sotomaru, Y.; Hirakawa, R.; Eto, T.; et al. Generation of transgenic non-human primates with germline transmission. Nature 2009, 459, 523–527. [Google Scholar] [CrossRef] [PubMed]
- Niu, Y.; Yu, Y.; Bernat, A.; Yang, S.; He, X.; Guo, X.; Chen, D.; Chen, Y.; Ji, S.; Si, W.; et al. Transgenic rhesus monkeys produced by gene transfer into early-cleavage-stage embryos using a simian immunodeficiency virus-based vector. Proc. Natl. Acad. Sci. USA 2010, 107, 17663–17667. [Google Scholar] [CrossRef] [PubMed]
- Lai, L.; Prather, R.S. Production of cloned pigs by using somatic cells as donors. Cloning Stem Cells 2003, 5, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Shen, B.; Zhang, J.; Wu, H.; Wang, J.; Ma, K.; Li, Z.; Zhang, X.; Zhang, P.; Huang, X. Generation of gene-modified mice via Cas9/RNA-mediated gene targeting. Cell Res. 2013, 23, 720–723. [Google Scholar] [CrossRef]
- Wang, H.; Yang, H.; Shivalila, C.S.; Dawlaty, M.M.; Cheng, A.W.; Zhang, F.; Jaenisch, R. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell 2013, 153, 910–918. [Google Scholar] [CrossRef]
- Southwell, A.L.; Warby, S.C.; Carroll, J.B.; Doty, C.N.; Skotte, N.H.; Zhang, W.; Villanueva, E.B.; Kovalik, V.; Xie, Y.; Pouladi, M.A.; et al. A fully humanized transgenic mouse model of Huntington disease. Hum. Mol. Genet. 2013, 22, 18–34. [Google Scholar] [CrossRef]
- Lawrence, A.D.; Sahakian, B.J.; Hodges, J.R.; Rosser, A.E.; Lange, K.W.; Robbins, T.W. Executive and mnemonic functions in early Huntington’s disease. Brain 1996, 119, 1633–1645. [Google Scholar] [CrossRef]


| Knock-In Mouse Model | ||||||
|---|---|---|---|---|---|---|
| Animal Model | Strain | CAG Repeat Length | Life Span | Behavioral and Neuronal Alterations | References | |
| Well characterized congenic. lines of C57BL/6 and CBA | CAG 140 | C57BL/6J | 140 | Normal life span | Hyperactivity in 1st month and hypoactivity at 4 months. Vertical pole, non-accelerating rotarod, running wheels—sensorimotor performance deficit (4 months), irregularities in gait (12 months). Anxiety-like phenotype, no depressive behavior, long-term memory recognition impairment. Nuclear and neuropil inclusions in the striatum, cortex, hippocampus, and cerebellum at the age of 2 months. Loss of neurons (23 months). At 20–26 months, the corpus callosum volume is reduced. | [48,114] |
| ZQ 175 | C57BL/6J | 198 | Reduced life Expectancy in (19 months) | Progressive reduction in open field (2 months), grip strength (1 month homozygous), phenotype (4 months homozygous), climbing activity (8 months homozygous), cylinder test (1 month heterozygous), nesting (heterozygous, 16 months). Two options for procedural learning loss Test swimming ability (homozygous, 10 months). Impairment of executive function test (7 months). Two-choice visual discrimination test for cognitive flexibility (7 months). Nuclear inclusions with a slowed circadian cycle (heterozygous). Atrophy of the striatum. Cortical thinning is a condition in which the cortex thins out. Reduced amounts of dopamine and BDNF | [104,106] | |
| HDhQ 92 | C57BL/6J | 92 | Normal life span | Testing for depressive-like phenotype in women with splash tests and forced swim tests. Open field test for anxiety-like phenotypes (male). Deficit in olfactory function due to an anxio-depressive-like phenotype. Discrimination in the workplace (males). The rotarod of motor learning has been altered. Memory impairment for long-term object recognition (4 months). Impairment of spatial memory (8 months). Working memory and reversal learning deficits non-matching to position tasks and delayed matching to position tasks (8 months). There is no evidence of striatal degeneration. At 4.5 months, Huntingtin protein translocation to nucleus and appears punctate. Striatal gliosis and gait problems at 24 months of age, no overt symptoms. There were no alterations in spontaneous locomotion that could be detected. Inclusions in mHtt (12 months). Loss of neurons (24 months). There is no striatal atrophy. | [115] | |
| HDhQ 111 | 111 | |||||
| HDhQ 20 | 20 | |||||
| HDhQ 50 | 50 | |||||
| HDhQ 80 | 80 | |||||
| Inconsistent CAG repeat transmission | HDhQ 50 | C57BL/6J | 50 | Normal life span | Learning problems in the spatial and reverse directions in the water maze with three stages (4 and 8 months, respectively). Performance issues with extra-dimensional shifts (6 months). Reduced sensitivity to startling stimuli (6 months homozygous). Balance beam performance is impaired (homozygous for 4 months, heterozygous for 12.5 months). Grip strength test with decreased muscular strength (20 months), irregularities in gait (15 months). On the rotarod, no impairment was seen. Water mazes with impaired spatial and reversal learning (4 and 8 months, respectively). Impaired performance in extra-dimensional shifts (6 months). Reduced sensitivity to startling stimuli (6 months homozygous). Intranuclear inclusions are a type of intranuclear inclusion (13 months). Gliosis (14 months, heterozygous). Loss of neurons (12.5 months). Atrophy of the striatum (12.5 months). Reduced D1 and D2 receptor binding (only homozygous). Two types of striatal and cortical astrogliosis (20 months). Abnormalities of the cerebellum Purkinje neurons. | [99,111,116] |
| HDhQ 100 | 100 | |||||
| HDhQ 150 | 150 | |||||
| HDhQ 200 | 200 | |||||
| HDhQ 250 | 250 | |||||
| HDhQ 315 | 315 | |||||
| HDhQ 365 | 365 | |||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nittari, G.; Roy, P.; Martinelli, I.; Bellitto, V.; Tomassoni, D.; Traini, E.; Tayebati, S.K.; Amenta, F. Rodent Models of Huntington’s Disease: An Overview. Biomedicines 2023, 11, 3331. https://doi.org/10.3390/biomedicines11123331
Nittari G, Roy P, Martinelli I, Bellitto V, Tomassoni D, Traini E, Tayebati SK, Amenta F. Rodent Models of Huntington’s Disease: An Overview. Biomedicines. 2023; 11(12):3331. https://doi.org/10.3390/biomedicines11123331
Chicago/Turabian StyleNittari, Giulio, Proshanta Roy, Ilenia Martinelli, Vincenzo Bellitto, Daniele Tomassoni, Enea Traini, Seyed Khosrow Tayebati, and Francesco Amenta. 2023. "Rodent Models of Huntington’s Disease: An Overview" Biomedicines 11, no. 12: 3331. https://doi.org/10.3390/biomedicines11123331
APA StyleNittari, G., Roy, P., Martinelli, I., Bellitto, V., Tomassoni, D., Traini, E., Tayebati, S. K., & Amenta, F. (2023). Rodent Models of Huntington’s Disease: An Overview. Biomedicines, 11(12), 3331. https://doi.org/10.3390/biomedicines11123331